Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes


Koltsov Institute of Developmental Biology, Russian Academy of Sciences, 119334 Moscow, Russia
*
Author to whom correspondence should be addressed.
†
The authors contributed equally to the manuscript.
Int. J. Mol. Sci. 2020, 21(5), 1602; https://doi.org/10.3390/ijms21051602
Submission received: 31 January 2020
/
Revised: 21 February 2020
/
Accepted: 24 February 2020
/
Published: 26 February 2020
(This article belongs to the Special Issue Rare Diseases: Molecular Mechanisms and Therapeutic Strategies (II))
Abstract
:Retinal development is under the coordinated control of overlapping networks of signaling pathways and transcription factors. The paper was conceived as a review of the data and ideas that have been formed to date on homeobox genes mutations that lead to the disruption of eye organogenesis and result in inherited eye/retinal diseases. Many of these diseases are part of the same clinical spectrum and have high genetic heterogeneity with already identified associated genes. We summarize the known key regulators of eye development, with a focus on the homeobox genes associated with monogenic eye diseases showing retinal manifestations. Recent advances in the field of genetics and high-throughput next-generation sequencing technologies, including single-cell transcriptome analysis have allowed for deepening of knowledge of the genetic basis of inherited retinal diseases (IRDs), as well as improve their diagnostics. We highlight some promising avenues of research involving molecular-genetic and cell-technology approaches that can be effective for IRDs therapy. The most promising neuroprotective strategies are aimed at mobilizing the endogenous cellular reserve of the retina.
1. Introduction
The development of the human eye is controlled by a morphogenetic process that requires precise spatial and temporal gene regulation [1,2]. Perturbation of early eye organogenesis due to genetic factors can result in halting of eye development or multiple eye tissues disorders, and among them degenerations of the retina occupies a special place [3,4,5]. Inherited eye diseases make up a clinically and genetically heterogeneous group of diseases and mutations in which over 260 genes have been proven to be causative. These genes include functionally heterogeneous groups [6]. This review highlights the role of the main homeobox genes associated with inherited eye diseases showing retinal manifestations. Mutations of these genes leading to vision loss in humans have been identified by genetic screenings. Homeobox genes from different classes include retina-specific regulatory genes accepted as critical for eye field specification and retinal cells type differentiation by a broad array of loss- or gain-of-function models. Among these genes are some that are known to cause inherited retinal diseases (IRDs) that disturb the development, function, and survival of rod and cone photoreceptors, ganglion cells, or retinal pigment epithelial cells [4,6,7,8]. In this review, we focused on IRDs associated with single homeobox gene malfunctions as a result of mutations. Mutations in a number of homeobox genes under consideration can manifest themselves the in retina as secondary effects due to impaired functioning of the other eye tissues. It is obvious that an integrated approach should keep in mind the multigenic and systemic nature of a number of retinal/eye diseases to chart the way for appropriate personalized genes and cells therapies technologies and pharmacologic neuroprotection [9,10,11]. We discuss the advantages and disadvantages of modern molecular genetic and cellular approaches, including those that show the most promise for the treatment of a number of retinal neurodegenerative diseases in some of the most striking cases.
2. Retinal Organization
The general plan of the retinal architecture is similar across all vertebrates and humans, despite the morphological and functional peculiarities [12]. The retina is composed of two parts: The single-layered retinal pigment epithelium (RPE) on the posterior side and the neuroretina on the anterior side of the eye. The neuroretina is a highly organized multilayered tissue that includes interconnecting layers of specialized cells: Six main types of neurons (photoreceptors, bipolar cells, horizontal cells, amacrine cells, ganglion cells, and interplexiform neurons) and four types of radial glia cells (Muller cells, astrocytes, microglia, and oligodendrocytes) (Figure 1).
Radial glia of the retina line the bottom and lateral surface of the optic cup, forming the radial layers [14,15]. Three nuclear layers, consisting of different types of sensory neurons, and two plexiform layers (outer and inner) representing synaptic connections between retinal neurons of the border nuclear layer, are distinguished in the retina. The outer nuclear layer (ONL) of the retina includes light-sensitive cells (rods and cones of photoreceptors). The outer segments of photoreceptors are in close interaction with the RPE (single row layer of intensely pigmented epithelial cells). RPE cells are located between the photoreceptors and the choroid and perform a number of physiological functions: Protection of photoreceptors from excess light, transduction of visual signal, retinal homeostasis (growth factor secretion, regulation of ion balance in subretinal space), and phagocytosis of exfoliated discs of outer segments of photoreceptors [16,17]. The RPE is underlain by Bruch’s membrane which consists of the components of the choroid endothelium and the fibrillar layer of the RPE basal plate [18]. The second (inner) nuclear retinal layer (INL) is formed by interneurons: bipolars and amacrine and horizontal cells. Bipolars participate in transmission of visual signal from the photoreceptors into the ganglion, and horizontal and amacrine cells connect all cells of the retina [15]. Ganglion cells are the neurons of the second order, forming the third ganglion nuclear layer. Their axons take part in the formation of the optic nerve [19]. The outer plexiform layer (OPL) is formed by synaptic contacts between rods/cones and bipolar cells, while the inner plexiform layer provides a connection between bipolar and ganglion neurons, as well as a horizontal connection between amacrine and horizontal neurons [20,21]. The INL is where the bodies of Muller glia cells are localized. These retinal macroglia permeates the entire retina from external to internal border basal membranes (formed by outgrowths of these cells) localized on the border of plexiform tissue layers. Muller glia performs structural and neurotrophic functions in the retina [22]. Functions of the other elements of retinal glia, such as astrocytes, are associated with maintaining the structure and metabolic activity of retinal neurons. Astrocytes can secrete vasoactive substances that regulate retinal vascular tone [23]. Retinal microglia present as a resident population of immunocompetent cells [24]. The retina blood supply comes from the central retinal artery feeding the inner retinal layers and choriocapillaries for the outer layers: photoreceptor layer, ONL, and OPL [25]. Retinal neurons, macroglia, and microglia, as well as the wall cells of the microvessels (endotheliocytes and pericytes) interact with each other and form a blood–retinal barrier that regulates the supply of oxygen and trophic factors to retinal neurons and is involved in recycling of metabolic products [26,27,28].
3. Homeobox Transcription Factors Expressed in Retina
Retinal development in vertebrate embryogenesis is under strict spatial and temporal regulation by the overlapping gene networks [1,29]. Studies on morphological and molecular characteristics of the eye tissues at different stages of human ontogenesis were conducted many years ago and have contributed to the accumulation of information about various aspects of homeobox genes expression [30,31,32,33,34,35].
Homeobox transcription factors, an evolutionarily conserved class among the transcription factors, are key regulators of developmental processes such as regional specification, cells migration, and differentiation and morphogenesis of the tissues and organs, by regulating the expression of specific sets of target genes. The developing retina is marked by distinct boundaries of homeobox gene expression at different developmental time points (Figure 1). During retinal development the homeobox genes play multiple roles such as regulation of patterning of the retina along the dorsoventral and nasotemporal axes. These genes are essential for the control of proliferation and the choice of cells fate, the order of differentiation of specific neuronal and glial subtypes through instruction signals from surrounding tissues, and retinal cells survival [36,37,38]. Transcription factors expressed in the retina belong to three major groups: basic helix-loop-helix (bHLH), forkhead box (FOX) and homeobox genes [6,39,40,41]. Among them are the eye field transcription factors (Rx1, Pax6, Otx2, Optx2, Six3, Chx10, Prox1, Dlx1-2, Pitx1-2, etc.) that are activated in the retina at the early stage of embryogenesis and control retinal cell type specialization [42,43,44].
Homeobox transcription factors control the expression of numerous classes of target genes. It is known that, as retinal differentiation proceeds, Atoh7, through activation of Pou4f2 and Isl1, leads to the differentiation of ganglion cells; Foxn4, via Neurod1, Neurod4, and Ptf1a, provides the fate of amacrine cells; Rax2 and Otx2 are responsible for the photoreceptor phenotype, and Vsx2 and Ascl1 for the appearance of bipolar cells, but Muller glia arises as a result of overexpression of Hes1, Hes5, and Hey2. A number of reviews considered the interactions between transcription factors to determine the phenotypes of neurons and glia of the retina as the cells are specializing [43]. Homeobox-containing genes continue to be expressed in adult eye tissues, the retina in particular, ensuring eye homeostasis and supporting axon function [4,45]. For example, PAX6 remains distinctly expressed throughout the lifespan of the human retina, suggesting a role for PAX6 in the retina after the completion of eye morphogenesis [46]. The role of homeobox genes, such as Prox1, as tumor suppressors is well known [47].
The general order of retinal cells differentiation in vertebrates is conserved [48], which makes it possible to model human retinal diseases in experimental animals. Despite the similarities, there are peculiar properties in the retinas of evolutionarily distant species due to the heterochronicity of cellular and molecular processes. These specific features are associated with the length of embryogenesis, adaptation to environmental conditions, the variety of neurons among the general retinal cell types, the expression patterns of key regulatory genes and their isoforms [49,50,51]. These features lead to limitations in modeling human eye/retinal diseases in animal models. Some genes have important differences in temporal expression, such as CRX [4,52,53,54]. It is obvious that these differences are associated with species-specific regulatory strategies in genetic programs.
Differences in gene expression levels, including variation in expression depending on age, gender, eye laterality, gene function, and age-by-gender interactions, have been characterized by custom human retinal microarray analysis. These factors contribute significantly to phenotype variation across normal adult retinas. The greater expression variability of the many key genes for retinal function (including photoreceptor-specific genes) may be partially explained by the dynamics of the vision process. Findings show that a significant fraction of gene expression variation in the normal human retina is attributable to identifiable biological factors. Such diversity may result in different levels of disease susceptibility, which is why exploring its sources may provide insights into the pathogenesis of retinal disease [55].
There are homeobox genes specific to the retina (CRX, VAX, etc.) and others that are widely expressed not only in retina (PAX, SOX, OTX2, etc.), but also in many other organs. Mutations of specific retinal genes are direct and cause primary retinal disorders [52,56,57,58]. The etiologies of many congenital diseases are multigenic in nature. The difficulties in characterizing and diagnosing specific retinal pathologies demonstrate that a number of mutations do not correlate with phenotypic manifestations and many have a pleiotropic effect. Mutations of the genes common to many tissues and organs can often cause the development of secondary eye pathologies (diabetic retinopathy, retinal degeneration in glaucoma, etc.). One well-known example is mutation of the homeobox gene Prox1, which causes pathology of the pancreas, and retinal degeneration occurs as a secondary effect [59]. Thus, mutations of different genes can lead to pathological changes in the retina characterized by similar phenotypic traits (age-related macular degeneration (AMD), glaucoma, retinitis pigmentosa (RP), etc). Recent evidence shows that the pathogenesis of anophthalmia and microphthalmia share several molecular factors [35]. Pathologies of other parts of the eye often affect the retina. A more advanced level of high myopia can lead to complications such as glaucoma, cataracts, and retinal detachment. Patients with pathological myopia are at increased risk of retinal detachment due to axial elongation of the globe and peripheral retinal thinning [60].
There has been significant progress in identifying the key genes associated with IRDs and other inherited eye disorders. In the review we mainly paid attention to IRDs associated with single homeobox gene malfunctions as a result of mutations. Disruption of key regulatory genes in the early stages of embryogenesis can result, in the ocular system, in halted eye formation, resulting in various pathological phenotypes of the eye tissues. Distinct clinical manifestations are associated with certain IRDs, but often the diagnosis is complicated by the fact that many genes give rise to more than one disorder [4,6]. In humans, mutations of homeobox genes expressed in the retina have been shown to result in multiple disorders. More than 260 homeobox genes are currently linked to human retinal diseases. This may reflect the essential functions of these homeobox genes throughout embryogenesis or the degree of functional redundancy during retinal development. This is illustrated in Table 1, which includes basic structural properties of reviewed homeobox genes and Table 2, which includes the names all the homeobox genes implicated in the most common IRDs (including some inherited eye diseases with retinal manifestations) and highlights the genes that are shared between different disorders.
It should be noted that mutations in many genes can be both dominant and recessive. In addition, many of these disorders are part of the same clinical spectrum and have high genetic heterogeneity with both identified and as yet unidentified associated genes [7].
3.1. Adnp
The activity-dependent neuroprotector (ADNP) gene family belongs to the zinc finger (ZF) homeobox gene class and includes two genes with significant homologies in human and mouse genomes (ADNP1/Adnp1, ADNP2/Adnp2). The human ADNP gene contains five exons of which the last three are translated. The predicted protein has nine zinc finger motifs, a proline-rich region, a bipartite nuclear localization signal, a partial homeobox domain, a glutaredoxin active site, and a leucine-rich nuclear export sequence [61].
ADNP is essential for embryonic brain development [62]. Its expression was demonstrated by RT PCR in adult rat retina [63]. One of the most important cellular processes associated with ADNP is autophagy. Secreted octapeptide NAP, which is derived from ADNP, was found to enhance autophagy, while protecting microtubules [64]. NAP protects retinal ganglion cells against damage induced by retinal ischemia and optic nerve crush [65]. Its neuroprotective actions seem to be mediated by the activation of mitogen-activated protein kinase/extracellular signal-regulated protein kinase (MAPK/ERK) and phosphatidylinositol-3-kinase (PI-3K)/AKT signaling [66].
A set of heterozygous truncating mutations in the ADNP gene was identified in patients with Helsmoortel–Van der Aa syndrome (HVDAS). They were characterized by intellectual disability with global developmental delay, gastrointestinal problems, structural brain anomalies, visual problems, and early tooth eruption. ADNP is classified as one of three most prevalent autism-causing genes [67]. Ophthalmic findings were remarkable for progressive nystagmus, macular pigment mottling, mild foveal hypoplasia with abnormal macular laminations, persistent rod dysfunction with electronegative waveform, and progressive cone degeneration [68]. HVDAS with ocular anomalies is associated with specific mutations clustering within the “bipartite nuclear localization signal” [69].
Adnp knockout in mice resulted in cranial neural tube closure failure and death at the time of neural tube closure (E8.5–9.5) [70]. The Adnp haploinsufficient mouse mimics the human ADNP syndrome in terms of synaptic density and gene expression patterns. Daily intranasal treatments with NAP showed significant effects on hippocampal and cerebral cortical expression of the presynaptic Slc17a7 gene encoding vesicular excitatory glutamate transporter 1 (VGLUT1) [71]. The same effect would be expected for photoreceptor cells that express a splicing variant of VGLUT1 associated with synaptic vesicles [72].
3.2. Alx Gene Family
The Alx gene family belongs to the PRD homeobox gene class and comprises three genes in human and mouse genomes (ALX1/Alx1, ALX2/Alx3, ALX3/Alx4). ALX1 and ALX3 genes contain four exons each, and encode proteins have a homeodomain and an OAR domain [73].
Vertebrate Alx genes are expressed during embryogenesis in forebrain mesenchyme, cranial arches, limb buds, and cartilage [74]. Mouse Alx genes show similar developmental expression patterns in the cranial regions of the embryo. From the early stages (E8.5–E9.5), they are markedly expressed in frontonasal head mesenchyme, and later in the first and second pharyngeal arches [74,75]. In the anterior part of the head expression of Alx1 is first detected in the optic vesicles and restricted to presumptive neural retina in zebrafish embryos. After the completion of retinal differentiation, its expression is restricted to germinal cells at the margin of the retina [76].
In humans, mutations in each of the ALX genes have been reported to be associated with congenital craniofacial malformation. There are at least three subtypes of frontonasal dysplasia in humans that are distinguished by their genetic causes (Table 2).
Mutations of ALX1 gene lead to frontonasal dysplasia type 3, including eyes that are missing (anophthalmia) or very small (microphthalmia) and low-set ears that are rotated backward [78]. In contrast, mutations of ALX3 and ALX4 cause milder forms of frontonasal dysplasia without significant eye manifestations. Individuals with homozygous mutations in the ALX3 gene have been found to present frontonasal dysplasia type 1, named frontorhiny, characterized by distinctive facial anomalies such as hypertelorism, wide nasal bridge, short nasal ridge, bifid nasal tip, and others [79]. Alx4 loss-of-function mutations result in frontonasal dysplasia type 2. Heterozygous mice show a single extra digit in the preaxial part of one of the hindlimbs, whereas homozygous mice show a complex phenotype including extensive preaxial polydactyly, tibial anomalies, craniofacial defects, and hypomorphic interfollicular epidermis with reduced suprabasal layers [80,81].
Ectopic expression of Alx1 in transgenic mice does not disturb development, whereas expression of the form of Alx1 with deleted OAR domain results in severe cranial and vertebral malformations. It was suggested that the OAR domain of Alx1 restrains its activity in vivo through its effect on DNA binding. The OAR domain of Alx3 appeared to have lost most of its function, and its mutations had not visible phenotypic manifestations [82]. Alx1 homozygous mutant mice are born alive with acrania and meroanencephaly but die soon after birth. This gene is implicated in craniofacial development and is necessary for survival and migration of cranial neural crest cells into the frontonasal primordia [83]. Alx3-deficient mice exhibit increased failure of cranial neural tube closure and increased cell death in the craniofacial region [84], but adult mutants do not show any apparent abnormalities, perhaps due the deaths of a number of Alx3-null mice during embryogenesis [84]. In contrast, severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice have been demonstrated [85]. Treatment with Alx1 antisense oligonucleotides show a reduction of the eye size as well as fewer cell layers in the retinas of zebrafish embryos [76]. Morpholino knock-down of zebrafish Alx1 expression causes a profound craniofacial phenotype including loss of the facial cartilages and defective ocular development [83]. All of these features correspond with frontonasal dysplasia, a syndrome in humans caused by neural tube closure defects, and suggest a crucial role of Arl genes in regulating the migration of cranial neural crest cells into the frontonasal primordia.
3.3. CerS2
The ceramide synthase (CerS) gene family belongs to the CERS homeobox gene class and comprises six genes in human and mouse genomes (CERS1–6/CerS1–6). They contain seven (CERS1) or ten (CERS2-6) coding exons and encode transmembrane proteins, each of which synthesizes ceramides with distinct acyl chain lengths [86]. They encode integral membrane proteins of the endoplasmic reticulum and in some cases are associated with mitochondria. All mammalian CerS genes contain a unique C-terminal TLC domain, six transmembrane domains, and a Hox-like domain (except CerS1), which is derived from DNA-binding homeodomain that lost the first 15 amino acids [87]. Thus, CerS proteins are not DNA-binding transcription factors, but enzymes that participate in lipid metabolism.
Usually more than one type of CerS protein is expressed in a given cell type. CerS2 transcripts are found at the highest level of all CerS, have the broadest tissue distribution and synthesize ceramides containing mainly C20–C26 fatty acids, with little or no synthesis of C16- and C18-ceramides [88].
CerS2 mRNA is ubiquitously expressed and is highly abundant in many tissues. The highest CerS2 expression was detected in liver and kidney. In mouse brain, CerS2 is predominantly expressed in oligodendrocytes and Schwann cells and is up-regulated duringthe period of active myelination. Moreover, CerS2 genes are located within chromosomal regions that are replicated early within the cell cycle and consist of other features typical of a housekeeping gene, although no other CerS genes exhibit these characteristics [86]. Moderate CerS1, CerS2, and CerS4 protein levels were found in the retina. In the cornea, CerS1 and CerS4 are weakly present, whereas CerS2 is strongly expressed. CerS4 is ubiquitously expressed in all retinal neurons (photoreceptors, especially cones, horizontal cells, ON and OFF bipolar cells, GABAergic amacrine cells, ganglion cells) and in Muller glia [89]. In the optic nerve, immunofluorescent reaction on CerS2 was found in the somata of oligodendrocytes, but not along myelinated axons or their myelins’ heath outside of oligodendrocyte somata [90].
Ceramides play a central role in the induction of apoptosis by death receptors and many stress stimuli such as gamma-irradiation and ultraviolet (UV) light [91]. Oxidative stress has also been shown to stimulate an increase in ceramide levels, inducing photoreceptor apoptosis [92].
In humans, rhegmatogenous retinal detachment was found to be associated with a missense coding single-nucleotide polymorphism in the Hox domain of CERS2, resulting in an elevated expression level of CERS2 [93]. A role of CERS genes in the progression of AMD was demonstrated, especially in the late stages. In aging, the function of the RPE declines, resulting in an accumulation of degraded photoreceptor outer segments in the form of lipofuscin granules, leading to oxidative stress and retinal inflammation. Oxidative stress-induced Cer biosynthesis genes are involved in photoreceptor cell death [94]. Increased Cer levels in RPE cells raise the levels of inflammatory factors and reactive oxygen species (ROS), which leads to activation of apoptosis [95]. Specific ceramide species were elevated in patients with late-stage AMD. Malondialdehyde–acetaldehyde adducts, oxidation products commonly found in AMD retinas, induced an increase in ceramide levels in retinoblastoma-derived cells in parallel with increased expression of CerS2 genes [96].
All CerS-deficient mice showed reduced electroretinogram amplitudes, with the most severe phenotype in CerS2 knockout (KO) mice, but had normal retinal morphology. CerS-deficient mice showed altered sphingolipid composition in the retina; moreover, retinal C16-sphingomyelins levels were elevated while C18-sphingomyelins levels were reduced [89]. Inhibition of ceramide synthesis by the sphingosine analog FTY720 protects rat retina from light-induced degeneration [97]. In Farber disease, there is accumulation of ceramide in the retina due to a deficiency of ceramidase. The pathological changes are observed in retinal ganglion cells with gross distention and inclusions [98,99]. It is highly possible that CerS genes also participate in the pathogenesis of Farber disease. In addition, CERS2 inactivation leads to myelin sheath defects in axons of the brain and peripheral nervous system [100].
3.4. Crx
The Crx gene belongs to the PRD class of homeobox genes and is a member of the Otx gene family, comprising three genes in human and mouse genomes (OTX1/Otx1, OTX2/Otx2, CRX/Crx). The human gene consists of four coding exons. The CRX protein includes a homeodomain as well as the WSP motif and the OTX tail [101,102].
In the mammalian retina, Crx is expressed predominantly in postmitotic developing and mature photoreceptors, more weakly in retinal bipolar cells, and in the pinealocytes of the pineal gland [52,103]. In zebrafish Crx is expressed in proliferating retinal progenitors and may be involved in patterning the early optic primordium and in promoting the differentiation of retinal progenitors [104].
The Crx homeodomain protein is a transactivator of many photoreceptor/pineal-specific genes in vivo, such as rhodopsin and the cone opsins [105,106]. Crx controls the expression of genes that encode presynaptic proteins associated with the cytomatrix active zone and synaptic vesicles, but not the formation of ribbon synapses, which connect rod and cone photoreceptors to bipolar neurons [107]. CRX also controls outer segment biogenesis and photoreceptor disk renewal [101].
Mutations of the human CRX gene are associated with diseases characterized by photoreceptor cell destruction: Autosomal-dominant cone-rod dystrophy 2, Leber’s congenital amaurosis-7 (LCA7) (both autosomal-recessive and -dominant patterns), and, in rare cases, autosomal-dominant RP [108,109,110,111,112]. These diseases are phenotypically and genetically heterogeneous. Cone-rod dystrophy is characterized by primary cone degeneration with significant secondary rod involvement, with additional progressive loss in peripheral vision and night blindness [113]. RP is characterized by progressive loss of rods and cones and causes severe visual dysfunction and eventual blindness in bilateral eyes. In addition to more than 3000 genetic mutations from about 70 genes, a wide genetic overlap with other types of retinal dystrophies has been reported with RP [114,115]. On the cellular level, RP correlates with a predominantly affected rod photoreceptor system. In later stages, the disease may further affect the cone photoreceptors, eventually causing complete blindness. The diseased photoreceptors undergo apoptosis, which is reflected in reduced ONL thickness within the retina, as well as lesions and/or retinal pigment deposits in the fundus [116]. LCA is the most severe childhood retinal dystrophy, characterized by a non detectable electroretinogram and other symptoms soon after birth. LCA is sometimes considered the most severe form of RP [117,118].
The retinas of postnatal day 21 (P21) Crx−/− mice were considerably thinner with almost complete absence of outer segments in photoreceptors [107]. In mice homozygous for a targeted null mutation of Crx, photoreceptors failed to form outer segments, and eventually degenerated, indicating that Crx function is required for their complete differentiation and survival [119]. Outer segment morphogenesis was found to be blocked at the elongation stage, leading to a failure in production of the phototransduction apparatus. Crx−/− photoreceptors demonstrated severely abnormal synaptic endings in the OPL [120]. Knockdown of Crx by antisense morpholino oligonucleotides resulted in delayed with drawal of RPC from the cell cycle and retardation of retinal differentiation in zebrafish embryos [104]. RNA-seq analysis of three Crx mutation knock-in mouse models (CrxR90W, CrxE168d2, and CrxE168d2neo) demonstrated a correlation of graded expression changes in shared gene sets with phenotype severity [121].
3.5. Hesx1
The Hesx gene family belongs to the PRD homeobox gene class and comprises one gene in human and mouse genomes (HESX1/Hesx1). The human gene consists of 4 coding exons encoding a 185-amino acid ORF that is highly conserved compared with the mouse and Xenopus, particularly across the homeodomain sharing 95% and 80% homology, respectively [122]. This gene encodes a conserved homeodomain protein that is required for normal development of the forebrain, eyes, and other anterior structures such as the olfactory placodes and pituitary gland [123]. Hesx1 is expressed in the rostral part of the chicken neural plate [124] and in the developing forebrain and Rathke’s pouch and is downregulated during pituitary cell differentiation in mice [125]. Hesx1 plays a role in the control of self-renewal and maintenance of the undifferentiated state in ESCs and mouse embryos [126]. It was demonstrated that it is also required to program hESC neural determination [127]. In normal development its expression is repressed by the RAX homeobox gene in presumptive retinal regions of mouse embryos [128].
A homozygous substitution of isoleucine at position 26 by threonine (I26T) in HESX1 has been associated with anterior pituitary hypoplasia in a human patient, with no forebrain or eye defects, but R160C mutation manifests septo-optic dysplasia characterized by a combination of optic nerve hypoplasia, pituitary hypoplasia, and abnormalities of midline structures in the forebrain [122,129,130].
The absence of Hesx1 leads to a posterior transformation of the anterior forebrain during mouse development. It is suggested that the mechanism underlying this transformation is the ectopic activation of Wnt/β-catenin signaling [131]. Hesx1 knockout mice have several defects of midline structures resembling human septo-optic dysplasia. They include a reduction in prospective forebrain tissue, craniofacial and optic nerve dysplasia, and abnormalities of the pituitary gland. Homozygous mutant mice show significant perinatal and postnatal mortality. Most mice surviving past birth display eye defects such as anophthalmia or microphthalmia [123,125].
3.6. Hmx1
The Hmx1 gene belongs to the ANTP class of homeobox genes and is a member of the HMX/NK5 gene family and comprises three genes in human and mouse genomes (HMX1/Hmx1, HMX2/Hmx2, HMX3/Hmx3). The human gene consists of two coding exons (ENSG00000215612, Ensembl version 98, September 2019). The HMX1 protein includes a homeobox domain [132].
Hmx1 is expressed in the eye (retina and lens), the craniofacial region, and various nerve ganglia in mice and humans [132,133,134]. In zebrafish embryos, Hmx1 transcripts were detected mainly in the nasal part of the ganglion cell layer and lens as well as optic vesicles and pharyngeal arches by 24–32 hpf. Before this stage, transcripts were more uniformly expressed in the optic vesicle [135].
A loss-of-function mutation in HMX1 causes an oculoauricular syndrome in humans. This syndrome is characterized by microphthalmia, microcornea, cataract, ocular coloboma, retinal pigment abnormalities, rod-cone dystrophy and anomalies of the external ear [132,136,137]. Homozygous missense mutation within the homeodomain of HMX1 was demonstrated to be associated with uveoretinal colobomas caused by a failure of ectodermal optic vesicle fissure closure [138].
A single C > T mutation that changes Glu65 to an amber stop codon in the Hmx1 gene displays microphthalmia, protruding ear and craniofacial malformations in mice [139]. Delayed withdrawal of retinal progenitors from the cell cycle resulting in retarded retinal differentiation and microphthalmia were observed after morpholino-mediated knockdown of Hmx1 in zebrafish [135].
3.7. Lmx1B
The Lmx gene family belongs to the LIM homeobox gene class and comprises two genes in human and mouse genomes (LMX1A/Lmx1a, LMX1B/Lmx1b). The human LMX1B gene contains eight exons and encodes protein featuring two LIM domains in their amino termini and a centrally located homeodomain, and a putative transcriptional activation domain at the COOH terminus. The LIM domain contains two tandemly repeated, cysteine-rich, double-zinc finger motifs that can be recognized by a number of co-factors [140,141].
In the mouse eye, Lmx1b is expressed in periocular mesenchyme and regulates anterior segment (cornea, iris, ciliary body, trabecular meshwork, and lens) development; mice lacking functional Lmx1b exhibit numerous abnormalities, including a lack of ciliary bodies and iris stroma, and corneal dysplasia [142].
Heterozygous mutations in the LIM homeodomain of LMX1B resulted in nail–patella syndrome (NPS), which is characterized by developmental defects of dorsal limb structures and nephropathy. Up to 50% of NPS patients exhibit clinical signs of primary open-angle glaucoma [143,144,145]. It was proposed that primary structural changes in the membranes of the lamina cribrosa and the trabecular meshwork may play a role in glaucomatous optic nerve damage in NPS and may result from disrupted collagen expression and/or an irregular arrangement or deposition of collagen fibrils [143].
It was strongly confirmed that haploinsufficiency is the principal pathogenetic mechanism of NPS in humans. A difference in dosage sensitivity for the LMX1B/Lmx1b gene between humans and mice was suggested [146].
An N-ethyl-N-nitrosourea-induced missense substitution, V265D, in the homeodomain of mouse Lmx1b abolishes DNA binding and causes glaucomatous eye defects in heterozygotes. A profound reduction in the number of retinal ganglion cells and optic nerve cupping was observed in some cases [147].
Anti-sense morpholinos against Lmx1b.1 and Lmx1b.2 isoforms resulted in defective migration of periocular mesenchymal cells around the eye and led to apoptosis of these cells. These defects in the periocular mesenchyme are correlated with a failure of fusion of the choroid fissure or, in some instances, more severe ventral optic cup morphogenesis phenotypes. The retinas of Lmx1b morphants showed defective nasotemporal patterning and lamination was delayed [148].
3.8. Meis1
The myeloid ecotropic insertion site 1 (Meis1) gene belongs to the three amino acid loop extension (TALE) class of homeobox genes and is a member of the Meis gene subfamily, comprising three genes in human and mouse genomes (MEIS1/Meis1, MEIS2/Meis2, and MEIS3/Meis3). The MEIS1 gene consists of 13 coding exons [149] (ENSG00000143995, Ensembl version 98, September 2019) and encodes protein containing a DNA-binding homeodomain toward the carboxy terminus and two protein–protein interaction domains toward the amino terminus (MEIS-A and MEIS-B). TALE homeodomain differs from the classical homeodomain by the insertion of three additional amino acids [150,151].
Meis1 is required to correctly specify both dorsoventral and nasotemporal identity in the zebrafish retina. It is initially expressed throughout the eye primordium. Later, Meis1 becomes repressed as neurogenesis is initiated, and its expression is confined to the ciliary margin, where the retinal stem population resides. Thus, Meis1 maintains RPC in a rapidly proliferating state. Cell cycle control is mediated through regulation of c-myc and/or cyclinD1 [152,153,154].
Morpholino knockdown of Meis1 causes a delay in the G1-to-S phase transition of retinal cells. Consequently, fish, chicks, or mice with compromised Meis1 function are microphthalmic [152,153,155]. Knockdown of Meis1 expression diminishes endogenous Foxn4 expression and affects development of horizontal and amacrine neurons in mice [156]. By combining an analysis of Meis1 loss of function and conditional Meis1 functional rescue with ChIP-seq and RNA-seq approaches, it was shown that in mice Meis1 coordinates, in a dose-dependent manner, retinal proliferation and differentiation by regulating genes responsible for human microphthalmia and components of the Notch signaling pathway [157].
Studies in animal models of the orthologous genes identified overlapping phenotypes for most factors, confirming the conservation of their function in vertebrate development. However, despite whole exome/genome testing more than half of patients currently remain without a molecular diagnosis. Anophthalmia and microphthalmia are part of the same clinical spectrum and have high genetic heterogeneity, with >90 identified associated genes. Currently, mutations in known genes explain less than 40% of these conditions therefore additional causative factors need to be discovered [35]. The MEIS1 gene is the best gap-filling candidate for decoding genetic pathways associated with anophthalmia and microphthalmia.
3.9. Msx2
The human MSX gene family consists of two members (MSX1, MSX2), but the mouse has an additional Msx3 gene. These genes belong to the ANTP class of homeobox genes. MSX2 consists of two coding exons and encodes proteins with homeodomain. These regulatory proteins function as transcriptional repressors and are widely expressed in many organs, particularly at the sites where epithelial–mesenchymal interactions take place [158,159]. In chick embryos, Msx2 was widely distributed in the dorsal neural retina and pigment epithelium as well as in the retro-ocular mesenchyme [160]. Mouse Msx3 is only expressed in the dorsal neural tube [161].
Msx2 drives osteogenic differentiation of multipotent mesenchymal progenitors and yet suppresses adipogenesis [162]. Direct interaction with DNA is not required for Msx2 suppressor function. Msx2 suppresses transcription via protein–protein interactions with components of the basal transcriptional machinery [163]. Msx2 controls diverse processes during brain development (e.g., apoptosis, neuronal specification and differentiation) mediating distinct stage-dependent roles of BMPs in dorsal neural-tube patterning [159].
A gain-of-function mutation in human MSX2 causes autosomal-dominant Boston-type craniosynostosis, characterized by overgrowth of skull bones [164,165], while loss-of-function mutations result in MSX2 haploinsufficiency and lead to reduced ossification of the parietal bones, enlarged parietal foramina and aberrant closure of the sagittal suture [166]. The case of a child with bicoronal synostosis and cutaneous syndactyly, who presented iridal and chorioretinal colobomas due to MSX2 gene duplication has been recently reported [167].
In mice, overexpression, misexpression, or deficiency of Msx2 impedes osteoblast differentiation and leads to many craniofacial deformities. Msx2 null mutation causes pleiotropic defects in bone growth and in the ectodermal organs, including the teeth, hair, and mammary glands, and impaired chondrogenesis [168]. These mice display defective skull ossification and marked reduction in bone formation associated with decreased osteoblast numbers, which is very similar to what is observed in humans. It has been shown that Msx2 promotes osteoblast differentiation independently of Runx2 and negatively regulates adipocyte differentiation through inhibition of PPAR and the C/EBP family [169]. Transgenic mice with a dominant gain-of-function mutation (Pro7His) of the Msx2 gene have been constructed. These mice exhibited precocious fusion of cranial bones and craniosynostosis and provided an excellent model of human craniosynostosis [170]. Overexpression of the wild-type form of the Msx2 gene in Xenopus resulted in embryos with a loss of anterior structures, including head and eyes [171].
Msx2 appears to play dual roles in promoting apoptosis and determining retinal fate. Overexpression of mouse Msx2 led to microphthalmia and optic nerve aplasia, suppressing Bmp4 expression in the optic vesicle before lens induction, whereas Bmp7 expression was upregulated. Retinal apoptosis together with an overall reduction in proliferation resulted in thinning of the retina and microphthalmia [172]. Antisense disruption of mouse Msx2 during early stages of neurulation produced hypoplasia of the maxillary, mandibular, and frontonasal prominences and somite, and neural tube, eye, and somite abnormalities. Eye defects consisted of enlarged optic vesicles, which may ultimately result in microphthalmia. Histological analysis revealed thinning of the neuroepithelium in the diencephalon and optic vesicle and hypoplasia of the mid- and forebrain regions [173]. Overexpression of Msx2 delayed the expression of RGC-specific differentiation markers (Math5 and Brn3b), which showed that it could affect the timing of RGC fate commitment and differentiation by delaying the timing of cell cycle exit of retinal progenitors [174]. Msx2 can suppress the differentiated state of RPE cells and promote their differentiation into neural cell types. Msx2-transfected cultures of chick RPE contained fewer cells expressing the RPE marker, Mitf, and more cells expressing class III b-tubulin, a neuronal marker. In addition, a small proportion of Msx2-transfected cells acquired a neural-like morphology [175].
3.10. Otx2
The Otx2 gene belongs to the PRD class of homeobox genes. This gene family includes three genes in human and mouse genomes (OTX1/Otx1, OTX2/Otx2, and CRX/Crx). The OTX2 gene consists of five exons (ENSG00000165588, Ensembl version 98, September 2019) and encodes protein with homeodomain [176]. The OTX2 protein contains a homeodomain responsible for DNA binding, SGQFTP and SIWSPA motifs involved in protein–protein interactions, and two C-terminal tandem OTX-tail motifs responsible for transactivation [177].
The Otx2 gene plays a critical role in forebrain and eye development. At later embryonic stages, the gene is expressed in several regions of the brain and in sensory organs such as the inner ear, retina, and olfactory epithelium. In the embryonic mouse, Otx2 was found in RPE cells and a restricted subset of retinal neurons, including ganglion cells. In the postnatal and adult eye, it was detected in the nuclei of RPE and bipolar cells, and restricted to a small volume at the inner periphery of nuclei of photoreceptors [178,179]. In the differentiating outer retina, Otx2 is expressed in progenitors of both photoreceptor and bipolar cells. Beyond developmental stages, Otx2 expression is abundantly maintained in RPE, photoreceptors, and bipolar cells [179].
A majority of the known OTX2 mutations involve homeobox and the last exon of the gene [177]. Heterozygous mutations of OTX2 cause severe ocular malformations, which range from bilateral anophthalmia to retinal defects resembling LCA and pigmentary retinopathy [180]. OTX2 loss-of-function mutations are frequently associated with coloboma, optic nerve hypoplasia or aplasia, microcephaly, brain, and pituitary anomalies and combined pituitary hormone deficiency [181,182,183]. Heterozygous mutations in OTX2 associated with early-onset retinal dystrophy with atypical maculopathy and bilateral microphthalmos have been reported [184]. Most OTX2 mutations reported to date originate from premature truncation of the protein product. As a rule, pituitary anomalies seem to be more strongly associated with mutations that occur in the second half of OTX2, after the homeodomain and SGQFTP motif [185].
Complete elimination of Otx2 in knockout mice results in the absence of the forebrain and embryonic fatality due to the absence of the rostral neuroectoderm fated to become forebrain, midbrain, and rostral hindbrain [186]. Otx2 hemizygotes survive until birth and demonstrate anomalies in RPE and retina development. Depending on the genetic background, Otx2+/− embryos show variable phenotype (acephaly, holoprosencephaly, short nose, anophthalmia/microphthalmia, agnathia/micrognathia, or normal phenotype) [187]. Otx2 conditional knockout mice exhibited a total absence of rods and cones in the retina due to their cell fate conversion to amacrine-like cells [188]. A number of mature bipolar cells were diminished in postnatal mice with bipolar cell-specific Otx2 conditional-knockout [189]. In the adult retina, loss of Otx2 protein after induced conditional knockout caused slow degeneration of photoreceptor cells due to dramatic changes of RPE consisting of reduction in melanin content, extensive vacuolization, and loss of RPE contact with disc-containing photoreceptor outer segments [190].
So, the gene is required for RPE specification and serves as a key regulator of photoreceptor genesis and differentiation, and is required after birth for bipolar cell terminal maturation and long-term maintenance of photoreceptors [177].
3.11. Pax2
The Pax2 gene belongs to the PRD homeobox gene class and is a member of the PAX258 gene subfamily (Pax group II). It comprises three genes in human and mouse genomes (PAX2/Pax2, PAX5/Pax5, and PAX8/Pax8). PAX2 contains 12 exons and encodes protein with a paired domain, an octapeptide domain, a partial homeodomain (which still has DNA-binding properties), and the transactivation domain. None of the known transcripts contain all 12 coding exons of the PAX2 gene lacking either exon 6 or exon 10 [191,192].
During human and mouse development, PAX2/Pax2 is expressed in the developing eye, ear, kidney, midbrain, hindbrain, and spinal cord. In the mouse eye, Pax2 expression begins in the optic vesicle and becomes restricted to the ventral half of the optic cup and stalk and later to the optic disc and nerve. After closure of the retinal fissure, Pax2 protein is lost from the ventral retina; however, it is localized mostly to the proximal regions, destined to contribute to the optic nerve and optic chiasm [193,194]. Pax2-expressing cells in the developing rat and human optic nerve are exclusively astrocyte precursors and astrocytes controlling oligodendrocyte differentiation in the optic stalk. Coloboma formation may result from impaired astrocyte differentiation during development [195,196]. In rodents, cells of astrocytic lineage migrate into the retina through the optic nerve head as a mixture of precursor cells and immature perinatal astrocytes, and then spread across the nerve fiber layer toward peripheral margins of the retina. The migration of astrocytes into the retina is followed by the invasion of endothelial cells to form the retinal vasculature, which in turn promotes astrocyte differentiation [197]. In the adult human retina, mature perinatal astrocytes were restricted to a region surrounding the optic nerve head, whereas adult astrocytes were apparent throughout the vascularized retina. A cluster of Pax2 cells was also present in a small region surrounding the optic nerve head at the ventricular surface of the developing retina, which suggests the existence of two distinct sites of astrocytic differentiation [195]. Pax2 is rapidly downregulated in explanted optic nerves that generate neurons, and its overexpression by electroporation in the optic nerve, or ectopically in the neural tube, is sufficient to block neuronal differentiation and allow glial development. It has been suggested that Pax2 is able to regulate a switch between neuronal and glial fates inhibiting neurogenesis or inducing gliogenesis in the optic nerve [198]. In chick and zebrafish embryos, Pax2 is expressed also by Muller glia cells in the central retina, but similar expression could not be detected in mouse, rat, guinea pig, rabbit, or pig retina [199,200]. Pax2 is expressed in astrocyte precursor cells and mature astrocytes of the optic stalk/nerve and glial cells of a vascular structure in avian and reptile species called the pecten [196,201].
The majority of mutations in PAX2 are pathogenic or likely pathogenic and expected to result in significant truncation of the PAX2 protein through a shift in the reading frame or the introduction of a premature stop codon. These mutations were located in all four known functional domains of this gene, but more often in exons 2–4 (encoding the paired domain) and exons 7–9 (encoding the transactivation domain) [192,202]. PAX2-related disorder was originally termed renal coloboma syndrome (also known as papillorenal syndrome) and characterized by renal (hypodysplastic kidneys) and optic disc anomalies. Abnormal renal structure or function is noted in 92% and ophthalmologic abnormalities in 77% of affected individuals [203]. Optic nerve malformations include optic nerve coloboma, optic nerve dysplasia, morning glory anomaly, and cystic malformation of the optic nerve. Other eye malformations may include retinal coloboma, microphthalmia, and macular dysplasia. Less common associated eye malformations include abnormal RPE, abnormal retinal vessels, and chorioretinal degeneration [204,205].
The developmental mechanism underlying the optic nerve abnormalities observed in PAX2-related disorder is under investigation in animal models; in both mouse and zebrafish, homozygous loss-of-function alleles of Pax2/Pax2a resulted in failed optic fissure closure [206]. Several mouse models of renal coloboma syndrome have been described. After deletion of exon 1 and 2 of the Pax2 gene, the optic tracts remained totally ipsilateral due to a genesis of the optic chiasma. These mutants showed extension of the pigmented retina into the optic stalks and failure of the optic fissure to close, resulting in coloboma [207]. A frame shift mutation (Pax21Neu) with a 1-bp insertion in the Pax2 gene has been described. The same mutation was in a human family with renal–coloboma syndrome. Heterozygous mutant mice exhibited defects in the kidney, optic nerve, and retinal layer of the eye. In homozygous mutant embryos, development of the optic nerve, kidney, and ventral regions of the inner ear was severely affected, as in the case of the same mutation in humans [208]. Mice heterozygous for a Pax2 missense mutation paired domain showed reduced optic nerve axon numbers [209]. The hypomorphic mutation Opdc for Pax2 has been reported. In homozygotes, the phenotypic consequence of this mutation on the development of the eye and ear was similar to that reported for null alleles of Pax2, but homozygotes had undisturbed kidney development under some strain backgrounds [210]. Nearly all homozygous mutant mice are anephric, with most not surviving past the immediate postnatal period. These mice have midbrain/hindbrain malformations, cranial neural tube closure defects (exencephaly), and absent cochlea. The ocular phenotype is characterized by a failure of closure of the optic fissure and failed basement membrane dissolution. Pax2 plays a critical role in optic chiasm development and in the guidance of axons along the optic tract. Null mutant Pax2 mice develop an abnormal chiasm in which all optic axons are prevented from projecting across the midline into the contralateral optic tract [207].
3.12. Pax6
The Pax6 gene belongs to the PRD homeobox gene class and is a member of the PAX46 gene subfamily (Pax group IV). It comprises two genes in human and mouse genomes (PAX4/Pax4, PAX6/Pax6). PAX6 contains 14 exons and encodes protein with two DNA binding domains, a paired domain and a homeodomain, and one proline/serine/threonine-rich transactivation domain [211]. The paired domain is a bipartite DNA binding structure composed of two helix–turn–helix motifs (the N-terminal PAI and C-terminal RED subdomains) that are separated by a flexible linker. These subdomains have tissue-specific effects on development and gene expression [212].
In vertebrates, this factor is essential for normal development of several organs, including the brain, pancreas, and eye [213]. Pax6 is expressed in the anterior neural plate in the cells that will give rise to the optic vesicle. Pax6 activity is required for the establishment and maintenance of dorsal and nasotemporal characteristics in the optic vesicle and, later, the optic cup [214]. Although Pax6 is not required for optic vesicle formation, it does play a role in subsequent steps of retinogenesis. At the optic cup stage, Pax6 seems to be required for cell proliferation and differentiation. Following optic cup formation, Pax6 is downregulated in the optic stalk and the RPE, but retained in the neuroretina. Pax6 plays a role in determining neuronal cell fate in the retina and, together with Pax2, directs the determination of RPE controlling the expression of the melanocyte determinant Mitf [215]. In differentiating RPE, Pax6 is required for promoting its melanogenic program [216]. Pax6 maintains the multipotency and proliferation of RPCs through the activation of proneural genes [217]. Expression in the retina is maintained in proliferating RPC, while it is downregulated in most cells upon differentiation. After evagination of the optic vesicle, Pax6 becomes restricted to all proliferating cells of the pigment epithelial and neural layers of the retina [218]. Pax6 is diffusely expressed in the undifferentiated retinal neuroepithelium as cells become postmitotic and arranged in a characteristic laminar pattern; however, Pax6 remains strongly expressed in putative ganglion, amacrine, and horizontal cells, while becoming conspicuously downregulated in photoreceptor progenitors and in the cells occupying the Muller/bipolar region of the INL [219,220]. Pax6 has been assumed to play a several roles during early retinal development: Proliferation of retinal cell progenitors [221], maintenance of multipotent progenitor potential [222], and regulation of timing of differentiation and cell fate determination [223]. In the adult human retina, PAX6 proteins are expressed in the ganglion cells and INL [46]. Pax6 has been detected immunochemically in amacrine and ganglion cells of adult retinas of rats and goldfish [224,225]. In the adult mouse, Pax6 was found to be essential for generating late-born glycinergic amacrine cells, along with most bipolar cell subtypes. Overexpression of Pax6 greatly increased the non-GABAergic/non-glycinergic amacrine cells, suppressed generation of both glycinergic amacrine cells and bipolar interneurons, and disrupted rod photoreceptor morphogenesis [226]. Pax6 has been proposed to regulate the maintenance of horizontal cells through the activation of ONECUT1 and ONECUT2 transcription factors [227]. It was demonstrated that development of the lens from the surface ectoderm requires a higher gene dose of Pax6 than development of the retina from the optic vesicle [228].
More than 500 different mutations have been described that affect PAX6 and its regulatory regions. The majority of PAX6 mutations result in null alleles and consequent PAX6 haploinsufficiency and are known to result in aniridia, an autosomal-dominant disorder that is marked by the complete or partial absence of the iris, often combined with cataracts, glaucoma, nystagmus, and foveal and optic nerve hypoplasia. Missense mutations reported in ~12% can potentially cause partial loss-of-function or gain-of-function generating single amino acid substitutions, and lead to less severe ocular abnormalities (foveal hypoplasia, Peters anomaly, congenital cataracts, microphthalmia, optic nerve hypoplasia, coloboma, and anterior segment dysgenesis) [229,230]. Patients with a PAX6 mutation occurring at the donor splice site of intron 4 have been reported to have congenital nystagmus with anterior segment anomalies (mainly iris hypoplasia or coloboma) associated with foveal hypoplasia [231]. Microphthalmia/anophthalmia/coloboma phenotypes are significantly associated with recurrent heterozygous PAX6 missense mutations in the paired domain that are likely to disrupt the PAX6–DNA interaction [232,233]. Anophthalmia is associated with homozygous PAX6 variants, where there is biallelic loss of function. The affected individuals are usually still born or die soon after birth with severe brain abnormalities [234,235]. A heterozygous, likely pathogenic, variant in PAX6 associated with microphthalmia was reported to have a highly conserved valine residue in the homeodomain [236].
Homozygous Pax6−/− mice, with two nonfunctional alleles, die at birth with no eyes or nose and with brain abnormalities [237,238]. Heterozygous Pax6+/− mice are viable and fertile but have a range of eye abnormalities, such as microphthalmia, iris hypoplasia, cataracts, a thin corneal epithelium with fewer cell layers, corneal opacity, failure of the lens to separate completely from the corneal epithelium, retinal dysplasia, coloboma, and adhesions between the lens and cornea or between the iris and cornea [239]. Embryos with putative null Pax6 mutations also show severe eye abnormalities and changes in brain development resembling human aniridia phenotypes [240]. In mutant mice with Pax6 activity ranging between 100% and 0%, the extent of eye development was progressively reduced as Pax6 activity decreased, to very early termination of eye development at the lowest levels of Pax6. Development of the lens and cornea is more sensitive to reduced levels of Pax6 activity than development of the retina [241]. Overexpression of Pax6 in PAX77+/+ mice prevents normal development of the retina from about E14.5 and results in microphthalmia, retinal dysplasia, and defective retinal ganglion cell axon guidance in postnatal life [242]. The Pax6-deficient mouse model of aniridia (Pax6Sey+/−) has been used for topical application of nonsense mutation suppression drugs on adult eyes. Nonsense suppression not only inhibited disease progression but also stably reversed corneal, lens, and retinal malformation defects and restored electrical and behavioral responses of the retina [243].
3.13. Rax
The Rax gene family belongs to the PRD class of homeobox genes and includes two genes in the human genome (RAX, RAX2) and only one gene in the mouse genome (Rax). RAX contains three exons and encodes a protein with an octapeptide, a DNA-binding paired-type homeodomain, an Rx domain, and an OAR domain [244,245]. The octapeptide functions in transcriptional repression through interaction with Groucho family corepressors [246].
Rax is initially expressed in the anterior neural region of developing mouse embryos, and later in the retina, pituitary gland, hypothalamus, and pineal gland [247]. In the early mouse embryo, Rax is expressed in the anterior neural fold, including areas that will give rise to the ventral forebrain and optic vesicles. Rax, mainly through activation of transcriptional repressors TLE2 and Hes4, is necessary and sufficient to inhibit endomesodermal gene expression in retinal precursors of the eye field [248]. Rax is expressed in all RPC in the neuroepithelium of the developing retina and in RPC in the ciliary margin of the mature retina in mice and frogs [249,250,251]. Its expression is progressively downregulated as neuronal differentiation proceeds, except for photoreceptors and Muller glia cells [249,250]. It has been demonstrated that Rax proteins are necessary for normal photoreceptor development and expression of normal levels of markers of differentiated photoreceptors in zebrafish and Xenopus [250,252]. Rax expression is retained in the ciliary marginal zone, which is a source of retinal stem cells in adult fish and amphibians [252].
RAX mutations in humans have been reported, including anophthalmia, microphthalmia, and eye coloboma [253,254], in some cases associated with sclerocornea [57] or severe cerebral malformation [255].
Rax-null homozygotes exhibit perinatal mortality, anophthalmia, and anterior nerve and craniofacial defects [256]. Rax−/− mouse embryos have no visible eye structures, while heterozygotes for the Rax mutation are apparently normal [257]. In zebrafish, knockdown of Rax over the period of eye organogenesis resulted in severely reduced proliferation of retinal progenitors, the loss of expression of transcription factor Pax6, delayed retinal neurogenesis, and extensive retinal cell death [258]. Knockdown of Rax using translation-inhibitory antisense morpholino oligonucleotides also resulted in anophthalmia [250,257]. Overexpression of Rax led to the loss of forebrain tissue and the ectopic formation of retinal tissue in zebrafish and Xenopus [257,259]. Moreover, for the same induction, expression of the Rax homeodomain alone is sufficient [260]. In summary, gain- and loss-of-function studies of Rax in different species have shown this gene to be necessary for the formation of anterior brain structures and eyes, and sufficient to induce retinal and neural tube hypertrophy [257,261,262].
3.14. Rax2
The Rax2 gene belongs to the PRD class of homeobox genes. RAX2 contains three exons and encodes a protein with a DNA-binding paired-type homeodomain, an Rx domain, and an OAR domain. Rax2 proteins of all tetrapods are shorter than Rax1 proteins and lack the octapeptide motif. There is no Rax2 gene in mouse and other rodent genomes [245,263]. Two Rax genes have been identified in chicks (cRax and cRaxL/cRax2) [264] and in Xenopus [265].
Rax2 are among the earliest markers of the eye field, being initially expressed in the anterior neural region of head-fold-stage embryos, but later becoming restricted to the neural retina and ventral hypothalamus [266]. Human RAX2 is expressed in the ONL and INL of the adult human retina and act synergistically with CRX and NRL to modulate the expression of photoreceptor genes [263]. RAX2 is one of the top expressed transcription factors in the adult human retina, which suggests a major role in the regulation of retinal transcription [267]. The chick RaxL/Rax2 gene is expressed in both RPC and early-developing photoreceptors. It is not sufficient to promote photoreceptor cell fate choice, but is required for cone photoreceptor cell differentiation [264]. In zebrafish embryos, the level of expression of Rax2/Zrx2 is greater than Rax/Zrx1 in immature photoreceptors at comparable stages, whereas expression of the latter is enhanced during maturation of photoreceptors. Expression of both genes continues in the adult retina, exclusively in cone, but not rod, photoreceptors [268].
A heterozygous mutation in RAX2 inherited in an autosomal-dominant fashion has been reported to be associated with mixed cone and rod dysfunction and AMD [263,269]. Biallelic RAX2 sequence and structural variants were found in patients with nonsyndromic autosomal-recessive RP [270].
The absence of Rax2 in mouse has made functional analysis and modeling of the associated human diseases more difficult. The expression of a putative dominant negative allele of a chick Rax2 gene caused a significant reduction in the level of expression of cone photoreceptor genes [264]. Morpholino-based knockdown of Rax2/Rx-L in Xenopus appeared to impair late retinogenesis and reduce photoreceptor-specific gene expression [271]. Animal models demonstrate that Rax2 is required for cell proliferation and differentiation within the retina by regulating the spatial expression of photoreceptor-specific genes in late retinogenesis.
3.15. Vax1
The Vax1 gene belongs to the ANTP class of homeobox genes. This gene family includes two genes in human and mouse genomes (VAX1/Vax1, VAX2/Vax2). VAX1 contains four exons and encodes a protein with DNA-binding paired-type homeodomain [272]. Vax1 protein can be secreted and penetrate into axons of RGC and other neurons [273].
In vivo, expression of Vax1 is highly restricted to ventral anterior regions of the developing CNS, including the glial precursor cells of the optic stalk and their descendent astrocytes of the optic nerve, and regions containing the glia that are thought to guide the formation of midline commissural tracts. At E18.5, Vax1 expression is visible in the ventral optic stalk, the glial cells of the optic nerve, the optic chiasm, and the rostral diencephalon [272,274,275]. Vax1 acts in concert with Vax2 to ventralize the developing eye field of mouse embryos. They allow for development of the optic nerve by inhibiting development of the retina through repression of the Pax6 and Rax genes and therefore are involved in the partitioning of the developing visual system in the eye and optic nerve [275,276]. In Danio rerio, Vax1 is expressed in the ventral portion of the developing eye [277]. It was discovered that Vax1 is secreted from ventral hypothalamic cells and diffuses to RGC axons, where it promotes axonal growth [273]. Microphthalmia associated with cleft lip and palate and agenesis of the corpus callosum caused by homozygous mutation in the Vax1 gene was reported [278].
VAX1 has been also identified as a candidate gene playing a role in human nonsyndromic cleft lip with or without cleft palate, which can be accompanied by brain anomalies, eye coloboma, and syndromic microphthalmia [279,280].
Vax1-null mice undergo neonatal mortality due to severe holoprosencephaly and cleft palate. The eyes of mutant homozygotes display a failure of the optic disks to close, leading to coloboma and loss of the eye–nerve boundary. Retinal axons fail to penetrate the brain and form an optic chiasm. The periphery of nearly the entire length of the Vax1 mutant nerve was frequently pigmented by cells of the RPE [272,275,276]. In Danio rerio, injection of antisense morpholinos against Vax1 resulted in colobomas and reduced retinal pigment at the site of the choroid fissure [277]. In chicks, ectopic expression of cVax and mVax2 led to ventralization of the early retina. Moreover, the projections of dorsal but not ventral ganglion cell axons onto the optic tectum showed profound targeting errors [281]. Overexpression of Xenopus Vax1/Xvax1 after injection of its mRNA into one or two blastomeres at the two-cell stage primarily affected eye development in a dose-dependent manner. Low doses resulted in only a slight reduction in eye diameter, whereas increasing doses produced cyclopic and microcephalic embryos and could inhibit head and eye formation completely [275].
3.16. Vax2
The Vax2 gene belongs to the ANTP class of homeobox genes. This gene family includes two genes in human and mouse genomes (VAX1/Vax1, VAX2/Vax2). VAX2 contains three exons and encodes a protein with DNA-binding paired-type homeodomain [282].
In mice, Vax2 transcripts are detected by whole-mount in situ hybridization almost exclusively in the ventral half of the optic vesicle from E9.0 onward. At E12.5, Vax2 is highly and almost exclusively expressed in the prospective inferior neural retina. Labeling seems to be equally strong in the deep and superficial layers of the differentiating neural retina. Expression is also detected at a lower level from the inferior neural retina along the entire optic nerve and stalk. Thus, at later stages the expression domains of Vax1 and Vax2 become segregated, with Vax1 predominantly found in the ventral optic stalk and Vax2 in the ventral retina [282,283]. Vax2 inactivation leads to altered expression of genes metabolizing retinoic acid and altered asymmetric expression of cone photoreceptor genes (S-Opsin and M-Opsin). Moreover, adult Vax2 mutant mice revealed disorganization of the nerve fiber layer within the retina. Therefore, Vax2 is confirmed to not only play a crucial role in the development of the embryonic ventral retina but also continue to function in the mature retina, and may play a role in the generation of the visual streak or the macula [284]. Vax2 was found to shuttle between the nucleus and cytoplasm in retinal differentiation. This shuttling is controlled by phosphorylation. Disruption of phosphorylation and constant nuclear localization of Vax2 protein in the chick optic vesicle results in constitutive repression of Pax6, and leads to the formation of an eyeless embryo [285].
It has been reported that VAX2 in humans has two isoforms; both forms are enriched in neuronal tissues, including the retina. In monkey retina, Vax2 was observed to localize either to the nucleus (ganglion cells) or to the cytoplasmic compartments (outer segment of cone photoreceptors) depending on the retinal cell type [286]. The VAX2 p.Leu139Arg variant, harbored by a cone dystrophy patient with loss of outer retinal tissue at the fovea, was identified. Mutant protein was mislocalized and degraded, forming aggresomes [287]. Bilateral rod/cone photoreceptor dystrophy and mild optic atrophy in the patient, with complete deletion of ATP6V1B1 and disruption of the VAX2 open reading frame, was revealed. Similar changes were not detected in an adult harboring a disruptive mutation in ATP6V1B1 usually associated with distal renal tubular acidosis [288].
Colobomas were rare and milder in Vax2 homozygous null mutants, but Vax1 and Vax2 double-mutant mice had severe colobomas that were fully penetrant [289,290]. The involvement of this gene in the development of ocular coloboma in humans was predicted [289]. Injections of antisense morpholinos against Vax2 or Vax1 resulted in colobomas and reduced retinal pigment at the site of the choroid fissure in Danio rerio [277]. Vax2 overexpression in mouse, frog and chick embryos ventralizes the eye. Furthermore, Vax2 overexpression induces a striking expansion of the optic stalk, a structure deriving from the most ventral region of the eye vesicle [282,283,289]. This ventralization is mediated, at least in part, by Vax2 and Vax1 repression of the Pax6 gene [290].
3.17. Vsx1
The Vsx1 gene belongs to the PRD class of homeobox genes. This gene family includes two genes in human and mouse genomes (VSX1/Vsx1, VSX2/Vsx2). VSX1 gene contains five exons and encodes a protein with paired-type homeodomain. In addition to the paired homeodomain, VSX1 contains an additional 54 amino acid conserved regions, termed the CVC domain, located immediately adjacent to the C-terminus of the homeodomain [291,292,293]. Vsx1 can function as a transcriptional repressor [294].
During postnatal development, Vsx1 is expressed in the presumptive INL, ganglion cells, and differentiating lens fibers, as well as in a few cells of the presumptive ONL (differentiating photoreceptor or horizontal cells). In the adult retina, Vsx1 is most likely expressed in cone bipolar cells [295]. Vsx1 is expressed in type 7 ON bipolar cells. Vsx1 is expressed only weakly in undifferentiated, presumptive neural retina of goldfish embryos and is then upregulated selectively in presumptive bipolar cells at early stages of differentiation before decreasing to an intermediate level, which is maintained in the differentiated adult retina. After completion of retinal lamination, Vsx1 expression is restricted to cells occupying the INL and to postmitotic, differentiating progenitor cells in the growth zone at the peripheral retina, where neurogenesis continues throughout life [291,296,297]. Vsx1 is known to mark the largest subsets of cone bipolar cells [298,299,300]. In Xenopus, Vsx1/Xvsx1 expression is maintained in retinal progenitors and in a peripheral region of the ciliary marginal zone, while in the central retina, it becomes restricted to differentiated bipolar cells [301]. It was found that Xvsx1 is initially transcribed but not translated in early retinal progenitors. Its translation requires cell cycle progression and is sequentially activated in bipolar cells [302].
Bipolar cell fate was not altered in the absence of Vsx1 function and expressed the pan-bipolar marker Chx10. The specification, number, and gross morphology of the subset of on-center and off-center cone bipolar cells were also normal in mutant mice. However, the terminal differentiation of OFF-CB cells was incomplete [300]. Vsx1 mutant retinal cells form but do not differentiate a mature cone bipolar cell phenotype. Electrophysiological studies demonstrated that mutant mice had defects in their cone visual pathway, whereas the rod visual pathway was unaffected [298]. Thus, Vsx1 is necessary for bipolar cell terminal differentiation and is required for the activation and repression of gene expression in OFF and ON bipolar cells, respectively.
Mutations in the VSX1 gene for distinct inherited corneal dystrophies, posterior polymorphous dystrophy and keratoconus, have been identified. These patients were found to have abnormal function of the inner retina, detected with electroretinography [303]. The mutations in the homeodomain and critical CVC domain of the VSX1 gene result in abnormal craniofacial features, absence of the roof of the sella turcica, and anomalous development of the corneal endothelium. This mutation also impacts the maintenance of cone bipolar cells of the visual system [304,305]. Nonetheless, the VSX1 gene has not been definitively demonstrated to play a causal role in keratoconus [306,307]. VSX1 mutations in humans have also been associated with visual signaling defects, and individuals with VSX1 mutations have exhibited abnormal ERG b-waves consistent with a defect in retinal bipolar interneuron function. H244R mutation of VSX1 was reported to be associated with selective cone ON bipolar cell dysfunction and macular degeneration in a family with posterior polymorphous corneal dystrophy [308].
Mice lacking Vsx1 exhibit ERG b-wave defects, decreased OFF visual signaling, and a perturbation of directional selectivity, all of which are thought to arise from dysfunctional cone bipolar cell signaling [298,300]. A mouse model for Vsx1 p.P247R did not have any corneal defects, but did exhibit an abnormal electroretinogram response [305].
3.18. Vsx2
The Vsx2 gene belongs to the PRD class of homeobox genes. This gene family includes two genes in human and mouse genomes (VSX1/Vsx1, VSX2/Vsx2). Like Vsx1, the VSX2/CHX10 gene contains five exons and encodes a protein with an octapeptide, homeodomain, CVC domain and OAR domain and functions as transcriptional repressor [309]. The CVC domain has been shown to be involved in ubiquitin-mediated control of VSX2 protein stability [310]. Additionally, the CVC domain is important for the strength of homeodomain-dependent DNA binding [311].
Vsx2 is first expressed in the part of the optic vesicle that gives rise to the neuroblasts of the optic cup [312]. Vsx2 is expressed in RPC during much of development and is important for the ability of RPCs to proliferate [313]. Vsx2 is not found in the ciliary marginal zone of Xenopus tadpoles, whereas it is expressed in RPC in warm-blooded vertebrates [314]. Vsx2 is required for progenitor cell proliferation and formation of bipolar cells. In mice and chicks, Vsx2/Chx10 is absent from all postmitotic cells except bipolar interneurons and a subpopulation of Muller glia [315,316]. Chx10 has also been implicated in G1-phase cell cycle regulation, and Chx10 mutations may cause cells to lengthen their cell cycle time [317]. Mitf (a transcriptional factor forced by an RPE cell identity) is expressed ectopically in the Chx10or-J/or-J neuroretina, demonstrating that Chx10 normally represses neuroretinal expression of Mitf. Ectopic expression of Mitf in the Chx10or-J/or-J retina diverges it to an RPE-like structure [318].
In humans, a single base substitution in the DNA-recognition helix of the VSX2 homeobox, a deletion of the homeobox, and a mutation of the CVC domain cause microphthalmia. A number of mutations of the VSX2 gene have been reported in patients from West and South Asia to be associated with autosomal-recessive anophthalmia/microphthalmia, with or without iris coloboma and other ocular anomalies. All affected individuals with homozygous mutations had defects of ocular tissues ranging from an abnormally small eye size to complete bilateral anophthalmia or severe microphthalmia [56,319,320,321].
Ocular retardation (Or) mice with a null mutation of Vsx2/Chx10 have microphthalmia, thin retinas, and optic nerve aplasia. The loss of Vsx2 leads to both reduced proliferation of RPC and the absence of differentiated bipolar cells [322]. Mutations in Chx10 cause reduced RPC proliferation and an absence of bipolar cells. Vsx2 directly represses Vsx1 transcription in the mouse retina, and Vsx1 mRNA is upregulated in the RPCs of Vsx2-deficient mice and zebrafish embryos injected with Vsx2 morpholino [323]. Overexpression of Vsx2/Chx10 after transfection with plasmid expressing this gene promoted differentiation of non-photoreceptor neurons while inhibiting differentiation of photoreceptor cells in dissociated retina of 5-day-old chick embryos [324]. In Chx10-null ocular retardation mice (Chx10or-J/or-J), delay of the normal temporal expression of genes essential for photoreceptor disc morphogenesis led to failure of correct rod and cone outer segment formation in the Chx10or-J/or-J mutant retina. In addition, the absence of Chx10 appears to affect the development of late-born cells more than that of early-born cells, in that a low number of rods develop, whereas formation of ganglion, amacrine, and cone cells is relatively unaffected [325].
4. Innovative Approaches of Modern Genomics and Cell Technology for IRDs Diagnostics
Current areas of study include the genetic basis of the etiopathogenesis of retinal diseases and how to improve the methods of genetic diagnostics that underlie technologies for directed editing of the differentiated cell genome and reprogramming [10]. Knowledge about the molecular aspects of inherited eye diseases, and particularly IRDs has increased tremendously in recent years. However, the main problem in their diagnostics is a very high degree of clinical and genetic heterogeneity. There are examples of significant differences between phenotypes (e.g., age at onset) in IRDs that carry the same mutations, both within and between families. The development of new technologies is aimed at finding the causative genetic variants, with special attention paid to next-generation sequencing and genome-wide SNP homozygosity mapping, which can combine molecular diagnostics and retinal disease gene identification [320,326]. Important aspects of disease diagnostics are the functional assessment of rare variants with RNA and protein effects that can only be predicted in silico and the assessment of putative splice defects. Very useful references on several key databases that provide extensive information about gene variants, phenotypic information that accompanies variants, and information about genotype–phenotype correlations were summarized [8]. Most databases make use of Human Genome Variation Society (HGVS) nomenclature to describe variants in a consistent manner (https://varnomen.hgvs.org). To study the mechanism of variable expression, there are high hopes for genome-wide analysis techniques such as whole exome sequencing (WES) and whole genome sequencing (WGS) [327,328].
The development of modern technologies of DNA sequencing of single cells and genome sequencing has allowed experiments to be carried out at the level of individual differentiating RPE and retinal cells from human fetuses [329,330]. Spatiotemporal characteristics and dynamics of the appearance and onset of functioning of one or another cell type are now obtained using single-cell RNA sequencing (RNA-seq), which makes it possible to obtain specific cellular “transcriptome landscapes” of retinas taken at weeks 5–24 of human fetal development [331]. This method, along with bioinformatics analysis and immunochemistry, make it possible to isolate the transcriptomes of individual retinal cells sequentially differentiating into the main cellular types—photoreceptors, interneurons, ganglion cells, and Muller glia cells—along with the specialization of RPE cells, which occurs in accordance with human neural retina maturation. Data obtained were clustered into classes corresponding to certain cell types, which, in turn, were identified on the basis of markers that were previously determined and turned out to be close to the cellular types of retinas of mice and humans [331].
Bioinformatics analysis of the main distinguished transcription factors shows that they have the highest activity in regulatory networks providing differentiation of RPE and neural retina separately. It was found that the RPE and retina of the human eye have differences in the networks of genetic expression quite early [332]. A set of expressed genes specific to prospective neural retina was highly characteristic of the development of the nervous system, while RPE cells expressed genes associated with retinol metabolism [333]. In addition to substantial detailing and refinement of existing information, this made it possible to come closer to understanding the functional role of the expression of certain genes in the development of these two major eye tissues. Until now, this has been a bottleneck against a backdrop of the rapid accumulation of data on the work of certain genes in the retinas of developing vertebrates, and particularly humans.
The genomics approaches clarified the genetic signature and time of appearance of certain cell types on the basis of sequencing of isolated cells [334]. It turned out that the owners of a particular phenotype in the prospective retina for a specific set of transcription factors may be present much earlier than the dates previously known for them, albeit in a small number, and only much later to reach peak expression. For example, individual ganglion cells, according to the profile of specific expression, are detected already at 5 weeks, but their maximum population is formed only at 8 weeks. It was also found that early RPC express genes specific for mature retinal rods, but not cones. A greater similarity in the set of transcription factors between cells of the late fetus and adult compared with the early stages of gestation is expected to be revealed. A comparison was made with the known data for the retina of macaque and mouse. It is interesting that a greater similarity in the transcriptome of fetal cell types of the retina of humans and mice than humans and monkeys revealed [332]. Analysis of the transcriptome of RPE cells in their development in humans revealed a steady decline in PAX6 expression and an increase in the expression of visual cycle genes such as RPE65 and LRAT. Moreover, proliferating RPE cells were detected at 5–6 weeks, and rhodopsin expression at 24 weeks, although other genes associated with the visual cycle had expression at 13 weeks of development. Thus, determining the cells of the developing human retina based on their individual transcriptome provided information on both transcription factors expression and structural proteins. These data allowed us to come closer to understanding the functional role of molecular participants in the regulation of spatiotemporal dynamics of gene expression [331]. Information has been made available on the development of a detailed gene map of the human retina as part of the Human Cell Atlas Project, a global project to create reference maps of all human cells [335]. The genetic sequences of more than 20,000 individual cells were examined in order to develop a gene profile of all major retinal cell types to support the function and stability of the retina. Constantly enriching the data on the human retina genetic map will help us to understand the molecular profile of individual retinal cell types that enable cells to keep functioning and contribute to healthy vision, and will help us to study how those genes impact different kinds of cells. From the other side, a molecular atlas of healthy cells with cells from retinal diseases and across different stages of human development will help us to understand the factors and signals that cause cells to stop functioning and lead to vision loss and blindness [335]. Variations in gene expression coupled with the gene’s biological sources provide insight into the complex pattern of retinal gene expression that underlies specific phenotypic tissue differences, and physiologic conditions within a tissue and between normal and disease states [55]. Large-scale genetic screening of IRDs patients using whole exome/genome sequencing will continue to improve the efficiency of diagnosis through identifying novel disease-associated genes and their isoforms associated with inherited retinal degeneration [327,328].
5. Gene-Based and Cellular Technologies in the Treatment of Inherited Retinal/Eye Diseases
Experimental approaches of modern genomics for the treatment of retinal pathologies associated with impaired functions of functionally significant genes are developing rapidly. Today, this work is based on the achievements of modern cellular technologies and closely related genetic approaches that are developed primarily on animal models in vivo and in vitro or cell cultures of human eye tissues [11,336,337,338,339].
In this review, we consider a few examples of experimental and clinical work that is being done to make up for significantly reduced or lost vision in humans. Genetically determined diseases of the retina associated with mutations in genes that regulate eye development are an incentive to improve and apply technologies for targeted genome editing. The use of directional genome editing using clustered regularly interspaced short palindromic repeats (CRISPR-Cas9) systems is a promising area in biomedicine and ophthalmology for the treatment of inherited eye diseases This approach is being worked out to eliminate genetic defects leading to retinal development pathologies and loss of vision, such as RP, in experimental animals [7,340] Earlier, there was evidence of successful gene therapy for LCA in mice, using an adenovirus to deliver the gene construct as a carrier [341]. Currently, work is actively ongoing in this area, using gene constructs to eliminate defects in the neural tissue of the eye. It is known that one of the reasons for the development of IRDs is dysfunction of RPE cells. The development of degenerative diseases of the retina—RP, and LCA—is associated with mutations in the retinal pigment epithelium 65 (RPE65) gene, which is specifically expressed in RPE. Clinical studies have begun on the treatment of these diseases by introducing the functional RPE65 gene [336].
For the first time, an attempt was made to treat RP in a mouse model using CRISPR-Cas9 technology. RP is a congenital disease, genetically variable, caused by disorders in more than 60 genes with 3000 mutations, which is reason to act with the help of gene therapy. It is known that RP is caused by the gradual death of rods in the retina (which not only are responsible for night vision, but also structurally and trophically support cones) in the beginning of disease, and then cones. Using a mouse eye model, CRISPR sgRNA, which targets the retinal transcription factor neural retina leucine zipper (Nrl), responsible for the maturation of rods in the developing retina and their maintenance in the adult retina, was developed [9]. Nrl-targeted sgRNA and Cas9 endonuclease were delivered directly to the retina of mice using two adeno-associated viral (AAV) vectors. The role of Nrl is to maintain the functioning of rods, and its elimination leads to a partial conversion of rods into cones and, much more significantly, a condition supporting the viability of cones. The proposed technology of direct delivery of the AAV design to mammalian and human retinas, mediated by CRISPR-Cas9, as shown by experiments on mice, works with different forms of retinal degeneration caused by PR. Injections were given to 2-week-old mice, and observations were made 3–4 and 6 months later. As a result, at first there was a significant decrease in Nrl expression, and then downregulation of rod genes and upregulation of cone cells, as well as inhibition of the death of cone cells [9]. However, in this case, which was an absolute success, where the experiments were carried out in a mouse model, with not one but 67 genes involved in the development of pathology, important questions remain. Answers to them must be obtained before the method is used in practice. In particular, it is still unknown to what extent the data can be extrapolated to the human retina, and in particular, if long-term expression of Cas9 is harmful to mammalian and human retinal cells.
Despite the advances in CRISPR-Cas9 technology, there are some limitations that should be taken into consideration and overcome for successful therapy. It is known that Cas9 induces double-strand breaks on sequences that differ by one or more base pairs from the gRNA sequence, which is the reason for the off-target therapeutic effects [342]. Strategies to overcome this problem include improving the specificity of the Cas9 molecules, that guide RNA design and the improving the delivery methods, because most Cas9 genes are too big to be delivered by a single AAV vector. Attempts are being made to solve this problem, or at least find approaches to solving it [343,344]. Smaller Cas9 molecules have been discovered that are small enough to be packaged within an AAV [345]. It opens up the prospect of directly administering the Cas9 gene into the retina for genome editing in vivo.
With the existing shortcomings, gene therapy holds great promise for developing a cure. The first gene therapy drug targeting IRDs caused by mutations in a single gene, Luxturna™, an AAV2/2 vector carrying the RPE65 gene for the treatment of RPE65-associated RP or LCA type 2 (voretigene neparvovec-rzyl; Spark Therapeutics, Inc., Philadelphia, PA, USA), has recently been approved. This product delivers a normal copy of the RPE65 gene to retinal cells for the treatment of biallelic RPE65 mutation–associated retinal dystrophy, a blinding disease [11].
Thanks to the successes of molecular genetics and experimental embryology, a new field of medicine has emerged, regenerative medicine, which combines molecular biological, pharmaceutical, cellular and tissue technologies to stimulate reparative processes in pathologies and retinal injuries [346]. The use of cellular technologies consists of attempts to experimentally reprogram source cells (embryonic stem cells, embryonic PRC, induced pluripotent cells (iPSC), poorly differentiated cells), obtain organoids based on them, and develop methods of transplantation, in an attempt to replace them with the help of pathology or damaged tissue of the eye (retina, RPE) [339,347,348,349,350].
Some of the developed approaches are aimed at creating specific models of autologous cells and tissues with corrected genotypes for use in cell replacement therapy and tissue engineering [351]. The most important achievement in technologies for producing pluripotent cells from differentiated cells of an adult organism was made during the last decade. The iPSCs were obtained from fibroblasts of human and mouse skin using transduction of four transcription factors: Oct3/4, Sox2, Klf4, and c-Myc [352]. In the field of ophthalmology, iPSC technology is used in the development of methods for treating retinal dystrophy. So, there were reports of decreases in the degenerative processes of the RPE of the monkey following the placement of pigmented epithelial cells obtained using iPSC technology from skin fibroblasts on the retina [353]. In 2013, Japan began clinical trials using iPSC-derived RPE cells to treat AMD, in which the destruction of central retinal cells occurs [354]. The application of stem cell technology to generate photoreceptor precursor cells from patient somatic cells, which can subsequently be used for RNA and protein studies, has been described [8,355]
Approaches to creating a functioning human retina with restoration of three-dimensional structure in vitro are being developed [355,356]. One study reported on the possibility of obtaining a fragment of the neural tissue of a human retina from human iPSCs in 3D in vitro (Johns Hopkins University School of Medicine, Baltimore, MD, USA) [356]. The emerging tissue, planted in cups on a special matrigel and in a medium providing neural differentiation, had certain developmental dynamics similar to ontogeny in humans and was maintained in vitro for 200 days. In this retina, all seven types of retinal cells expressing specific molecular markers were identified morphologically. The first steps were to obtain morphologically mature photoreceptors from human iPSCs that had all the attributes of functional specialization: the inner and outer segments of the photoreceptor cell. These cells contained optic opsin pigments, transmembrane proteins responsible for transmitting a signal from captured light photons to a physiological response, which was recorded by electrochemical assay [356]. This work demonstrated the possibility of further growth and differentiation of photoreceptors obtained from human iPSCs, followed by the formation of tissue containing cells not only in the desired ratio and orientation, but also with functional activity. In that study, the autonomous mechanism of the output of artificially grown retinal cells into differentiation was a surprise. It was probably due to the presence of as yet unknown endogenic instructions accompanying the development of the retina in the absence of other eye tissues. The results of this study are a definite achievement, since so far it has not been possible to bring the resulting cells into deep differentiation physiologically close to a complete retina. The authors of that study warned that a significant amount of time is still needed until such developed cellular products can be used in clinical practice.
The other study describes the failure of stem cell transplant attempts in three patients, older women with AMD. Adipose cells taken from patients were used to obtain stem cells, but they were not fully tested for their properties and possible pathogenicity. Transplantation of these cells to the AMD patients led to serious complications and loss of vision in all three cases. The article also notes that this case is only the tip of the iceberg, since it is known that transplants of unverified cellular material are carried out in 600 US clinics in the same way. The result of AMD treatment is still small, the absence of further visual impairment for a short period of time, about 1 year [351].
In all attempts at transplantation with iPSCs or other sources of cells, cell aggregates, or fragments of retinal tissue, the most difficult and most important aspects (since we are talking about sensory tissue) are successfully integrating the transplants into the tissues of recipients and establishing the correct contact both with other cells and with the visual analyzer [346]. The results of attempts related to cell transplantation are very modest due to the complexity of the structure of the retina, the occurrence of gliosis, and the formation of a scar (in the case of proliferative or diabetic retinopathy) [357].
6. Pharmacologic Neuroprotection and Activation of Endogenous Cell Potential as an Alternative to Genetic Engineering Methods
Significant progress in the field of pharmacologic neuroprotection for mammals and humans is driven in large part by advances in approaches aimed at suppressing gliosis and fibrosis and activating internal regenerative reserves, which remains an urgent problem [358]. The main directions in studies of the regenerative potential of the retina are the search for exogenous and endogenous cellular sources to replenish the pool of cells and the development of pharmacologic drugs for the treatment/restoration of the retina, in situations where this may be feasible [359].
Despite the high incidence of retinal diseases and the complexity of mechanisms involved, several promising neuroprotective treatments provide hope to prevent blindness [360]. Since transplantation of exogenous cellular sources for the retina is not always successful, endogenous stimulation has many advantages. In this regard, the search continues for opportunities to stimulate potential endogenous cellular sources, directing control of the differentiation of cells to restore tissue and the normal functioning of the organ of vision [360,361,362]. Studies show that in damaged neurons of the retina, activation of the transcription program is observed, which promotes initial recovery and partial restoration of function. Existing strategies for the use of pharmacologic antioxidants for the pathologies of neural tissues are directed to allow them to activate their own regenerative potential and antioxidant cell systems and maintain neurogenesis. The ADNP gene is one example of homeobox genes used for neuroprotection. This approach uses stimulation of secretion of the ADNP-derived octapeptide NAP, which enhances autophagy and protects ganglion neurons in conditions of retinal ischemia and optic nerve crush [65,71].
In mammalian and human retinas, cells with potential properties of stem cells are Muller glia and RPE, which, with damage or pathology, can dedifferentiate to form a population of progenitor cells developing in the neuronal direction [363]. The data on the hypomethylated status of DNA in fish Muller glia indicate the possibility that these cells can undergo phenotype change under the influence of internal and external factors, and can take on roles in epigenetic reprogramming mechanisms and regenerative responses [364]. In the retinas of mice damaged as a result of the action of neurotoxin, it is possible to stimulate cell division of Muller glia and neurogenesis as a result of overexpression of the Ascl1 gene [365]. Muller glia cells of the adult retina, as well as the RPE, in the in vitro system are characterized by the activity of transcription factors characteristic of RPC PAX6, SOX2, and CHX10 [366]. Various experimental approaches to the regulation of proliferation and activation of the neurogenesis program in Muller glia cells are based on stimulating proliferation with exogenous signaling proteins Wnt, Notch, and Hedgehog [367] or activation of endogenous transcription factors [368,369]. Exogenous neurotrophic factors can be delivered to neuronal tissue to promote neuronal survival and regeneration [370,371]. In a number of studies, it was shown that ciliary neurotrophic factor (CNTF) [372,373], pigment epithelium derived factor (PEDF) [374], and hepatocyte growth factor (HGF) can act as stimulants of reparative processes [375].
Growth factors can be secreted by glial cells, including microglia and macroglia, like the retinal astrocytes and Muller glia. Muller glia release factors such as PEDF, transforming growth factor β (TGFβ), vascular endothelial growth factor (VEGF), BDNF, and NGF [373]. In retinal pathologies, the release of these growth factors declines, confirming the hypothesis that supporting these factors to normal levels enhances neuronal survival. Some factors, such as BDNF, are critical for synaptic plasticity, i.e., the strengthening of synaptic transmission after the activation of neuronal firing, in addition to promoting neuron survival [376,377]. Muller glia cells can be induced to de-differentiate and be brought into a pluripotent state in vitro when incubated with extracellular vesicles (exosomes) obtained from ESCs, which may indicate the possibility of their activation to restore the retina in vivo [378].
It is known that application of neurotrophic factors for neuroprotection has significant limitations due to the fact that most these factors are hydrophilic and monomeric or dimeric proteins. This can be explained by their inability to easily pass through the blood–brain, blood–retinal, or blood–cerebrospinal barrier. In addition, it was noted that neurotrophic factors are characterized by short in vivo half-lives and poor pharmacokinetics [379]. The local delivery of neuroprotective factors to the eye avoids some of these limitations.
Another approach that is still being worked out on experimental animal models involves the use of synthetic analogues of angiogenesis stimulants such as epoxyeicosotrienic acids (EETs) in the retina. It is known that vascular endothelial cells when damaged produce EETs that stimulate angiogenesis by activating the synthesis of VEGF [380].
An alternative neuroprotective strategy is based on the activation of potential endogenous mechanisms through the use of neurotransmitters and their antagonists, and targeting of the corresponding receptors [381,382]. For example, this strategy involves the use of purinergic P2X receptor antagonists and A3R adenosine receptor agonists as neuroprotectors and regulators of retinal repair [383]. Stimulation of RPE cells by a nicotinic acetylcholine receptor agonist also contributes to the activation of Muller glia proliferation and neural differentiation in adult mice [384].
Among the many neuroprotective drugs in AMD and glaucoma are targets of survival pathways, including bile acids such as ursodeoxycholic acid (UDCA) and its taurine-conjugated derivative tauroursodeoxycholic acid (TUDCA), neurotrophic factors, and therapies that target retinal dopamine [359]. The success of these approaches depends largely on the delivery of dopamine-targeted treatments to the retina, focusing on sustained release, microneedles, and molecules that are specifically tagged for retinal delivery.
7. Conclusions/Insights
We have shown that most of the key homeobox genes are involved in both retinal development and IRDs. Homeobox genes perform many functions in the retina, so their mutations are characterized by diversity among different diseases. The range of phenotypes associated with variants in regulatory genes cause phenotypic heterogeneity [385]. IRDs have in some cases multigenic etiologies, which make them difficult to treat [7]. Besides, normal eye tissue interactions and metabolic disorders, and vascular changes might also be primarily causative of pathological degenerative changes in the retina appearing as secondary effects. All of these multiple factors should not be overlooked, particularly if they become altered under the influence of changed pathological conditions. Comprehensive characterization of the pleiotropic genetic effects could improve our understanding of the etiologies of eye diseases and their mechanisms [386], as the choice of strategies requires considering the systemic nature of these retinal pathologies [387].
Advanced sequencing methods and the use of cell type-specific transcriptome and proteome and high-throughput screening systems of phenotypic and pleiotropic genetic effects analysis are aimed at identifying target genes associated with inherited diseases and provide prerequisites for better diagnostics, appropriate therapy technologies, and pharmacologic retinal protection [331,335,388,389].
Much of the success in using iPS technologies and gene modifications, as we see, has been with research conducted on laboratory animals, but translating the results into medical practice is more complicated. The iPCS-based models provide many benefits for modeling retinal pathologies, but certainly have some limitations, since it is not always possible to draw a direct connection from model animals to human IRDs [390,391,392]. In attempts to transmit data from experimental animal models to the human eye, it is important to take into account species barriers and tissue-specific features of regulating the expression of regulatory transcription factors, among which are the homeobox genes [391]. The patient-specific iPSC for deriving retinal lineages are a powerful alternative tool for discovering new disease-causing mutations and developing personalized cell therapy [393].
Cellular and tissue-like 3-D-models of inherited eye diseases in general, and IRDs in particular, in combination with genome-editing technologies such as CRISPR-Cas9, will further advance our understanding of the function of homeobox genes, their place in the regulatory hierarchy of underlying retinal pathogenesis, and how their pathogenic alleles result in retinal disorders in order to create therapies for IRDs. The use of genome-editing technology can lead to major breakthroughs in therapy, but using it safely is impossible without knowledge of the genetic nature of the cell, the characteristics of tissues, cellular responses, and compliance with ethical and social factors. The attitude of the International Society for Stem Cell Research (ISSCR) toward research based on the use of the listed gene technologies is ambiguous [8,351,355,356]. In parallel with the study of functions and their place in the genetic regulation program, a large-scale search for molecular targets is being carried out [327,328,394].
Optimization of viral delivery methods is aimed at revealing new vectors with improved characteristics and using less invasive routes of administration (e.g., intravitreal or suprachoroidal injection) for more efficient transduction of target tissues [345,346]. Efforts in the area of nonviral delivery also continue with the EyeCET electroporation technology currently being evaluated [11]. One way to maintain retinal function may be to attempt to compensate gene function by activating synergistic genes.
Neuroprotective therapeutic approaches have the potential to prevent vision loss effects with eye diseases, but despite encouraging preclinical data, the translation of neuroprotective approaches into the clinic has been fraught with failure [360]. The problems of using latent exogenous cells with stem properties for neuroprotection and restoration of the retina in humans are still far from being resolved, which is due to both the complexity of the highly organized structure of the retina and its persistent tendency to form scar tissue [346]. Currently, cell transplantation and pharmacologic antioxidant drugs still do not allow the structure of the retina in adult mammals and humans to be fully restored in vivo. These approaches make it possible to delay the death of the retinal cells and maintain cell viability for some time [347,349,395].
Activation of endogenous retinal regenerative potential seems to be the most promising approach for regenerative medicine and ophthalmology [396,397,398]. It is obvious that successes in therapy for human degenerative retinal diseases are in line with the efficiency and safety of the biomedical approaches, and thorough preclinical studies are required before treatments to avoid potential failure.
Author Contributions
Y.M. and V.S. conceived the review, wrote the text, contributed to bibliographic research and to the discussion. V.S. prepared tables and figure. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Government Program of Basic Research of Koltzov Institute of Developmental Biology of the Russian Academy of Science (IDB RAS) in 2020.
Conflicts of Interest
The authors declare no conflict of interest. The authors state that the manuscript has not been published previously.
References
- Fuhrmann, S. Eye morphogenesis and patterning of the optic vesicle. Curr. Top. Dev. Biol. 2010, 93, 61–84. [Google Scholar] [PubMed] [Green Version]
- Sinn, R.; Wittbrodt, J. An eye on eye development. Mech. Dev. 2013, 130, 347–358. [Google Scholar] [CrossRef]
- Fitzpatrick, D.R.; Van Heyningen, V. Developmental eye disorders. Curr. Opin. Genet. Dev. 2005, 15, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Zagozewski, J.L.; Zhang, Q.; Eisenstat, D.D. Genetic regulation of vertebrate eye development. Clin. Genet. 2014, 86, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Plaisancié, J.; Ceroni, F.; Holt, R.; Zazo Seco, C.; Calvas, P.; Chassaing, N.; Ragge, N.K. Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia. Hum. Genet. 2019, 138, 799–830. [Google Scholar] [CrossRef]
- Zagozewski, J.L.; Zhang, Q.; Pinto, V.I.; Wigle, J.T.; Eisenstat, D.D. The role of homeobox genes in retinal development and disease. Dev. Biol. 2014, 393, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Fadaie, Z.; Cornelis, S.S.; Cremers, F.P.M.; Roosing, S. Identification and Analysis of Genes Associated with Inherited Retinal Diseases. Methods Mol. Biol. 2019, 1834, 3–27. [Google Scholar]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef] [Green Version]
- Mellough, C.B.; Bauer, R.; Collin, J.; Dorgau, B.; Zerti, D.; Dolan, D.W.P.; Jones, C.M.; Izuogu, O.G.; Yu, M.; Hallam, D.; et al. An integrated transcriptional analysis of the developing human retina. Development 2019, 146, dev169474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, G.A.; Shalaev, E.; Karami, T.K.; Cunningham, J.; Slater, N.K.H.; Rivers, H.M. Pharmaceutical Development of AAV-Based Gene Therapy Products for the Eye. Pharm. Res. 2018, 36, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H. Simple Anatomy of the Retina. In Webvision: The Organization of the Retina and Visual System [Internet]; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 2005. [Google Scholar]
- Li, F.; Jiang, D.; Samuel, M.A. Microglia in the developing retina. Neural Dev. 2019, 14, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, Y. Morphological Survey from Neurons to Circuits of the Mouse Retina. Methods Mol. Biol. 2018, 1753, 3–25. [Google Scholar]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Mazzolini, M.; Facchetti, G.; Andolfi, L.; Proietti Zaccaria, R.; Tuccio, S.; Treu, J.; Altafini, C.; Di Fabrizio, E.M.; Lazzarino, M.; Rapp, G.; et al. The phototransduction machinery in the rod outer segment has a strong efficacy gradient. Proc. Natl. Acad. Sci. USA 2015, 112, E2715–E2724. [Google Scholar] [CrossRef] [Green Version]
- Amram, B.; Cohen-Tayar, Y.; David, A.; Ashery-Padan, R. The retinal pigmented epithelium—from basic developmental biology research to translational approaches. Int. J. Dev Biol. 2017, 61, 225–234. [Google Scholar] [CrossRef]
- Sanes, J.R.; Masland, R.H. The types of retinal ganglion cells: Current status and implications for neuronal classification. Annu. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef]
- Kolb, H. Outer Plexiform Layer. In Webvision: The Organization of the Retina and Visual System [Internet]; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Scisence. Center: Salt Lake City, UT, USA, 2005. [Google Scholar]
- Kolb, H. Inner Plexiform Layer. In Webvision: The Organization of the Retina and Visual System [Internet]; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 2001. [Google Scholar]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A journey into the retina: Müller glia commanding survival and death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef]
- Kimelberg, H.K. Functions of mature mammalian astrocytes: A current view. Neuroscientist 2010, 16, 79–106. [Google Scholar] [CrossRef]
- Genini, S.; Beltran, W.A.; Stein, V.M.; Aguirre, G.D. Isolation and ex vivo characterization of the immunophenotype and function of microglia/macrophage populations in normal dog retina. Adv. Exp. Med. Biol. 2014, 801, 339–345. [Google Scholar] [PubMed] [Green Version]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-retinal barrier. Eur. J. Ophthalmol. 2011, 21 (Suppl. 6), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Coránguez, M.; Ramos, C.; Antonetti, D.A. The inner blood-retinal barrier: Cellular basis and development. Vision Res. 2017, 139, 123–137. [Google Scholar] [CrossRef]
- Trost, A.; Bruckner, D.; Rivera, F.J.; Reitsamer, H.A. Pericytes in the Retina. Adv. Exp. Med. Biol. 2019, 1122, 1–26. [Google Scholar]
- Gregory-Evans, C.Y.; Wallace, V.A.; Gregory-Evans, K. Gene networks: Dissecting pathways in retinal development and disease. Prog. Retin. Eye Res. 2013, 33, 40–66. [Google Scholar] [CrossRef]
- Mann, I. The Development of the Human Eye, 2nd ed.; Grune and Stratton: New York, NY, USA, 1950; p. 312. [Google Scholar]
- Nishina, S.; Kohsaka, S.; Yamaguchi, Y.; Handa, H.; Kawakami, A.; Fujisawa, H.; Azuma, N. PAX6 expression in the developing human eye. Br. J. Ophthalmol. 1999, 83, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Semina, E.V.; Brownell, I.; Mintz-Hittner, H.A.; Murray, J.C.; Jamrich, M. Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum. Mol. Genet. 2001, 10, 231–236. [Google Scholar] [CrossRef]
- Markitantova, Y.V.; Smirnova, Y.A.; Panova, I.G.; Sukhikh, R.D.; Zinov’eva, V.I.; Mitashov, V.I. Analysis of expression of regulatory genes Pax6, Prox1, and Pitx2 in differentiating eye cells in human fetus. Biol. Bull. 2006, 33, 339–346. [Google Scholar] [CrossRef]
- Markitantova, Y.V.; Zinovieva, R.D. Intracellular localization of transcription factor PROX1 in the human retina in ontogeny. Biol. Bull. 2014, 41, 103–108. [Google Scholar] [CrossRef]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, M.A.; Livesey, F.J.; Cepko, C.L.; Oliver, G. Prox1 function controls progenitor cell proliferation and horizontal cell genesis in the mammalian retina. Nat. Genet. 2003, 34, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Wigle, J.T.; Eisenstat, D.D. Homeobox genes in vertebrate forebrain development and disease. Clin. Genet. 2008, 73, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, G.; Giannaccini, M.; Biasci, D.; Mariotti, S.; Degl’innocenti, A.; Perrotta, M.; Barsacchi, G.; Andreazzoli, M. Characterization of the Rx1-dependent transcriptome during early retinal development. Dev. Dyn. 2014, 243, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.T.; Ma, W.; Liang, L.; Wang, S.Z. bHLH genes and retinal cell fate specification. Mol. Neurobiol. 2005, 32, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, R.; Kageyama, R. Regulation of retinal cell fate specification by multiple transcription factors. Brain Res. 2008, 1192, 90–98. [Google Scholar] [CrossRef]
- Xiang, M. Intrinsic control of mammalian retinogenesis. Cell. Mol. Life Sci. 2013, 70, 2519–2532. [Google Scholar] [CrossRef] [Green Version]
- Cid, E.; Santos-Ledo, A.; Parrilla-Monge, M.; Lillo, C.; Arévalo, R.; Lara, J.M.; Aijón, J.; Velasco, A. Prox1 expression in rod precursors and Müller cells. Exp. Eye Res. 2010, 90, 267–276. [Google Scholar] [CrossRef]
- Heavner, W.; Pevny, L. Eye development and retinogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008391. [Google Scholar] [CrossRef] [Green Version]
- Buono, L.; Martinez-Morales, J.R. Retina Development in Vertebrates: Systems Biology Approaches to Understanding Genetic Programs: On the Contribution of Next-Generation Sequencing Methods to the Characterization of the Regulatory Networks Controlling Vertebrate Eye Development. Bioessays 2020, e1900187. [Google Scholar] [CrossRef]
- Miles, A.; Tropepe, V. Coordinating progenitor cell cycle exit and differentiation in the developing vertebrate retina. Neurogenesis (Austin) 2016, 3, e1161697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanescu, D.; Iseli, H.P.; Schwerdtfeger, K.; Ittner, L.M.; Remé, C.E.; Hafezi, F. Continuous expression of the homeobox gene Pax6 in the ageing human retina. Eye (Lond) 2007, 21, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yoshimoto, T.; Shimoda, M.; Kono, T.; Koizumi, M.; Yazumi, S.; Shimada, Y.; Doi, R.; Chiba, T.; Kubo, H. Loss of function of the candidate tumor suppressor prox1 by RNA mutation in human cancer cells. Neoplasia 2006, 8, 1003–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.T.; Kim, J.W. Compartmentalization of vertebrate optic neuroephithelium: External cues and transcription factors. Mol. Cells 2012, 33, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Lamb, T.D.; Collin, S.P.; Pugh, E.N. Evolution of the vertebrate eye: Opsins, photoreceptors, retina and eye cup. Nat. Rev. Neurosci. 2007, 8, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Wässle, H.; Puller, C.; Müller, F.; Haverkamp, S. Cone contacts, mosaics, and territories of bipolar cells in the mouse retina. J. Neurosci. 2009, 29, 106–117. [Google Scholar] [CrossRef]
- Farkas, M.H.; Grant, G.R.; White, J.A.; Sousa, M.E.; Consugar, M.B.; Pierce, E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genom. 2013, 14, 486. [Google Scholar] [CrossRef] [Green Version]
- Bibb, L.C.; Holt, J.K.; Tarttelin, E.E.; Hodges, M.D.; Gregory-Evans, K.; Rutherford, A.; Lucas, R.J.; Sowden, J.C.; Gregory-Evans, C.Y. Temporal and spatial expression patterns of the CRX transcription factor and its downstream targets. Critical differences during human and mouse eye development. Hum. Mol. Genet. 2001, 10, 1571–1579. [Google Scholar] [CrossRef]
- Hartl, D.; Krebs, A.R.; Jüttner, J.; Roska, B.; Schübeler, D. Cis-regulatory landscapes of four cell types of the retina. Nucleic Acids Res. 2017, 45, 11607–11621. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.E.; Enright, J.M.; Myers, C.A.; Shen, S.Q.; Corbo, J.C. Cell Type-Specific Epigenomic Analysis Reveals A Uniquely Closed Chromatin Architecture in Mouse Rod Photoreceptors Scientific Reports. Sci Rep. 2017, 7, 43184. [Google Scholar] [CrossRef] [Green Version]
- Ferda Percin, E.; Ploder, L.A.; Yu, J.J.; Arici, K.; Horsford, D.J.; Rutherford, A.; Bapat, B.; Cox, D.W.; Duncan, A.M.; Kalnins, V.I.; et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat. Genet. 2000, 25, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Liu, D.; Farkas, R.H.; Gunatilaka, T.L.; Hackam, A.S.; Bernstein, S.L.; Campochiaro, P.A.; Parmigiani, G.; Zack, D.J. Gene expression variation in the adult human retina. Hum. Mol. Genet. 2003, 12, 2881–2893. [Google Scholar] [CrossRef] [PubMed]
- Voronina, V.A.; Kozhemyakina, E.A.; O’Kernick, C.M.; Kahn, N.D.; Wenger, S.L.; Linberg, J.V.; Schneider, A.S.; Mathers, P.H. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum. Mol. Genet. 2004, 13, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Hever, A.M.; Williamson, K.A.; Van Heyningen, V. Developmental malformations of the eye: The role of PAX6, SOX2 and OTX2. Clin. Genet. 2006, 69, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Westmoreland, J.J.; Kilic, G.; Sartain, C.; Sirma, S.; Blain, J.; Rehg, J.; Harvey, N.; Sosa-Pineda, B. Pancreas-specific deletion of Prox1 affects development and disrupts homeostasis of the exocrine pancreas. Gastroenterology 2012, 142, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Moisseiev, E.; Yiu, G. Retinal detachment in severe myopia. Lancet 2017, 389, 1133. [Google Scholar] [CrossRef]
- Zamostiano, R.; Pinhasov, A.; Gelber, E.; Steingart, R.A.; Seroussi, E.; Giladi, E.; Bassan, M.; Wollman, Y.; Eyre, H.J.; Mulley, J.C.; et al. Cloning and characterization of the human activity-dependent neuroprotective protein. J. Biol. Chem. 2001, 276, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Furman, S.; Steingart, R.A.; Mandel, S.; Hauser, J.M.; Brenneman, D.E.; Gozes, I. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 2004, 1, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Teuchner, B.; Dimmer, A.; Humpel, C.; Amberger, A.; Fischer-Colbrie, R.; Nemeth, J.; Waschek, J.A.; Kieselbach, G.; Kralinger, M.; Schmid, E.; et al. VIP, PACAP-38, BDNF and ADNP in NMDA-induced excitotoxicity in the rat retina. Acta Ophthalmol. 2011, 7, 670–675. [Google Scholar] [CrossRef]
- Sragovich, S.; Merenlender-Wagner, A.; Gozes, I. ADNP Plays a Key Role in Autophagy: From Autism to Schizophrenia and Alzheimer’s Disease. Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Jehle, T.; Dimitriu, C.; Auer, S.; Knoth, R.; Vidal-Sanz, M.; Gozes, I.; Lagrèze, W.A. The neuropeptide NAP provides neuroprotection against retinal ganglion cell damage after retinal ischemia and optic nerve crush. Graefes Arch. Clin. Exp. Ophthalmol. 2008, 246, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Guerri, C. The peptide NAP promotes neuronal growth and differentiation through extracellular signal-regulated protein kinase and Akt pathways, and protects neurons co-cultured with astrocytes damaged by ethanol. J. Neurochem. 2007, 103, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; Van den Ende, J.; Schuurs-Hoeijmakers, J.H.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, M.J.; Titus, H.E.; Harman, G.A.; Alabduljalil, T.; Dennis, A.; Wilson, J.L.; Koeller, D.M.; Finanger, E.; Blasco, P.A.; Chiang, P.W.; et al. Longitudinal ophthalmic findings in a child with Helsmoortel-Van der Aa Syndrome. Am. J. Ophthalmol. Case Rep. 2018, 10, 244–248. [Google Scholar] [CrossRef]
- Pascolini, G.; Agolini, E.; Majore, S.; Novelli, A.; Grammatico, P.; Digilio, M.C. Helsmoortel-Van der Aa Syndrome as emerging clinical diagnosis in intellectually disabled children with autistic traits and ocular involvement. Eur. J. Paediatr. Neurol. 2018, 22, 552–557. [Google Scholar] [CrossRef]
- Pinhasov, A.; Mandel, S.; Torchinsky, A.; Giladi, E.; Pittel, Z.; Goldsweig, A.M.; Servoss, S.J.; Brenneman, D.E.; Gozes, I. Activity-dependent neuroprotective protein: A novel gene essential for brain formation. Brain Res. Dev. Brain Res. 2003, 144, 83–90. [Google Scholar] [CrossRef]
- Sragovich, S.; Malishkevich, A.; Piontkewitz, Y.; Giladi, E.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Gozes, I. The autism/neuroprotection-linked ADNP/NAP regulate the excitatory glutamatergic synapse. Transl. Psychiatry 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, S.; Iharada, M.; Omote, H.; Moriyama, Y.; Hiasa, M. Function and expression of a splicing variant of vesicular glutamate transporter 1. Biochim. Biophys. Acta Biomembr. 2017, 1859, 931–940. [Google Scholar] [CrossRef]
- McGonnell, I.M.; Graham, A.; Richardson, J.; Fish, J.L.; Depew, M.J.; Dee, C.T.; Holland, P.W.; Takahashi, T. Evolution of the Alx homeobox gene family: Parallel retention and independent loss of the vertebrate Alx3 gene. Evol. Dev. 2011, 13, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Behringer, R.R.; De Crombrugghe, B. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat. Genet. 1996, 13, 275–283. [Google Scholar] [CrossRef]
- Ten Berge, D.; Brouwer, A.; El Bahi, S.; Guénet, J.L.; Robert, B.; Meijlink, F. Mouse Alx3: An aristaless-like homeobox gene expressed during embryogenesis in ectomesenchyme and lateral plate mesoderm. Dev. Biol. 1998, 199, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabino, S.M.; Spada, F.; Cotelli, F.; Boncinelli, E. Inactivation of the zebrafish homologue of Chx10 by antisense oligonucleotides causes eye malformations similar to the ocular retardation phenotype. Mech. Dev. 1997, 63, 133–143. [Google Scholar] [CrossRef]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111–1126.e5. [Google Scholar] [CrossRef] [PubMed]
- Uz, E.; Alanay, Y.; Aktas, D.; Vargel, I.; Gucer, S.; Tuncbilek, G.; Von Eggeling, F.; Yilmaz, E.; Deren, O.; Posorski, N.; et al. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: Expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am. J. Hum. Genet. 2010, 86, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twigg, S.R.; Versnel, S.L.; Nürnberg, G.; Lees, M.M.; Bhat, M.; Hammond, P.; Hennekam, R.C.; Hoogeboom, A.J.; Hurst, J.A.; Johnson, D.; et al. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am. J. Hum. Genet. 2009, 84, 698–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayserili, H.; Uz, E.; Niessen, C.; Vargel, I.; Alanay, Y.; Tuncbilek, G.; Yigit, G.; Uyguner, O.; Candan, S.; Okur, H.; et al. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum. Mol. Genet. 2009, 18, 4357–4366. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Chen, L.; Zhou, Y.; Mi, T.; Chen, D.Y.; Chen, L.; Yin, J.; Xue, Z.F. Multiple abnormalities due to a nonsense mutation in the Alx4 gene. Genet. Mol. Res. 2013, 12, 2771–2778. [Google Scholar] [CrossRef]
- Brouwer, A.; Ten Berge, D.; Wiegerinck, R.; Meijlink, F. The OAR/aristaless domain of the homeodomain protein Cart1 has an attenuating role in vivo. Mech. Dev. 2003, 120, 241–252. [Google Scholar] [CrossRef]
- Dee, C.T.; Szymoniuk, C.R.; Mills, P.E.; Takahashi, T. Defective neural crest migration revealed by a Zebrafish model of Alx1-related frontonasal dysplasia. Hum. Mol. Genet. 2013, 22, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Lakhwani, S.; García-Sanz, P.; Vallejo, M. Alx3-deficient mice exhibit folic acid-resistant craniofacial midline and neural tube closure defects. Dev. Biol. 2010, 344, 869–880. [Google Scholar] [CrossRef]
- Beverdam, A.; Brouwer, A.; Reijnen, M.; Korving, J.; Meijlink, F. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development 2001, 128, 3975–3986. [Google Scholar] [PubMed]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; Tidhar, R.; Futerman, A.H. Ceramide synthases: Roles in cell physiology and signaling. Adv. Exp. Med. Biol. 2010, 688, 60–71. [Google Scholar] [PubMed]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef] [Green Version]
- Brüggen, B.; Kremser, C.; Bickert, A.; Ebel, P.; Vom Dorp, K.; Schultz, K.; Dörmann, P.; Willecke, K.; Dedek, K. Defective ceramide synthases in mice cause reduced amplitudes in electroretinograms and altered sphingolipid composition in retina and cornea. Eur. J. Neurosci. 2016, 44, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Kremser, C.; Klemm, A.L.; Van Uelft, M.; Imgrund, S.; Ginkel, C.; Hartmann, D.; Willecke, K. Cell-type-specific expression pattern of ceramide synthase 2 protein in mouse tissues. Histochem. Cell Biol. 2013, 140, 533–547. [Google Scholar] [CrossRef]
- Schenck, M.; Carpinteiro, A.; Grassmé, H.; Lang, F.; Gulbins, E. Ceramide: Physiological and pathophysiological aspects. Arch. Biochem. Biophys. 2007, 462, 171–175. [Google Scholar] [CrossRef]
- German, O.L.; Miranda, G.E.; Abrahan, C.E.; Rotstein, N.P. Ceramide is a mediator of apoptosis in retina photoreceptors. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1658–1668. [Google Scholar] [CrossRef] [Green Version]
- Kirin, M.; Chandra, A.; Charteris, D.G.; Hayward, C.; Campbell, S.; Celap, I.; Bencic, G.; Vatavuk, Z.; Kirac, I.; Richards, A.J.; et al. Genome-wide association study identifies genetic risk underlying primary rhegmatogenous retinal detachment. Hum. Mol. Genet. 2013, 22, 3174–3185. [Google Scholar] [CrossRef] [Green Version]
- Barak, A.; Morse, L.S.; Goldkorn, T. Ceramide: A potential mediator of apoptosis in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 247–254. [Google Scholar]
- Kannan, R.; Jin, M.; Gamulescu, M.A.; Hinton, D.R. Ceramide-induced apoptosis: Role of catalase and hepatocyte growth factor. Free Radic. Biol. Med. 2004, 37, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Lereis, L.M.; Liebisch, G.; Schick, T.; Lin, Y.; Grassmann, F.; Uchida, K.; Zipfel, P.F.; Fauser, S.; Skerka, C.; Weber, B.H.F. Evaluation of serum sphingolipids and the influence of genetic risk factors in age-related macular degeneration. PLoS ONE 2018, 13, e0200739. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tran, J.T.; Eckerd, A.; Huynh, T.P.; Elliott, M.H.; Brush, R.S.; Mandal, N.A. Inhibition of de novo ceramide biosynthesis by FTY720 protects rat retina from light-induced degeneration. J. Lipid Res. 2013, 54, 1616–1629. [Google Scholar] [CrossRef] [Green Version]
- Zarbin, M.A.; Green, W.R.; Moser, H.W.; Morton, S.J. Farber’s disease. Light and electron microscopic study of the eye. Arch. Ophthalmol. 1985, 103, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, M.A.; Green, W.R.; Moser, A.B.; Tiffany, C. Increased levels of ceramide in the retina of a patient with Farber’s disease. Arch. Ophthalmol. 1988, 106, 1163. [Google Scholar] [CrossRef] [PubMed]
- Imgrund, S.; Hartmann, D.; Farwanah, H.; Eckhardt, M.; Sandhoff, R.; Degen, J.; Gieselmann, V.; Sandhoff, K.; Willecke, K. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J. Biol. Chem. 2009, 284, 33549–33560. [Google Scholar] [CrossRef] [Green Version]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.A.; Duncan, A.; et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Morrow, E.M.; Cepko, C.L. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 1997, 91, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, Q.L.; Nie, Z.; Sun, H.; Lennon, G.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Zack, D.J. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 1997, 19, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.C.; Raymond, P.A. Zebrafish cone-rod (crx) homeobox gene promotes retinogenesis. Dev. Biol. 2004, 269, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Livesey, F.J.; Furukawa, T.; Steffen, M.A.; Church, G.M.; Cepko, C.L. Microarray analysis of the transcriptional network controlled by the photoreceptor homeobox gene Crx. Curr. Biol. 2000, 10, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, A.; Koike, C.; Lippincott, P.; Cepko, C.L.; Furukawa, T. The mouse Crx 5’-upstream transgene sequence directs cell-specific and developmentally regulated expression in retinal photoreceptor cells. J. Neurosci. 2002, 22, 1640–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assawachananont, J.; Kim, S.Y.; Kaya, K.D.; Fariss, R.; Roger, J.E.; Swaroop, A. Cone-rod homeobox CRX controls presynaptic active zone formation in photoreceptors of mammalian retina. Hum. Mol. Genet. 2018, 27, 3555–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivolta, C.; Berson, E.L.; Dryja, T.P. Dominant Leber congenital amaurosis, cone-rod degeneration, and retinitis pigmentosa caused by mutant versions of the transcription factor CRX. Hum. Mutat. 2001, 18, 488–498. [Google Scholar] [CrossRef]
- Kawamura, T.; Ohtsubo, M.; Mitsuyama, S.; Ohno-Nakamura, S.; Shimizu, N.; Minoshima, S. KMeyeDB: A graphical database of mutations in genes that cause eye diseases. Hum. Mutat. 2010, 31, 667–674. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Guo, X.; Zhang, Q. CRX variants in cone-rod dystrophy and mutation overview. Biochem. Biophys. Res. Commun. 2012, 426, 498–503. [Google Scholar] [CrossRef]
- Ibrahim, M.T.; Alarcon-Martinez, T.; Lopez, I.; Fajardo, N.; Chiang, J.; Koenekoop, R.K. A complete, homozygous CRX deletion causing nullizygosity is a new genetic mechanism for Leber congenital amaurosis. Sci. Rep. 2018, 8, 5034. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Tan, H.; Zeng, J.; Tao, D.; Ma, Y.; Liu, Y. A novel CRX variant (p.R98X) is identified in a Chinese family of Retinitis pigmentosa with atypical and mild manifestations. Genes Genom. 2019, 41, 359–366. [Google Scholar] [CrossRef]
- Kohl, S.; Kitiratschky, V.; Papke, M.; Schaich, S.; Sauer, A.; Wissinger, B. Genes and mutations in autosomal dominant cone and cone-rod dystrophy. Adv. Exp. Med. Biol. 2012, 723, 337–343. [Google Scholar]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; Da Silva Cunha, A.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Verbakel, S.K.; Van Huet, R.A.C.; Boon, C.J.F.; Den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog, Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.M.; Sieving, P.A. Assessment of foveal cone photoreceptors in Stargardt’s macular dystrophy using a small dot detection task. Vision Res. 1993, 33, 1509–1524. [Google Scholar] [CrossRef] [Green Version]
- Coppieters, F.; De Wilde, B.; Lefever, S.; De Meester, E.; De Rocker, N.; Van Cauwenbergh, C.; Pattyn, F.; Meire, F.; Leroy, B.P.; Hellemans, J.; et al. Massively parallel sequencing for early molecular diagnosis in Leber congenital amaurosis. Genet. Med. 2012, 14, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weleber, R.G.; Francis, P.J.; Trzupek, K.M.; Beattie, C. Leber Congenital Amaurosis. In Source Gene Reviews® [Internet]; University of Washington: Seattle, WA, USA, 2004. [Google Scholar]
- Furukawa, T.; Morrow, E.M.; Li, T.; Davis, F.C.; Cepko, C.L. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat. Genet. 1999, 23, 466–470. [Google Scholar] [CrossRef]
- Morrow, E.M.; Furukawa, T.; Raviola, E.; Cepko, C.L. Synaptogenesis and outer segment formation are perturbed in the neural retina of Crx mutant mice. BMC Neurosci. 2005, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Ruzycki, P.A.; Tran, N.M.; Kefalov, V.J.; Kolesnikov, A.V.; Chen, S. Graded gene expression changes determine phenotype severity in mouse models of CRX-associated retinopathies. Genome Biol. 2015, 16, 171. [Google Scholar] [CrossRef] [Green Version]
- Kelberman, D.; Dattani, M.T. Genetics of septo-optic dysplasia. Pituitary 2007, 10, 393–407. [Google Scholar] [CrossRef]
- Dattani, M.T.; Martinez-Barbera, J.P.; Thomas, P.Q.; Brickman, J.M.; Gupta, R.; Martensson, I.L.; Toresson, H.; Fox, M.; Wales, J.K.; Hindmarsh, P.C.; et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat. Genet. 1998, 19, 125–133. [Google Scholar] [CrossRef]
- Fernández-Garre, P.; Rodríguez-Gallardo, L.; Gallego-Díaz, V.; Alvarez, I.S.; Puelles, L. Fate map of the chicken neural plate at stage 4. Development 2002, 129, 2807–2822. [Google Scholar]
- Thomas, P.Q.; Dattani, M.T.; Brickman, J.M.; McNay, D.; Warne, G.; Zacharin, M.; Cameron, F.; Hurst, J.; Woods, K.; Dunger, D.; et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum. Mol. Genet. 2001, 10, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, S.; Bowling, S.; Apps, J.; Brickman, J.M.; Rodriguez, T.A.; Martinez-Barbera, J.P. Genetic Deletion of Hesx1 Promotes Exit from the Pluripotent State and Impairs Developmental Diapause. Stem Cell Rep. 2019, 13, 970–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, R.; Qiao, N.; Peng, G.; Zhang, K.; Tang, K.; Han, J.J.; Jing, N. Transcriptome analysis reveals determinant stages controlling human embryonic stem cell commitment to neuronal cells. J. Biol. Chem. 2017, 292, 19590–19604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, M.B.; Nakayama, T.; Fisher, M.; Hirsch, N.; Cox, A.; Reeder, R.; Carruthers, S.; Hall, A.; Stemple, D.L.; Grainger, R.M. Xenopus mutant reveals necessity of rax for specifying the eye field which otherwise forms tissue with telencephalic and diencephalic character. Dev. Biol. 2014, 395, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, L.; McNally, R.J.; Harrison, E.; Lloyd, I.C.; Clayton, P.E. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J. Pediatr. 2006, 148, 85–88. [Google Scholar] [CrossRef]
- Sajedi, E.; Gaston-Massuet, C.; Signore, M.; Andoniadou, C.L.; Kelberman, D.; Castro, S.; Etchevers, H.C.; Gerrelli, D.; Dattani, M.T.; Martinez-Barbera, J.P. Analysis of mouse models carrying the I26T and R160C substitutions in the transcriptional repressor HESX1 as models for septo-optic dysplasia and hypopituitarism. Dis. Model. Mech. 2008, 1, 241–254. [Google Scholar] [CrossRef]
- Andoniadou, C.L.; Signore, M.; Sajedi, E.; Gaston-Massuet, C.; Kelberman, D.; Burns, A.J.; Itasaki, N.; Dattani, M.; Martinez-Barbera, J.P. Lack of the murine homeobox gene Hesx1 leads to a posterior transformation of the anterior forebrain. Development 2007, 134, 1499–1508. [Google Scholar] [CrossRef] [Green Version]
- Schorderet, D.F.; Nichini, O.; Boisset, G.; Polok, B.; Tiab, L.; Mayeur, H.; Raji, B.; De la Houssaye, G.; Abitbol, M.M.; Munier, F.L. Mutation in the human homeobox gene NKX5-3 causes an oculo-auricular syndrome. Am. J. Hum. Genet. 2008, 82, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Yoshiura, K.; Leysens, N.J.; Reiter, R.S.; Murray, J.C. Cloning, characterization, and mapping of the mouse homeobox gene Hmx1. Genomics 1998, 50, 61–68. [Google Scholar] [CrossRef]
- Wang, W.; Lo, P.; Frasch, M.; Lufkin, T. Hmx: An evolutionary conserved homeobox gene family expressed in the developing nervous system in mice and Drosophila. Mech. Dev. 2000, 99, 123–137. [Google Scholar] [CrossRef]
- Boisset, G.; Schorderet, D.F. Zebrafish hmx1 promotes retinogenesis. Exp. Eye Res. 2012, 105, 34–42. [Google Scholar] [CrossRef]
- Vaclavik, V.; Schorderet, D.F.; Borruat, F.X.; Munier, F.L. Retinal dystrophy in the oculo-auricular syndrome due to HMX1 mutation. Ophthalmic Genet. 2011, 32, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, G.M.H.; Abdel-Hamid, M.S.; Mehrez, M.I.; Kamal, A.M.; Taher, M.B.; Afifi, H.H. Further delineation of the oculoauricular syndrome phenotype: A new family with a novel truncating HMX1 mutation. Ophthalmic Genet. 2018, 39, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.L.; Urquhart, J.; Lovell, S.C.; Biswas, S.; Parry, N.R.; Schorderet, D.F.; Lloyd, I.C.; Clayton-Smith, J.; Black, G.C. Abrogation of HMX1 function causes rare oculoauricular syndrome associated with congenital cataract, anterior segment dysgenesis, and retinal dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munroe, R.J.; Prabhu, V.; Acland, G.M.; Johnson, K.R.; Harris, B.S.; O’Brien, T.P.; Welsh, I.C.; Noden, D.M.; Schimenti, J.C. Mouse H6 Homeobox 1 (Hmx1) mutations cause cranial abnormalities and reduced body mass. BMC Dev. Biol. 2009, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunston, J.A.; Hamlington, J.D.; Zaveri, J.; Sweeney, E.; Sibbring, J.; Tran, C.; Malbroux, M.; O’Neill, J.P.; Mountford, R.; McIntosh, I. The human LMX1B gene: Transcription unit, promoter, and pathogenic mutations. Genomics 2004, 84, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, C.; Hu, X. LIM homeobox transcription factors, a novel subfamily which plays an important role in cancer (review). Oncol. Rep. 2014, 31, 1975–1985. [Google Scholar] [CrossRef] [Green Version]
- Pressman, C.L.; Chen, H.; Johnson, R.L. LMX1B, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis 2000, 26, 15–25. [Google Scholar] [CrossRef]
- Bongers, E.M.; Huysmans, F.T.; Levtchenko, E.; De Rooy, J.W.; Blickman, J.G.; Admiraal, R.J.; Huygen, P.L.; Cruysberg, J.R.; Toolens, P.A.; Prins, J.B.; et al. Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur. J. Hum. Genet. 2005, 13, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Millá, E.; Hernan, I.; Gamundi, M.J.; Martínez-Gimeno, M.; Carballo, M. Novel LMX1B mutation in familial nail-patella syndrome with variable expression of open angle glaucoma. Mol. Vis. 2007, 13, 639–648. [Google Scholar]
- Choquet, H.; Paylakhi, S.; Kneeland, S.C.; Thai, K.K.; Hoffmann, T.J.; Yin, J.; Kvale, M.N.; Banda, Y.; Tolman, N.G.; Williams, P.A.; et al. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat. Commun. 2018, 9, 2278. [Google Scholar] [CrossRef] [Green Version]
- Bongers, E.M.; De Wijs, I.J.; Marcelis, C.; Hoefsloot, L.H.; Knoers, N.V. Identification of entire LMX1B gene deletions in nail patella syndrome: Evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur. J. Hum. Genet. 2008, 16, 1240–1244. [Google Scholar] [CrossRef] [Green Version]
- Cross, S.H.; Macalinao, D.G.; McKie, L.; Rose, L.; Kearney, A.L.; Rainger, J.; Thaung, C.; Keighren, M.; Jadeja, S.; West, K.; et al. A dominant-negative mutation of mouse Lmx1b causes glaucoma and is semi-lethal via LDB1-mediated dimerization. PLoS Genet. 2014, 10, e1004359. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.; Gestri, G.; Wilson, S.W.; Link, B.A. Lmx1b is essential for survival of periocular mesenchymal cells and influences Fgf-mediated retinal patterning in zebrafish. Dev. Biol. 2009, 332, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, L.; Catoire, H.; Dion, P.; Gaspar, C.; Lafrenière, R.G.; Girard, S.L.; Levchenko, A.; Rivière, J.B.; Fiori, L.; St-Onge, J.; et al. MEIS1 intronic risk haplotype associated with restless legs syndrome affects its mRNA and protein expression levels. Hum. Mol. Genet. 2009, 18, 1065–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longobardi, E.; Penkov, D.; Mateos, D.; De Florian, G.; Torres, M.; Blasi, F. Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev. Dyn. 2014, 243, 59–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, D.; Geerts, D. MEIS transcription factors in development and disease. Development 2019, 146, dev174706. [Google Scholar] [CrossRef] [Green Version]
- Bessa, J.; Tavares, M.J.; Santos, J.; Kikuta, H.; Laplante, M.; Becker, T.S.; Gómez-Skarmeta, J.L.; Casares, F. Meis1 regulates cyclin D1 and c-myc expression; and controls the proliferation of the multipotent cells in the early developing zebrafish eye. Development 2008, 135, 799–803. [Google Scholar] [CrossRef] [Green Version]
- Heine, P.; Dohle, E.; Bumsted-O’Brien, K.; Engelkamp, D.; Schulte, D. Evidence for an evolutionary conserved role of homothorax/Meis1/2 during vertebrate retina development. Development 2008, 135, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Erickson, T.; French, C.R.; Waskiewicz, A.J. Meis1 specifies positional information in the retina and tectum to organize the zebrafish visual system. Neural Dev. 2010, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Hisa, T.; Spence, S.E.; Rachel, R.A.; Fujita, M.; Nakamura, T.; Ward, J.M.; Devor-Henneman, D.E.; Saiki, Y.; Kutsuna, H.; Tessarollo, L.; et al. Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. EMBO J. 2004, 23, 450–459. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Li, Y.; Luo, H.; Xiang, M.; Cai, L. Meis1 regulates Foxn4 expression during retinal progenitor cell differentiation. Biol. Open. 2013, 2, 1125–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos, S.; González-Lázaro, M.; Beccari, L.; Carramolino, L.; Martin-Bermejo, M.J.; Aarie, O.; Mateos-San Martín, D.; Torroja, C.; Bogdanović, O.; Doohan, R.; et al. Meis1 coordinates a network of genes implicated in eye development and microphthalmia. Development 2015, 142, 3009–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alappat, S.; Zhang, Z.Y.; Chen, Y.P. Msx homeobox gene family and craniofacial development. Cell Res. 2003, 13, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Robert, B. msh/Msx gene family in neural development. Trends Genet. 2005, 21, 624–632. [Google Scholar] [CrossRef]
- Trousse, F.; Esteve, P.; Bovolenta, P. Bmp4 mediates apoptotic cell death in the developing chick eye. J. Neurosci. 2001, 21, 1292–1301. [Google Scholar] [CrossRef]
- Wang, W.; Chen, X.; Xu, H.; Lufkin, T. Msx3: A novel murine homologue of the Drosophila msh homeobox gene restricted to the dorsal embryonic central nervous system. Mech. Dev. 1996, 58, 203–215. [Google Scholar] [CrossRef]
- Cheng, S.L.; Shao, J.S.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. MSX2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J. Biol. Chem. 2003, 278, 45969–45977. [Google Scholar] [CrossRef] [Green Version]
- Newberry, E.P.; Latifi, T.; Battaile, J.T.; Towler, D.A. Structure-function analysis of Msx2-mediated transcriptional suppression. Biochemistry 1997, 36, 10451–10462. [Google Scholar] [CrossRef]
- Jabs, E.W.; Müller, U.; Li, X.; Ma, L.; Luo, W.; Haworth, I.S.; Klisak, I.; Sparkes, R.; Warman, M.L.; Mulliken, J.B.; et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 1993, 75, 443–450. [Google Scholar] [CrossRef]
- Florisson, J.M.; Verkerk, A.J.; Huigh, D.; Hoogeboom, A.J.; Swagemakers, S.; Kremer, A.; Heijsman, D.; Lequin, M.H.; Mathijssen, I.M.; Van der Spek, P.J. Boston type craniosynostosis: Report of a second mutation in MSX2. Am. J. Med. Genet. A. 2013, 161A, 2626–2633. [Google Scholar] [CrossRef]
- Wilkie, A.O.; Tang, Z.; Elanko, N.; Walsh, S.; Twigg, S.R.; Hurst, J.A.; Wall, S.A.; Chrzanowska, K.H.; Maxson, R.E. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat. Genet. 2000, 24, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Plaisancié, J.; Collet, C.; Pelletier, V.; Perdomo, Y.; Studer, F.; Fradin, M.; Schaefer, E.; Speeg-Schatz, C.; Bloch-Zupan, A.; Flori, E.; et al. MSX2 Gene Duplication in a Patient with Eye Development Defects. Ophthalmic Genet 2015, 36, 353–358. [Google Scholar]
- Satokata, I.; Ma, L.; Ohshima, H.; Bei, M.; Woo, I.; Nishizawa, K.; Maeda, T.; Takano, Y.; Uchiyama, M.; Heaney, S.; et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 2000, 24, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Ichida, F.; Nishimura, R.; Hata, K.; Matsubara, T.; Ikeda, F.; Hisada, K.; Yatani, H.; Cao, X.; Komori, T.; Yamaguchi, A.; et al. Reciprocal roles of MSX2 in regulation of osteoblast and adipocyte differentiation. J. Biol. Chem. 2004, 279, 34015–34022. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.H.; Kundu, R.; Wu, L.; Luo, W.; Ignelzi, M.A.; Snead, M.L.; Maxson, R.E. Premature suture closure and ectopic cranial bone in mice expressing Msx2 transgenes in the developing skull. Proc. Natl. Acad. Sci. USA 1995, 92, 6137–6141. [Google Scholar] [CrossRef] [Green Version]
- Homon, J.A.; Gong, S.G. A statistical analysis of the overexpression of the msx2 RNA in Xenopus laevis. Arch. Oral. Biol. 1999, 44, 795–803. [Google Scholar] [CrossRef]
- Wu, L.Y.; Li, M.; Hinton, D.R.; Guo, L.; Jiang, S.; Wang, J.T.; Zeng, A.; Xie, J.B.; Snead, M.; Shuler, C.; et al. Microphthalmia resulting from MSX2-induced apoptosis in the optic vesicle. Invest. Ophthalmol. Vis. Sci. 2003, 44, 2404–2412. [Google Scholar] [CrossRef] [Green Version]
- Foerst-Potts, L.; Sadler, T.W. Disruption of Msx-1 and Msx-2 reveals roles for these genes in craniofacial, eye, and axial development. Dev. Dyn. 1997, 209, 70–84. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Wang, J.T. Msx2 alters the timing of retinal ganglion cells fate commitment and differentiation. Biochem. Biophys. Res. Commun. 2010, 395, 524–529. [Google Scholar] [CrossRef]
- Holme, R.H.; Thomson, S.J.; Davidson, D.R. Ectopic expression of Msx2 in chick retinal pigmented epithelium cultures suggests a role in patterning the optic vesicle. Mech. Dev. 2000, 91, 175–187. [Google Scholar] [CrossRef]
- Dateki, S.; Kosaka, K.; Hasegawa, K.; Tanaka, H.; Azuma, N.; Yokoya, S.; Muroya, K.; Adachi, M.; Tajima, T.; Motomura, K.; et al. Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J. Clin. Endocrinol. Metab. 2010, 95, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beby, F.; Lamonerie, T. The homeobox gene Otx2 in development and disease. Exp. Eye Res. 2013, 111, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Baas, D.; Bumsted, K.M.; Martinez, J.A.; Vaccarino, F.M.; Wikler, K.C.; Barnstable, C.J. The subcellular localization of Otx2 is cell-type specific and developmentally regulated in the mouse retina. Mol. Brain Res. 2000, 78, 26–37. [Google Scholar] [CrossRef]
- Fossat, N.; Le Greneur, C.; Béby, F.; Vincent, S.; Godement, P.; Chatelain, G.; Lamonerie, T. A new GFP-tagged line reveals unexpected Otx2 protein localization in retinal photoreceptors. BMC Dev. Biol. 2007, 7, 122. [Google Scholar] [CrossRef] [Green Version]
- Ragge, N.K.; Brown, A.G.; Poloschek, C.M.; Lorenz, B.; Henderson, R.A.; Clarke, M.P.; Russell-Eggitt, I.; Fielder, A.; Gerrelli, D.; Martinez-Barbera, J.P.; et al. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005, 76, 1008–1022. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.; Bakrania, P.; Bunyan, D.J.; Osborne, R.J.; Crolla, J.A.; Salt, A.; Ayuso, C.; Newbury-Ecob, R.; Abou-Rayyah, Y.; Collin, J.R.; et al. Novel heterozygous OTX2 mutations and whole gene deletions in anophthalmia; microphthalmia and coloboma. Hum. Mutat. 2008, 29, E278–E283. [Google Scholar] [CrossRef]
- Ashkenazi-Hoffnung, L.; Lebenthal, Y.; Wyatt, A.W.; Ragge, N.K.; Dateki, S.; Fukami, M.; Ogata, T.; Phillip, M.; Gat-Yablonski, G. A novel loss-of-function mutation in OTX2 in a patient with anophthalmia and isolated growth hormone deficiency. Hum. Genet. 2010, 12, 721–729. [Google Scholar] [CrossRef]
- Catania, A.; Legati, A.; Peverelli, L.; Nanetti, L.; Marchet, S.; Zanetti, N.; Lamperti, C.; Ghezzi, D. Homozygous variant in OTX2 and possible genetic modifiers identified in a patient with combined pituitary hormone deficiency, ocular involvement, myopathy, ataxia, and mitochondrial impairment. Am. J. Med. Genet. A 2019, 179, 827–831. [Google Scholar] [CrossRef]
- Abdalla-Elsayed, M.E.; Schatz, P.; Neuhaus, C.; Khan, A.O. Heterozygous mutation in OTX2 associated with early-onset retinal dystrophy with atypical maculopathy. Mol. Vis. 2017, 23, 778–784. [Google Scholar]
- Schilter, K.F.; Schneider, A.; Bardakjian, T.; Soucy, J.F.; Tyler, R.C.; Reis, L.M.; Semina, E.V. OTX2 microphthalmia syndrome: Four novel mutations and delineation of a phenotype. Clin. Genet. 2011, 79, 158–168. [Google Scholar] [CrossRef] [Green Version]
- Acampora, D.; Mazan, S.; Lallemand, Y.; Avantaggiato, V.; Maury, M.; Simeone, A.; Brûlet, P. Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development 1995, 121, 3279–3290. [Google Scholar] [PubMed]
- Matsuo, I.; Kuratani, S.; Kimura, C.; Takeda, N.; Aizawa, S. Mouse Otx2 functions in the formation and patterning of rostral head. Genes. Dev. 1995, 9, 2646–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, A.; Furukawa, A.; Koike, C.; Tano, Y.; Aizawa, S.; Matsuo, I.; Furukawa, T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci. 2003, 6, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Koike, C.; Nishida, A.; Ueno, S.; Saito, H.; Sanuki, R.; Sato, S.; Furukawa, A.; Aizawa, S.; Matsuo, I.; Suzuki, N.; et al. Functional roles of Otx2 transcription factor in postnatal mouse retinal development. Mol. Cell. Biol. 2007, 27, 8318–8329. [Google Scholar] [CrossRef] [Green Version]
- Béby, F.; Housset, M.; Fossat, N.; Le Greneur, C.; Flamant, F.; Godement, P.; Lamonerie, T. Otx2 gene deletion in adult mouse retina induces rapid RPE dystrophy and slow photoreceptor degeneration. PLoS ONE 2010, 5, e11673. [Google Scholar] [CrossRef] [Green Version]
- Sanyanusin, P.; Norrish, J.H.; Ward, T.A.; Nebel, A.; McNoe, L.A.; Eccles, M.R. Genomic structure of the human PAX2 gene. Genomics 1996, 35, 258–261. [Google Scholar] [CrossRef]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef]
- Nornes, H.O.; Dressler, G.R.; Knapik, E.W.; Deutsch, U.; Gruss, P. Spatially and temporally restricted expression of Pax2 during murine neurogenesis. Development 1990, 109, 797–809. [Google Scholar]
- Otteson, D.C.; Shelden, E.; Jones, J.M.; Kameoka, J.; Hitchcock, P.F. Pax2 expression and retinal morphogenesis in the normal and Krd mouse. Dev. Biol. 1998, 193, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Hughes, S.; Chan-Ling, T. Differentiation and migration of astrocyte precursor cells and astrocytes in human fetal retina: Relevance to optic nerve coloboma. FASEB J. 2001, 15, 2013–2015. [Google Scholar] [CrossRef]
- Chan-Ling, T.; Chu, Y.; Baxter, L.; Weible, M., II; Hughes, S. In vivo characterization of astrocyte precursor cells (APCs) and astrocytes in developing rat retinae: Differentiation, proliferation, and apoptosis. Glia 2009, 57, 39–53. [Google Scholar] [CrossRef]
- Tao, C.; Zhang, X. Development of astrocytes in the vertebrate eye. Dev. Dyn. 2014, 243, 1501–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soukkarieh, C.; Agius, E.; Soula, C.; Cochard, P. Pax2 regulates neuronal-glial cell fate choice in the embryonic optic nerve. Dev. Biol. 2007, 303, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Boije, H.; Ring, H.; López-Gallardo, M.; Prada, C.; Hallböök, F. Pax2 is expressed in a subpopulation of Müller cells in the central chick retina. Dev. Dyn. 2010, 239, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Stanke, J.; Moose, H.E.; El-Hodiri, H.M.; Fischer, A.J. Comparative study of Pax2 expression in glial cells in the retina and optic nerve of birds and mammals. J. Comp. Neurol. 2010, 518, 2316–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, R.; Karcavich, R.; Carlson, S.; Belecky-Adams, T.L. Ectopic Pax2 expression in chick ventral optic cup phenocopies loss of Pax2 expression. Dev. Biol. 2008, 319, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bower, M.; Eccles, M.; Heidet, L.; Schimmenti, L.A. Clinical utility gene card for: Renal coloboma (Papillorenal) syndrome. Eur. J. Hum. Genet. 2011, 19, 1017. [Google Scholar] [CrossRef] [Green Version]
- Bower, M.A.; Schimmenti, L.A.; Eccles, M.R. PAX2-Related Disorder. In Source Gene Reviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2007. [Google Scholar]
- Schimmenti, L.A.; Manligas, G.S.; Sieving, P.A. Optic nerve dysplasia and renal insufficiency in a family with a novel PAX2 mutation, Arg115X: Further ophthalmologic delineation of the renal-coloboma syndrome. Ophthalmic Genet. 2003, 24, 191–202. [Google Scholar] [CrossRef]
- Schimmenti, L.A. Renal coloboma syndrome. Eur. J. Hum. Genet. 2011, 19, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Schimmenti, L.A. Genetic and developmental basis of renal coloboma (papillorenal) syndrome. Exp Rev. Ophthalmol. 2009, 4, 135–144. [Google Scholar] [CrossRef]
- Torres, M.; Gómez-Pardo, E.; Gruss, P. Pax2 contributes to inner ear patterning and optic nerve trajectory. Development 1996, 122, 3381–3391. [Google Scholar] [PubMed]
- Favor, J.; Sandulache, R.; Neuhäuser-Klaus, A.; Pretsch, W.; Chatterjee, B.; Senft, E.; Wurst, W.; Blanquet, V.; Grimes, P.; Spörle, R.; et al. The mouse Pax2(1Neu) mutation is identical to a human PAX2 mutation in a family with renal-coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proc. Natl. Acad. Sci. USA 1996, 93, 13870–13875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alur, R.P.; Cox, T.A.; Crawford, M.A.; Gong, X.; Brooks, B.P. Optic nerve axon number in mouse is regulated by PAX2. J. AAPOS 2008, 12, 117–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, S.H.; McKie, L.; West, K.; Coghill, E.L.; Favor, J.; Bhattacharya, S.; Brown, S.D.; Jackson, I.J. The Opdc missense mutation of Pax2 has a milder than loss-of-function phenotype. Hum. Mol. Genet. 2011, 20, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, T.; Walton, D.S.; Maas, R.L. Genomic structure; evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat. Genet. 1992, 2, 232–239. [Google Scholar] [CrossRef]
- Mayran, A.; Pelletier, A.; Drouin, J. Pax factors in transcription and epigenetic remodelling. Semin. Cell. Dev. Biol. 2015, 44, 135–144. [Google Scholar] [CrossRef]
- Cvekl, A.; Callaerts, P. PAX6: 25th anniversary and more to learn. Exp. Eye Res. 2017, 156, 10–21. [Google Scholar] [CrossRef]
- Bäumer, N.; Marquardt, T.; Stoykova, A.; Ashery-Padan, R.; Chowdhury, K.; Gruss, P. Pax6 is required for establishing naso-temporal and dorsal characteristics of the optic vesicle. Development 2002, 129, 4535–4545. [Google Scholar]
- Bäumer, N.; Marquardt, T.; Stoykova, A.; Spieler, D.; Treichel, D.; Ashery-Padan, R.; Gruss, P. Retinal pigmented epithelium determination requires the redundant activities of Pax2 and Pax6. Development 2003, 130, 2903–2915. [Google Scholar] [CrossRef] [Green Version]
- Cavodeassi, F.; Bovolenta, P. New functions for old genes: Pax6 and Mitf in eye pigment biogenesis. Pigment Cell Melanoma Res. 2014, 27, 1005–1007. [Google Scholar] [CrossRef]
- Ashery-Padan, R.; Gruss, P. Pax6 lights-up the way for eye development. Curr. Opin. Cell. Biol. 2001, 13, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, R.; Wilson, S.W. Distribution of Pax6 protein during eye development suggests discrete roles in proliferative and differentiated visual cells. Dev. Genes Evol. 1997, 206, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Belecky-Adams, T.; Tomarev, S.; Li, H.S.; Ploder, L.; McInnes, R.R.; Sundin, O.; Adler, R. Pax-6, Prox 1, and Chx10 homeobox gene expression correlates with phenotypic fate of retinal precursor cells. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1293–1303. [Google Scholar]
- Canto-Soler, M.V.; Huang, H.; Romero, M.S.; Adler, R. Transcription factors CTCF and Pax6 are segregated to different cell types during retinal cell differentiation. Dev. Dyn. 2008, 237, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Sunderland, M.E.; Coles, B.L.; Kam, A.; Holowacz, T.; Ashery-Padan, R.; Marquardt, T.; McInnes, R.R.; Van der Kooy, D. The proliferation and expansion of retinal stem cells require functional Pax6. Dev. Biol. 2007, 304, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, T.; Ashery-Padan, R.; Andrejewski, N.; Scardigli, R.; Guillemot, F.; Gruss, P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell 2001, 105, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Philips, G.T.; Stair, C.N.; Young Lee, H.; Wroblewski, E.; Berberoglu, M.A.; Brown, N.L.; Mastick, G.S. Precocious retinal neurons: Pax6 controls timing of differentiation and determination of cell type. Dev. Biol. 2005, 279, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock, P.F.; Macdonald, R.E.; VanDeRyt, J.T.; Wilson, S.W. Antibodies against Pax6 immunostain amacrine and ganglion cells and neuronal progenitors, but not rod precursors, in the normal and regenerating retina of the goldfish. J. Neurobiol. 1996, 29, 399–413. [Google Scholar] [CrossRef]
- Rath, M.F.; Bailey, M.J.; Kim, J.S.; Coon, S.L.; Klein, D.C.; Møller, M. Developmental and daily expression of the Pax4 and Pax6 homeobox genes in the rat retina: Localization of Pax4 in photoreceptor cells. J. Neurochem. 2009, 108, 285–294. [Google Scholar] [CrossRef]
- Remez, L.A.; Onishi, A.; Menuchin-Lasowski, Y.; Biran, A.; Blackshaw, S.; Wahlin, K.J.; Zack, D.J.; Ashery-Padan, R. Pax6 is essential for the generation of late-born retinal neurons and for inhibition of photoreceptor-fate during late stages of retinogenesis. Dev. Biol. 2017, 432, 140–150. [Google Scholar] [CrossRef]
- Klimova, L.; Antosova, B.; Kuzelova, A.; Strnad, H.; Kozmik, Z. Onecut1 and Onecut2 transcription factors operate downstream of Pax6 to regulate horizontal cell development. Dev. Biol. 2015, 402, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasue, A.; Kono, H.; Habuta, M.; Bando, T.; Sato, K.; Inoue, J.; Oyadomari, S.; Noji, S.; Tanaka, E.; Ohuchi, H. Relationship between somatic mosaicism of Pax6 mutation and variable developmental eye abnormalities-an analysis of CRISPR genome-edited mouse embryos. Sci. Rep. 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Tadokoro, K.; Asaka, A.; Kawase, E.; Yamada, M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am. J. Hum. Genet. 2003, 72, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, B.K.; Yang, Y.; Cveklová, K.; Cvekl, A. Functional properties of natural human PAX6 and PAX6(5a) mutants. Invest. Ophthalmol. Vis. Sci. 2004, 45, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.C.; Gallai, R.; Olivier, D.; Speeg-Schatz, C.; Flament, J.; Calvas, P.; Dollfus, H. Variable phenotype related to a novel PAX 6 mutation (IVS4 + 5G > C) in a family presenting congenital nystagmus and foveal hypoplasia. Am. J. Ophthalmol. 2004, 138, 1016–1021. [Google Scholar] [CrossRef]
- Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef]
- Williamson, K.A.; Hall, H.N.; Owen, L.J.; Livesey, B.J.; Hanson, I.M.; Adams, G.G.W.; Bodek, S.; Calvas, P.; Castle, B.; Clarke, M.; et al. Recurrent heterozygous PAX6 missense variants cause severe bilateral microphthalmia via predictable effects on DNA-protein interaction. Genet. Med. 2019. published online 08 November 2019. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.S.; Fitzpatrick, D.R. Anophthalmia and microphthalmia. Orphanet, J. Rare Dis. 2007, 2, 47. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Sidor, B.; Szymańska, K.; Williamson, K.; Van Heyningen, V.; Roszkowski, T.; Wierzba-Bobrowicz, T.; Zaremba, J. Malformations of the brain in two fetuses with a compound heterozygosity for two PAX6 mutations. Folia Neuropathol. 2009, 47, 372–382. [Google Scholar]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2016, 24, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Hogan, B.L.; Horsburgh, G.; Cohen, J.; Hetherington, C.M.; Fisher, G.; Lyon, M.F. Small eyes (Sey): A homozygous lethal mutation on chromosome 2 which affects the differentiation of both lens and nasal placodes in the mouse. J. Embryol. Exp. Morphol. 1986, 97, 95–110. [Google Scholar] [PubMed]
- Kaufman, M.H.; Chang, H.H.; Shaw, J.P. Craniofacial abnormalities in homozygous Small eye (Sey/Sey) embryos and newborn mice. J. Anat. 1995, 186, 607–617. [Google Scholar]
- Graw, J.; Löster, J.; Puk, O.; Münster, D.; Haubst, N.; Soewarto, D.; Fuchs, H.; Meyer, B.; Nürnberg, P.; Pretsch, W.; et al. Three novel Pax6 alleles in the mouse leading to the same small-eye phenotype caused by different consequences at target promoters. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4671–4683. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, T.; Fisher, M.; Nakajima, K.; Odeleye, A.O.; Zimmerman, K.B.; Fish, M.B.; Yaoita, Y.; Chojnowski, J.L.; Lauderdale, J.D.; Netland, P.A.; et al. Xenopus pax6 mutants affect eye development and other organ systems; and have phenotypic similarities to human aniridia patients. Dev. Biol. 2015, 408, 328–344. [Google Scholar] [CrossRef] [Green Version]
- Favor, J.; Gloeckner, C.J.; Neuhäuser-Klaus, A.; Pretsch, W.; Sandulache, R.; Saule, S.; Zaus, I. Relationship of Pax6 activity levels to the extent of eye development in the mouse, Mus musculus. Genetics 2008, 179, 1345–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuel, M.; Pratt, T.; Liu, M.; Jeffery, G.; Price, D.J. Overexpression of Pax6 results in microphthalmia; retinal dysplasia and defective retinal ganglion cell axon guidance. BMC Dev. Biol. 2008, 8, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory-Evans, C.Y.; Wang, X.; Wasan, K.M.; Zhao, J.; Metcalfe, A.L.; Gregory-Evans, K. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J. Clin. Investig. 2014, 124, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, T.; Kozak, C.A.; Cepko, C.L. Rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc. Natl. Acad. Sci. USA 1997, 94, 3088–3093. [Google Scholar] [CrossRef] [Green Version]
- Orquera, D.P.; De Souza, F.S.J. Evolution of the Rax family of developmental transcription factors in vertebrates. Mech. Dev. 2017, 144, 163–170. [Google Scholar] [CrossRef]
- Chen, G.; Courey, A.J. Groucho/TLE family proteins and transcriptional repression. Gene 2000, 249, 1–16. [Google Scholar] [CrossRef]
- Muranishi, Y.; Terda, K.; Furukawa, T. An essential role for Rax in retina and neuroendocrine system development. Dev. Growth Differ. 2012, 54, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Giannaccini, M.; Giudetti, G.; Biasci, D.; Mariotti, S.; Martini, D.; Barsacchi, G.; Andreazzoli, M. Brief report: Rx1 defines retinal precursor identity by repressing alternative fates through the activation of TLE2 and Hes4. Stem Cells 2013, 31, 2842–2847. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Mukherjee, S.; Bao, Z.Z.; Morrow, E.M.; Cepko, C.L. rax, Hes1, and notch1 promote the formation of Müller glia by postnatal retinal progenitor cells. Neuron 2000, 26, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Martinez-De Luna, R.I.; Lou, C.H.; Nekkalapudi, S.; Kelly, L.E.; Sater, A.K.; El-Hodiri, H.M. Regulation of photoreceptor gene expression by the retinal homeobox (Rx) gene product. Dev. Biol. 2010, 339, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, R.; Tachibana, N.; Touahri, Y.; Zinyk, D.; Logan, C.; Schuurmans, C. Gene expression is dynamically regulated in retinal progenitor cells prior to and during overt cellular differentiation. Gene Expr. Patterns 2014, 14, 42–54. [Google Scholar] [CrossRef]
- Harris, W.A.; Perron, M. Molecular recapitulation: The growth of the vertebrate retina. Int. J. Dev. Biol. 1998, 42, 299–304. [Google Scholar] [PubMed]
- Lequeux, L.; Rio, M.; Vigouroux, A.; Titeux, M.; Etchevers, H.; Malecaze, F.; Chassaing, N.; Calvas, P. Confirmation of RAX gene involvement in human anophthalmia. Clin. Genet. 2008, 74, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rodriguez, J.; Pelcastre, E.L.; Tovilla-Canales, J.L.; Garcia-Ortiz, J.E.; Amato-Almanza, M.; Villanueva-Mendoza, C.; Espinosa-Mattar, Z.; Zenteno, J.C. Mutational screening of CHX10, GDF6, OTX2, RA.X and SOX2 genes in 50 unrelated microphthalmia-anophthalmia-coloboma (MAC) spectrum cases. Br. J. Ophthalmol. 2010, 94, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Abouzeid, H.; Youssef, M.A.; Bayoumi, N.; ElShakankiri, N.; Marzouk, I.; Hauser, P.; Schorderet, D.F. RAX and anophthalmia in humans: Evidence of brain anomalies. Mol. Vis. 2012, 18, 1449–1456. [Google Scholar]
- Voronina, V.A.; Kozlov, S.; Mathers, P.H.; Lewandoski, M. Conditional alleles for activation and inactivation of the mouse Rx homeobox gene. Genesis 2005, 41, 160–164. [Google Scholar] [CrossRef]
- Bailey, T.J.; El-Hodiri, H.; Zhang, L.; Shah, R.; Mathers, P.H.; Jamrich, M. Regulation of vertebrate eye development by Rx genes. Int. J. Dev. Biol. 2004, 48, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, S.M.; Park, L.; Stenkamp, D.L. Retinal homeobox 1 is required for retinal neurogenesis and photoreceptor differentiation in embryonic zebrafish. Dev. Biol. 2009, 328, 24–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreazzoli, M.; Gestri, G.; Angeloni, D.; Menna, E.; Barsacchi, G. Role of Xrx1 in Xenopus eye and anterior brain development. Development 1999, 126, 2451–2460. [Google Scholar] [PubMed]
- Chuang, J.C.; Raymond, P.A. Zebrafish genes rx1 and rx2 help define the region of forebrain that gives rise to retina. Dev. Biol. 2001, 231, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Winkler, S.; Loosli, F.; Henrich, T.; Wakamatsu, Y.; Wittbrodt, J. The conditional medaka mutation eyeless uncouples patterning and morphogenesis of the eye. Development 2000, 127, 1911–1919. [Google Scholar]
- Pan, Y.; Kelly, L.E.; El-Hodiri, H.M. Identification of retinal homeobox (rax) gene-dependent genes by a microarray approach: The DNA endoglycosylase neil3 is a major downstream component of the rax genetic pathway. Dev. Dyn. 2018, 247, 1199–1210. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.L.; Chen, S.; Esumi, N.; Swain, P.K.; Haines, H.S.; Peng, G.; Melia, B.M.; McIntosh, I.; Heckenlively, J.R.; Jacobson, S.G.; et al. QRX, a novel homeobox gene, modulates photoreceptor gene expression. Hum. Mol. Genet. 2004, 13, 1025–1040. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.M.; Cepko, C.L. The chicken RaxL gene plays a role in the initiation of photoreceptor differentiation. Development 2002, 129, 5363–5375. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.F.; Holland, P.W. The dynamics of vertebrate homeobox gene evolution: Gain and loss of genes in mouse and human lineages. BMC Evol. Biol. 2011, 11, 169. [Google Scholar]
- Sánchez-Arrones, L.; Ferán, J.L.; Rodríguez-Gallardo, L.; Puelles, L. Incipient forebrain boundaries traced by differential gene expression and fate mapping in the chick neural plate. Dev. Biol. 2009, 335, 43–65. [Google Scholar] [CrossRef] [Green Version]
- Pinelli, M.; Carissimo, A.; Cutillo, L.; Lai, C.H.; Mutarelli, M.; Moretti, M.N.; Singh, M.V.; Karali, M.; Carrella, D.; Pizzo, M.; et al. An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res. 2016, 44, 5773–5784. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Mathers, P.H.; Raymond, P.A. Expression of three Rx homeobox genes in embryonic and adult zebrafish. Mech. Dev. 1999, 84, 195–198. [Google Scholar] [CrossRef]
- Yang, P.; Chiang, P.W.; Weleber, R.G.; Pennesi, M.E. Autosomal Dominant Retinal Dystrophy with Electronegative Waveform Associated with a Novel RAX2 Mutation. JAMA Ophthalmol. 2015, 133, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Sompele, S.; Smith, C.; Karali, M.; Corton, M.; Van Schil, K.; Peelman, F.; Cherry, T.; Rosseel, T.; Verdin, H.; Derolez, J.; et al. Biallelic sequence and structural variants in RAX2 are a novel cause for autosomal recessive inherited retinal disease. Genet. Med. 2019, 21, 1319–1329. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Y.; Perron, M.; Hollemann, T. The role of Xenopus Rx-L in photoreceptor cell determination. Dev. Biol. 2009, 327, 352–365. [Google Scholar] [CrossRef]
- Bertuzzi, S.; Hindges, R.; Mui, S.H.; O’Leary, D.D.; Lemke, G. The homeodomain protein vax1 is required for axon guidance and major tract formation in the developing forebrain. Genes Dev. 1999, 13, 3092–3105. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Min, K.W.; Kang, K.H.; Lee, E.J.; Kim, H.T.; Moon, K.; Choi, J.; Le, D.; Lee, S.H.; Kim, J.W. Regulation of retinal axon growth by secreted Vax1 homeodomain protein. Elife 2014, 3, e02671. [Google Scholar] [CrossRef]
- Hallonet, M.; Hollemann, T.; Wehr, R.; Jenkins, N.A.; Copeland, N.G.; Pieler, T.; Gruss, P. Vax1 is a novel homeobox-containing gene expressed in the developing anterior ventral forebrain. Development 1998, 125, 2599–2610. [Google Scholar]
- Hallonet, M.; Hollemann, T.; Pieler, T.; Gruss, P. Vax1, a novel homeobox-containing gene, directs development of the basal forebrain and visual system. Genes Dev. 1999, 13, 3106–3114. [Google Scholar] [CrossRef] [Green Version]
- Mui, S.H.; Kim, J.W.; Lemke, G.; Bertuzzi, S. Vax genes ventralize the embryonic eye. Genes Dev. 2005, 19, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Take-uchi, M.; Clarke, J.D.; Wilson, S.W. Hedgehog signalling maintains the optic stalk-retinal interface through the regulation of Vax gene activity. Development 2003, 130, 955–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavotinek, A.M.; Chao, R.; Vacik, T.; Yahyavi, M.; Abouzeid, H.; Bardakjian, T.; Schneider, A.; Shaw, G.; Sherr, E.H.; Lemke, G.; et al. VAX1 mutation associated with microphthalmia, corpus callosum agenesis, and orofacial clefting: The first description of a VAX1 phenotype in humans. Hum. Mutat. 2012, 33, 364–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butali, A.; Suzuki, S.; Cooper, M.E.; Mansilla, A.M.; Cuenco, K.; Leslie, E.J.; Suzuki, Y.; Niimi, T.; Yamamoto, M.; Ayanga, G.; et al. Replication of genome wide association identified candidate genes confirm the role of common and rare variants in PAX7 and VAX1 in the etiology of nonsyndromic CL(P). Am. J. Med. Genet. A. 2013, 161A, 965–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setó-Salvia, N.; Stanier, P. Genetics of cleft lip and/or cleft palate: Association with other common anomalies. Eur. J. Med. Genet. 2014, 57, 381–393. [Google Scholar] [CrossRef]
- Schulte, D.; Furukawa, T.; Peters, M.A.; Kozak, C.A.; Cepko, C.L. Misexpression of the Emx-related homeobox genes cVax and mVax2 ventralizes the retina and perturbs the retinotectal map. Neuron 1999, 24, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Ohsaki, K.; Morimitsu, T.; Ishida, Y.; Kominami, R.; Takahashi, N. Expression of the Vax family homeobox genes suggests multiple roles in eye development. Genes Cells 1999, 4, 267–276. [Google Scholar] [CrossRef]
- Barbieri, A.M.; Lupo, G.; Bulfone, A.; Andreazzoli, M.; Mariani, M.; Fougerousse, F.; Consalez, G.G.; Borsani, G.; Beckmann, J.S.; Barsacchi, G.; et al. A homeobox gene, vax2, controls the patterning of the eye dorsoventral axis. Proc. Natl. Acad. Sci. USA 1999, 96, 10729–10734. [Google Scholar] [CrossRef] [Green Version]
- Alfano, G.; Conte, I.; Caramico, T.; Avellino, R.; Arnò, B.; Pizzo, M.T.; Tanimoto, N.; Beck, S.C.; Huber, G.; Dollé, P.; et al. Vax2 regulates retinoic acid distribution and cone opsin expression in the vertebrate eye. Development 2011, 138, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Lemke, G. Hedgehog-regulated localization of Vax2 controls eye development. Genes Dev. 2006, 20, 2833–2847. [Google Scholar] [CrossRef] [Green Version]
- Alfano, G.; Shah, A.Z.; Jeffery, G.; Bhattacharya, S.S. First insights into the expression of VAX2 in humans and its localization in the adult primate retina. Exp. Eye Res. 2016, 148, 24–29. [Google Scholar] [CrossRef]
- Alfano, G.; Waseem, N.H.; Webster, A.R.; Bhattacharya, S.S. Identification and characterization of the VAX2 p.Leu139Arg variant: Possible involvement of VAX2 in cone dystrophy. Ophthalmic Genet. 2018, 39, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Norgett, E.E.; Yii, A.; Blake-Palmer, K.G.; Sharifian, M.; Allen, L.E.; Najafi, A.; Kariminejad, A.; Karet Frankl, F.E. A role for VAX2 in correct retinal function revealed by a novel genomic deletion at 2p13.3 causing distal Renal Tubular Acidosis: Case report. BMC Med. Genet. 2015, 16, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, A.M.; Broccoli, V.; Bovolenta, P.; Alfano, G.; Marchitiello, A.; Mocchetti, C.; Crippa, L.; Bulfone, A.; Marigo, V.; Ballabio, A.; et al. Vax2 inactivation in mouse determines alteration of the eye dorsal-ventral axis; misrouting of the optic fibres and eye coloboma. Development 2002, 129, 805–813. [Google Scholar] [PubMed]
- Mui, S.H.; Hindges, R.; O’Leary, D.D.; Lemke, G.; Bertuzzi, S. The homeodomain protein Vax2 patterns the dorsoventral and nasotemporal axes of the eye. Development 2002, 129, 797–804. [Google Scholar] [PubMed]
- Levine, E.M.; Passini, M.; Hitchcock, P.F.; Glasgow, E.; Schechter, N. Vsx-1 and Vsx-2: Two Chx10-like homeobox genes expressed in overlapping domains in the adult goldfish retina. J. Comp. Neurol. 1997, 387, 439–448. [Google Scholar] [CrossRef]
- Hayashi, T.; Huang, J.; Deeb, S.S. RINX(VSX1), a novel homeobox gene expressed in the inner nuclear layer of the adult retina. Genomics 2000, 67, 128–139. [Google Scholar] [CrossRef]
- Semina, E.V.; Mintz-Hittner, H.A.; Murray, J.C. Isolation and characterization of a novel human paired-like homeodomain-containing transcription factor gene, VSX1, expressed in ocular tissues. Genomics 2000, 63, 289–293. [Google Scholar] [CrossRef]
- Dorval, K.M.; Bobechko, B.P.; Ahmad, K.F.; Bremner, R. Transcriptional activity of the paired-like homeodomain proteins CHX10 and VSX1. J. Biol. Chem. 2005, 280, 10100–10108. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Huang, J.; Deeb, S.S. Expression of rinx/vsx1 during postnatal eye development in cone-bipolar, differentiating ganglion, and lens fiber cells. Jpn. J. Ophthalmol. 2005, 49, 93–105. [Google Scholar] [CrossRef]
- Levine, E.M.; Hitchcock, P.F.; Glasgow, E.; Schechter, N. Restricted expression of a new paired-class homeobox gene in normal and regenerating adult goldfish retina. J. Comp. Neurol. 1994, 348, 596–606. [Google Scholar] [CrossRef]
- Passini, M.A.; Levine, E.M.; Canger, A.K.; Raymond, P.A.; Schechter, N. Vsx-1 and Vsx-2: Differential expression of two paired-like homeobox genes during zebrafish and goldfish retinogenesis. J. Comp. Neurol. 1997, 388, 495–505. [Google Scholar] [CrossRef]
- Ohtoshi, A.; Wang, S.W.; Maeda, H.; Saszik, S.M.; Frishman, L.J.; Klein, W.H.; Behringer, R.R. Regulation of retinal cone bipolar cell differentiation and photopic vision by the CVC homeobox gene Vsx1. Curr. Biol. 2004, 14, 530–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, R.L.; Snow, B.; Novak, J.; Looser, J.; Freund, C.; Vidgen, D.; Ploder, L.; McInnes, R.R. Vsx1, a rapidly evolving paired-like homeobox gene expressed in cone bipolar cells. Mech. Dev. 2001, 109, 315–322. [Google Scholar] [CrossRef]
- Chow, R.L.; Volgyi, B.; Szilard, R.K.; Ng, D.; McKerlie, C.; Bloomfield, S.A.; Birch, D.G.; McInnes, R.R. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proc. Natl. Acad. Sci. USA 2004, 101, 1754–1759. [Google Scholar] [CrossRef] [Green Version]
- D’Autilia, S.; Decembrini, S.; Casarosa, S.; He, R.Q.; Barsacchi, G.; Cremisi, F.; Andreazzoli, M. Cloning and developmental expression of the Xenopus homeobox gene Xvsx1. Dev. Genes Evol. 2006, 216, 829–834. [Google Scholar] [CrossRef]
- Decembrini, S.; Andreazzoli, M.; Vignali, R.; Barsacchi, G.; Cremisi, F. Timing the generation of distinct retinal cells by homeobox proteins. PLoS Biol. 2006, 4, e272. [Google Scholar] [CrossRef] [Green Version]
- Héon, E.; Greenberg, A.; Kopp, K.K.; Rootman, D.; Vincent, A.L.; Billingsley, G.; Priston, M.; Dorval, K.M.; Chow, R.L.; McInnes, R.R.; et al. VSX1: A gene for posterior polymorphous dystrophy and keratoconus. Hum. Mol. Genet. 2002, 11, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Mintz-Hittner, H.A.; Semina, E.V.; Frishman, L.J.; Prager, T.C.; Murray, J.C. VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, and abnormal retinal and auditory bipolar cells. Ophthalmology 2004, 111, 828–836. [Google Scholar] [CrossRef]
- Litke, A.M.; Samuelson, S.; Delaney, K.R.; Sauvé, Y.; Chow, R.L. Investigating the Pathogenicity of VSX1 Missense Mutations and Their Association with Corneal Disease. Invest. Ophthalmol. Vis. Sci. 2018, 59, 5824–5835. [Google Scholar] [CrossRef] [Green Version]
- Aldave, A.J.; Yellore, V.S.; Salem, A.K.; Yoo, G.L.; Rayner, S.A.; Yang, H.; Tang, G.Y.; Piconell, Y.; Rabinowitz, Y.S. No VSX1 gene mutations associated with keratoconus. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2820–2822. [Google Scholar] [CrossRef] [Green Version]
- Tanwar, M.; Kumar, M.; Nayak, B.; Pathak, D.; Sharma, N.; Titiyal, J.S.; Dada, R. VSX1 gene analysis in keratoconus. Mol. Vis. 2010, 16, 2395–2401. [Google Scholar] [PubMed]
- Valleix, S.; Nedelec, B.; Rigaudiere, F.; Dighiero, P.; Pouliquen, Y.; Renard, G.; Le Gargasson, J.F.; Delpech, M. H244R VSX1 is associated with selective cone ON bipolar cell dysfunction and macular degeneration in a PPCD family. Invest. Ophthalmol. Vis. Sci. 2006, 47, 48–54. [Google Scholar] [CrossRef]
- Liang, L.; Sandell, J.H. Focus on molecules: Homeobox protein Chx10. Exp Eye Res. 2008, 86, 541–542. [Google Scholar] [CrossRef]
- Dorval, K.M.; Bobechko, B.P.; Fujieda, H.; Chen, S.; Zack, D.J.; Bremner, R. CHX10 targets a subset of photoreceptor genes. J. Biol. Chem. 2006, 281, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Levine, E.M. Vsx2 controls eye organogenesis and retinal progenitor identity via homeodomain and non-homeodomain residues required for high affinity DNA binding. PLoS Genet. 2012, 8, e1002924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, I.S.; Chen, J.D.; Ploder, L.; Vidgen, D.; Van der Kooy, D.; Kalnins, V.I.; McInnes, R.R. Developmental expression of a novel murine homeobox gene (Chx10): Evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron 1994, 13, 377–393. [Google Scholar] [CrossRef]
- Rowan, S.; Cepko, C.L. A POU factor binding site upstream of the Chx10 homeobox gene is required for Chx10 expression in subsets of retinal progenitor cells and bipolar cells. Dev. Biol. 2005, 281, 240–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Comiskey, D.F.; Kelly, L.E.; Chandler, D.S.; El-Hodiri, H.M. Regulation of photoreceptor gene transcription via a highly conserved transcriptional regulatory element by vsx gene products. Mol. Vis. 2016, 22, 1421–1428. [Google Scholar] [PubMed]
- Rowan, S.; Chen, C.M.; Young, T.L.; Fisher, D.E.; Cepko, C.L. Transdifferentiation of the retina into pigmented cells in ocular retardation mice defines a new function of the homeodomain gene Chx10. Development 2004, 131, 5139–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, B.; Singhal, S.; Lawrence, J.M.; Khaw, P.T.; Limb, G.A. Distribution of Müller stem cells within the neural retina: Evidence for the existence of a ciliary margin-like zone in the adult human eye. Exp. Eye Res. 2009, 89, 373–382. [Google Scholar] [CrossRef]
- Green, E.S.; Stubbs, J.L.; Levine, E.M. Genetic rescue of cell number in a mouse model of microphthalmia: Interactions between Chx10 and G1-phase cell cycle regulators. Development 2003, 130, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsford, D.J.; Nguyen, M.T.; Sellar, G.C.; Kothary, R.; Arnheiter, H.; McInnes, R.R. Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development 2005, 132, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.; Al-Mureikhi, M.S.; Peltekova, I.; Tsui, L.C.; Teebi, A.S. Mutations in the CHX10 gene in non-syndromic microphthalmia/anophthalmia patients from Qatar. Clin. Genet. 2007, 72, 164–166. [Google Scholar] [CrossRef]
- Iseri, S.U.; Wyatt, A.W.; Nürnberg, G.; Kluck, C.; Nürnberg, P.; Holder, G.E.; Blair, E.; Salt, A.; Ragge, N.K. Use of genome-wide SNP homozygosity mapping in small pedigrees to identify new mutations in VSX2 causing recessive microphthalmia and a semidominant inner retinal dystrophy. Hum. Genet. 2010, 128, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Burkitt Wright, E.M.; Perveen, R.; Bowers, N.; Ramsden, S.; McCann, E.; O’Driscoll, M.; Lloyd, I.C.; Clayton-Smith, J.; Black, G.C. VSX2 in microphthalmia: A novel splice site mutation producing a severe microphthalmia phenotype. Br. J. Ophthalmol. 2010, 94, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.; Novak, J.; Liang, M.Y.; Basu, S.; Ploder, L.; Hawes, N.L.; Vidgen, D.; Hoover, F.; Goldman, D.; Kalnins, V.I.; et al. Ocular retardation mouse caused by Chx10 homeobox null allele: Impaired retinal progenitor proliferation and bipolar cell differentiation. Nat. Genet. 1996, 12, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Yun, S.; Veien, E.S.; Wu, Y.Y.; Chow, R.L.; Dorsky, R.I.; Levine, E.M. Negative regulation of Vsx1 by its paralog Chx10/Vsx2 is conserved in the vertebrate retina. Brain Res. 2008, 1192, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Toy, J.; Norton, J.S.; Jibodh, S.R.; Adler, R. Effects of homeobox genes on the differentiation of photoreceptor and nonphotoreceptor neurons. Invest. Ophthalmol. Vis. Sci. 2002, 43, 3522–3529. [Google Scholar]
- Rutherford, A.D.; Dhomen, N.; Smith, H.K.; Sowden, J.C. Delayed expression of the Crx gene and photoreceptor development in the Chx10-deficient retina. Invest. Ophthalmol. Vis. Sci. 2004, 45, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.J.; Jiang, P.; Howden, S.; Barney, P.; Min, J.; York, N.W.; Chu, L.F.; Capowski, E.E.; Cash, A.; Jain, S.; et al. A Novel Approach to Single Cell RNA-Sequence Analysis Facilitates In Silico Gene Reporting of Human Pluripotent Stem Cell-Derived Retinal Cell Types. Stem Cells 2018, 36, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Bryant, L.; Lozynska, O.; Maguire, A.M.; Aleman, T.S.; Bennett, J. Prescreening whole exome sequencing results from patients with retinal degeneration for variants in genes associated with retinal degeneration. Clin. Ophthalmol. 2017, 12, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, S.; Sun, W.; Xiao, X.; Jia, X.; Liu, M.; Xu, L.; Long, Y.; Zhang, Q. An Ophthalmic Targeted Exome Sequencing Panel as a Powerful Tool to Identify Causative Mutations in Patients Suspected of Hereditary Eye Diseases. Transl. Vis. Sci. Technol. 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazen, J.L.; Faust, G.G.; Rodriguez, A.R.; Ferguson, W.C.; Shumilina, S.; Clark, R.A.; Boland, M.J.; Martin, G.; Chubukov, P.; Tsunemoto, R.K.; et al. The Complete Genome Sequences, Unique Mutational Spectra, and Developmental Potency of Adult Neurons Revealed by Cloning. Neuron 2016, 89, 1223–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Calton, M.A.; Abell, N.S.; Benchorin, G.; Gloudemans, M.J.; Chen, M.; Hu, J.; Li, X.; Balliu, B.; Bok, D.; et al. Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms. Commun. Biol. 2019, 2, 186. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Hu, B.; Mao, Y.; Chen, Y.; Yan, L.; Yong, J.; Dong, J.; Wei, Y.; Wang, W.; et al. Dissecting the transcriptome landscape of the human fetal neural retina and retinal pigment epithelium by single-cell RNA-seq analysis. PLoS Biol. 2019, 17, e3000365. [Google Scholar] [CrossRef]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, J.; Liu, Z.; Li, J.; Yao, K.; Wu, Y. Potential Therapeutic Agents Against Retinal Diseases Caused by Aberrant Metabolism of Retinoids. Invest. Ophthalmol. Vis. Sci. 2016, 57, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Lodato, M.A.; Woodworth, M.B.; Lee, S.; Evrony, G.D.; Mehta, B.K.; Karger, A.; Lee, S.; Chittenden, T.W.; D’Gama, A.M.; Cai, X.; et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015, 350, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Lukowski, S.W.; Lo, C.Y.; Sharov, A.A.; Nguyen, Q.; Fang, L.; Hung, S.S.; Zhu, L.; Zhang, T.; Grünert, U.; Nguyen, T.; et al. A single-cell transcriptome atlas of the adult human retina. EMBO J. 2019, 38, e100811. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Aleman, T.S.; Boye, S.L.; Schwartz, S.B.; Kaushal, S.; Roman, A.J.; Pang, J.J.; Sumaroka, A.; Windsor, E.A.; Wilson, J.M.; et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA 2008, 105, 15112–15117. [Google Scholar] [CrossRef] [Green Version]
- Bacchi, N.; Casarosa, S.; Denti, M.A. Splicing-correcting therapeutic approaches for retinal dystrophies: Where endogenous gene regulation and specificity matter. Invest. Ophthalmol. Vis. Sci. 2014, 55, 3285–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelinger, L.; Swaroop, A. RNA Biology in Retinal Development and Disease. Trends Genet. 2018, 34, 341–351. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Rep. 2018, 10, 300–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjurjo-Soriano, C.; Kalatzis, V. Guiding Lights in Genome Editing Inherited Retinal Disorders: Implications for Gene and Cell Therapy. Neural Plast. 2018, 2018, 5056279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennicelli, J.; Wright, J.F.; Komaromy, A.; Jacobs, J.B.; Hauck, B.; Zelenaia, O.; Mingozzi, F.; Hui, D.; Chung, D.; Rex, T.S.; et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol. Ther. 2008, 16, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Wu, Y.; Gembrling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 29, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Kulcsár, P.I.; Tálas, A.; Huszár, K.; Ligeti, Z.; Tóth, E.; Weinhardt, N.; Fodor, E.; Welker, E. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017, 18, 190. [Google Scholar] [CrossRef] [Green Version]
- Fagerlund, R.D.; Staals, R.H.; Fineran, P.C. The Cpf1 CRISPR-Cas protein expands genome-editing tools. Genome Biol. 2015, 16, 251. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.H.; Tian, Y.; Funderburgh, J.; Pellegrini, G.; Zhang, K.; Goldberg, J.L.; Ali, R.R.; Young, M.; Xie, Y.; Temple, S. Regenerating Eye Tissues to Preserve and Restore Vision. Cell Stem Cell 2018, 22, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Qi, S.; Su, G. Retinal organotypic culture—A candidate for research on retinas. Tissue Cell 2018, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuzzani, O.; Binette, F.; Sternberg, H.; West, M.D.; Nasonkin, I.O. Pluripotent Stem Cells for Retinal Tissue Engineering: Current Status and Future Prospects. Stem Cell Rev. Rep. 2018, 14, 463–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.Y.; Kaya, K.D.; Dong, L.; Swaroop, A. Three-dimensional retinal organoids from mouse pluripotent stem cells mimic in vivo development with enhanced stratification and rod photoreceptor differentiation. Mol. Vis. 2016, 22, 1077–1094. [Google Scholar] [PubMed]
- Kuriyan, A.E.; Albini, T.A.; Townsend, J.H.; Rodriguez, M.; Pandya, H.K.; Leonard, R.E.; Parrott, M.B.; Rosenfeld, P.J.; Flynn, H.W.; Goldberg, J.L. Vision Loss after Intravitreal Injection of Autologous "Stem Cells" for AMD. N. Engl. J. Med. 2017, 376, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Takahashi, M. Induction of retinal pigment epithelial cells from monkey iPS cells. Invest. Ophthalmol. Vis. Sci. 2011, 52, 8785–8790. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Lee, M.J.; Palczewska, G.; Marsili, S.; Tesar, P.J.; Palczewskim, K.; Takahashi, M.; Maeda, A. Retinal pigmented epithelial cells obtained from human induced pluripotent stem cells possess functional visual cycle enzymes in vitro and in vivo. J. Biol. Chem. 2013, 288, 34484–34493. [Google Scholar] [CrossRef] [Green Version]
- Welby, E.; Lakowski, J.; Di Foggia, V.; Budinger, D.; Gonzalez-Cordero, A.; Lun, A.T.L.; Epstein, M.; Patel, A.; Cuevas, E.; Kruczek, K.; et al. Isolation and Comparative Transcriptome Analysis of Human Fetal and iPSC-Derived Cone Photoreceptor Cells. Stem Cell Rep. 2017, 9, 1898–1915. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Amin, S.; Roy, S. Retinal fibrosis in diabetic retinopathy. Exp. Eye. Res. 2016, 142, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusciano, D.; Pezzino, S.; Mutolo, M.G.; Giannotti, R.; Librando, A.; Pescosolido, N. Neuroprotection in Glaucoma: Old and New Promising Treatments. Adv. Pharmacol. Sci. 2017, 2017, 4320408. [Google Scholar] [CrossRef] [PubMed]
- Pardue, M.T.; Allen, R.S. Neuroprotective strategies for retinal disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef] [PubMed]
- Wubben, T.J.; Besirli, C.G.; Johnson, M.W.; Zacks, D.N. Retinal Neuroprotection: Overcoming the Translational Roadblocks. Am. J. Ophthalmol. 2018, 192, xv–xxii. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, E.; Novikova, Y.; Kilina, O.; Philippov, P. New antioxidant SkQ1 is an effective protector of rat neural retina under conditions of long-term organotypic cultivation. Adv. Aging Res. 2013, 2, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennis, A.; Gorgels, T.G.; Ten Brink, J.B.; Van der Spek, P.J.; Bossers, K.; Heine, V.M.; Bergen, A.A. Comparison of Mouse and Human Retinal Pigment Epithelium Gene Expression Profiles: Potential Implications for Age-Related Macular Degeneration. PLoS ONE 2015, 10, e0141597. [Google Scholar] [CrossRef]
- Powell, C.; Grant, A.R.; Cornblath, E.; Goldman, D. Analysis of DNA methylation reveals a partial reprogramming of the Müller glia genome during retina regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 19814–19819. [Google Scholar] [CrossRef] [Green Version]
- Jorstad, N.L.; Wilken, M.S.; Grimes, W.N.; Wohl, S.G.; VandenBosch, L.S.; Yoshimatsu, T.; Wong, R.O.; Rieke, F.; Reh, T.A. Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 2017, 548, 103–107. [Google Scholar] [CrossRef]
- Bhatia, B.; Singhal, S.; Jayaram, H.; Khaw, P.T.; Limb, G.A. Adult retinal stem cells revisited. Open Ophthalmol. J. 2010, 4, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Hamon, A.; Roger, J.E.; Yang, X.J.; Perron, M. Müller glial cell-dependent regeneration of the neural retina: An overview across vertebrate model systems. Dev. Dyn. 2016, 245, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Boda, E.; Nato, G.; Buffo, A. Emerging pharmacological approaches to promote neurogenesis from endogenous glial cells. Biochem. Pharmacol. 2017, 141, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.J.; Hyde, D.R. Opportunities for CRISPR/Cas9 Gene Editing in Retinal Regeneration Research. Front. Cell Dev. Biol. 2017, 5, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgetts, S.I.; Harvey, A.R. Neurotrophic Factors Used to Treat Spinal Cord Injury. Vitam. Horm. 2017, 104, 405–457. [Google Scholar]
- Önger, M.E.; Delibaş, B.; Türkmen, A.P.; Erener, E.; Altunkaynak, B.Z.; Kaplan, S. The role of growth factors in nerve regeneration. Drug Discov. Ther. 2017, 10, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Leaver, S.G.; Cui, Q.; Plant, G.W.; Arulpragasam, A.; Hisheh, S.; Verhaagen, J.; Harvey, A.R. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006, 13, 1328–1341. [Google Scholar] [CrossRef] [Green Version]
- Logan, A.; Ahmed, Z.; Baird, A.; Gonzalez, A.M.; Berry, M. Neurotrophic factor synergy is required for neuronal survival and disinhibited axon regeneration after CNS injury. Brain 2006, 129, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Vigneswara, V.; Esmaeilim, M.; Deer, L.; Berry, M.; Logan, A.; Ahmed, Z. Eye drop delivery of pigment epithelium-derived factor-34 promotes retinal ganglion cell neuroprotection and axon regeneration. Mol. Cell. Neurosci. 2015, 68, 212–221. [Google Scholar] [CrossRef]
- Tönges, L.; Ostendorf, T.; Lamballe, F.; Genestine, M.; Dono, R.; Koch, J.C.; Bähr, M.; Maina, F.; Lingor, P. Hepatocyte growth factor protects retinal ganglion cells by increasing neuronal survival and axonal regeneration in vitro and in vivo. J. Neurochem. 2011, 117, 892–903. [Google Scholar] [CrossRef]
- Leal, G.; Comprido, D.; Duarte, C.B. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology 2014, 76, 639–656. [Google Scholar] [CrossRef] [Green Version]
- Sasi, M.; Vignoli, B.; Canossa, M.; Blum, R. Neurobiology of local and intercellular BDNF signaling. Pflugers Arch. 2017, 469, 593–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsman, D.; Stackpole, E.J.; Domin, D.R.; Farber, D.B. Embryonic stem cell-derived microvesicles induce gene expression changes in Müller cells of the retina. PLoS ONE 2012, 7, e50417. [Google Scholar] [CrossRef] [PubMed]
- Thorne, R.G.; Frey, W.H. Delivery of neurotrophic factors to the central nervous system: Pharmacokinetic considerations. Clin. Pharmacokinet. 2001, 40, 907–946. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Dexter, D.T. Neuroprotective and symptomatic effects of targeting group III mGlu receptors in neurodegenerative disease. J. Neurochem. 2014, 129, 4–20. [Google Scholar] [CrossRef]
- Blackiston, D.J.; Vien, K.; Levin, M. Serotonergic stimulation induces nerve growth and promotes visual learning via posterior eye grafts in a vertebrate model of induced sensory plasticity. NPJ Regen. Med. 2017, 2, 8. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, M.; Laties, A.M.; Mitchell, C.H. Balance of purines may determine life or death of retinal ganglion cells as A3 adenosine receptors prevent loss following P2X7 receptor stimulation. J. Neurochem. 2006, 98, 566–575. [Google Scholar] [CrossRef]
- Webster, M.K.; Barnett, B.J.; Stanchfield, M.L.; Paris, J.R.; Webster, S.E.; Cooley-Themm, C.A.; Levine, E.M.; Otteson, D.C.; Linn, C.L. Stimulation of Retinal Pigment Epithelium With an α7 nAChR Agonist Leads to Müller Glia Dependent Neurogenesis in the Adult Mammalian Retina. Invest. Ophthalmol. Vis. Sci. 2019, 60, 570–579. [Google Scholar] [CrossRef]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Grassmann, F.; Kiel, C.; Zimmermann, M.E.; Gorski, M.; Grassmann, V.; Stark, K.; International AMD Genomics Consortium (IAMDGC); Heid, I.M.; Weber, B.H. Genetic pleiotropy between age-related macular degeneration and 16 complex diseases and traits. Genome Med. 2017, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W.J. Molecular Therapies for Inherited Retinal Diseases-Current Standing, Opportunities and Challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef] [Green Version]
- Faber, S.; Roepman, R. Balancing the Photoreceptor Proteome: Proteostasis Network Therapeutics for Inherited Retinal Disease. Genes 2019, 10, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.M.; Buck, T.M.; Mulder, A.A.; Ohonin, C.; Alves, C.H.; Vos, R.M.; Bialecka, M.; Van Herwaarden, T.; Van Dijk, E.H.C.; Talib, M.; et al. Human iPSC-Derived Retinas Recapitulate the Fetal CRB1 CRB2 Complex Formation and Demonstrate that Photoreceptors and Müller Glia Are Targets of AAV5. Stem Cell Rep. 2019, 12, 906–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacheiro, P.; Haendel, M.A.; Smedley, D.; International Mouse Phenotyping Consortium and the Monarch Initiative. New models for human disease from the International Mouse Phenotyping Consortium. Mamm. Genome 2019, 30, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, A.; Hoshino, A.; Finkbeiner, C.R.; Chitsazan, A.; Dai, L.; Haugan, A.K.; Eschenbacher, K.M.; Jackson, D.L.; Trapnell, C.; Bermingham-McDonogh, O.; et al. Single-Cell Transcriptomic Comparison of Human Fetal Retina, hPSC-Derived Retinal Organoids, and Long-Term Retinal Cultures. Cell Rep. 2020, 30, 1644–1659. [Google Scholar] [CrossRef]
- Chen, F.K.; McLenachan, S.; Edel, M.; Da Cruz, L.; Coffey, P.J.; Mackey, D.A. iPS Cells for Modelling and Treatment of Retinal Diseases. J. Clin Med. 2014, 3, 1511–1541. [Google Scholar] [CrossRef] [Green Version]
- Delyfer, M.N.; Raffelsberger, W.; Mercier, D.; Korobelnik, J.F.; Gaudric, A.; Charteris, D.G.; Tadayoni, R.; Metge, F.; Caputo, G.; Barale, P.O.; et al. Transcriptomic analysis of human retinal detachment reveals both inflammatory response and photoreceptor death. PLoS ONE 2011, 6, e28791. [Google Scholar] [CrossRef]
- Gasparini, S.J.; Llonch, S.; Borsch, O.; Ader, M. Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog. Retin. Eye Res. 2019, 69, 1–37. [Google Scholar] [CrossRef]
- Karl, M.O.; Reh, T.A. Regenerative medicine for retinal diseases: Activating endogenous repair mechanisms. Trends Mol. Med. 2010, 16, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, C.; Su, G. Cellular Signaling in Müller Glia: Progenitor Cells for Regenerative and Neuroprotective Responses in Pharmacological Models of Retinal Degeneration. J. Ophthalmol. 2019, 2019, 5743109. [Google Scholar] [CrossRef] [PubMed]
- Markitantova, Y.V.; Simirskii, V.N. The Role of the Redox System in Initiation of Neural Eye Tissues Regenerative Response in Vertebrates. Russian J. Dev. Biol. 2020, 51, 16–30. [Google Scholar]
Figure 1.
Expression of homeobox genes in the adult mouse retina. The retinal architecture is detailed in the review. The cell-specific expression of genes is detected by single-cell RNA sequencing (data from https://eyeintegration.nei.nih.gov). The only genes which are known to associate with eye/retinal malformations in humans are shown. On the left: Retinal layers; on the right: homeobox genes indicated in the same color as the cell types expressed them. Abbreviations in parentheses show corresponding cell types. RPE, retinal pigment epithelium; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; C, cone; R, rod; HC, horizontal cell; AC, amacrine cell; cBC, cone bipolar cell; rBC, rod bipolar cell; MG, Muller glia; RGC, retinal ganglion cell; Mi, microglial cell. Modified from [13]. License to reproduce: https://creativecommons.org/licenses/by/4.0/.
Figure 1.
Expression of homeobox genes in the adult mouse retina. The retinal architecture is detailed in the review. The cell-specific expression of genes is detected by single-cell RNA sequencing (data from https://eyeintegration.nei.nih.gov). The only genes which are known to associate with eye/retinal malformations in humans are shown. On the left: Retinal layers; on the right: homeobox genes indicated in the same color as the cell types expressed them. Abbreviations in parentheses show corresponding cell types. RPE, retinal pigment epithelium; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; C, cone; R, rod; HC, horizontal cell; AC, amacrine cell; cBC, cone bipolar cell; rBC, rod bipolar cell; MG, Muller glia; RGC, retinal ganglion cell; Mi, microglial cell. Modified from [13]. License to reproduce: https://creativecommons.org/licenses/by/4.0/.


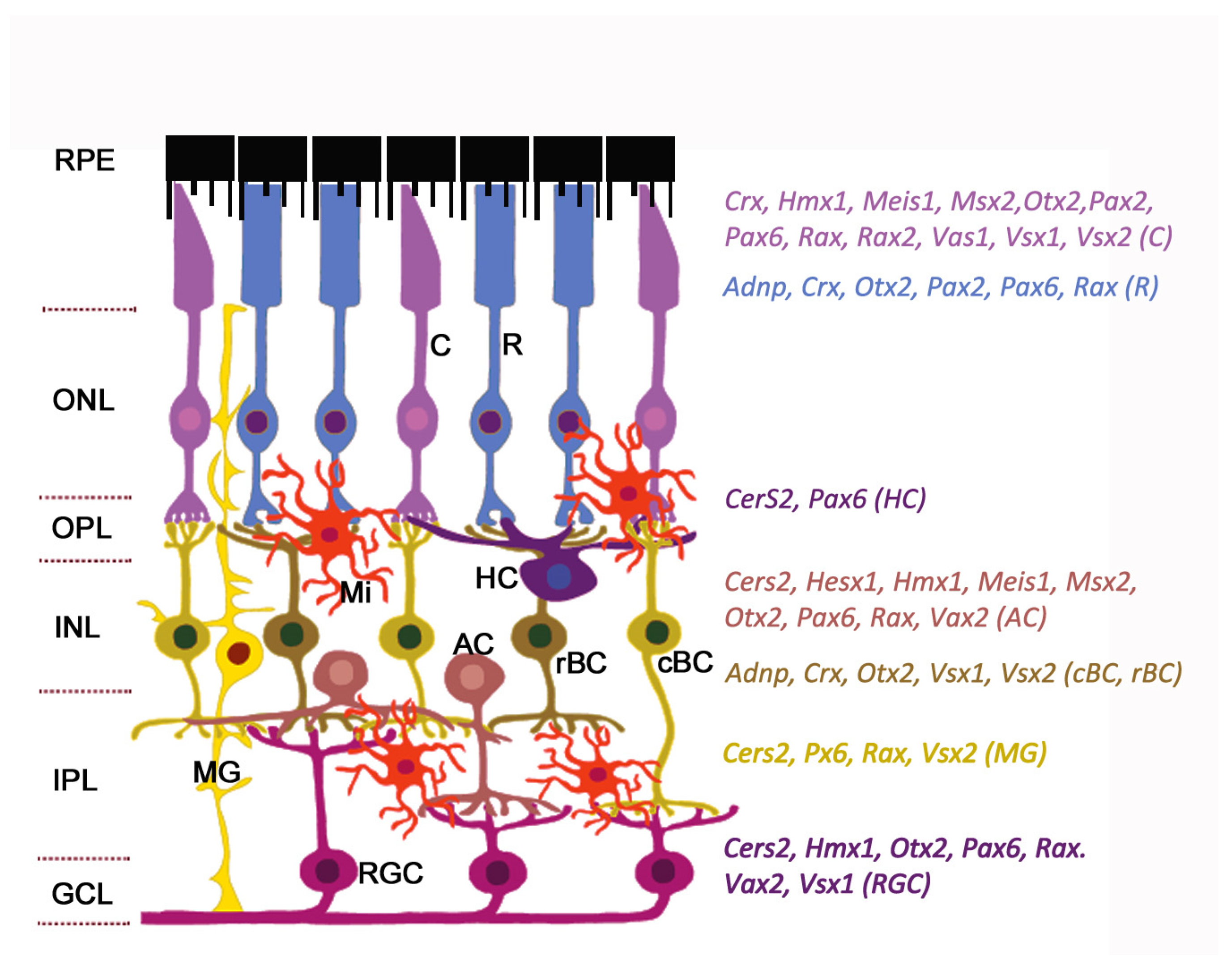{kind=link}
Table 1.
Properties of human homeobox genes associated with inherited retinal diseases (IRDs) and secondary retinal malformations.
Table 1.
Properties of human homeobox genes associated with inherited retinal diseases (IRDs) and secondary retinal malformations.
| Gene | Full Name | Synonyms | Ensembl ID | Location | Transcripts (Total/Protein-Coding) (Ensembl) | Homeobox Class | Expression in the Retina | |
|---|---|---|---|---|---|---|---|---|
| RNA (Microarray) (BioGPS) | Protein (ProteomicsDB) | |||||||
| ADNP | Activity-dependent neuroprotector homeobox | ADNP1, KIAA0784 | ENSG00000101126 | 20q13.13 | 9/8 | ZF | 20 | 4 |
| ALX1 | Aristaless-like homeobox 1 | CART1, HEL23 | ENSG00000180318 | 12q21.31 | 1/1 | PRD | 3 | 0 |
| CERS2 | Ceramide synthase 2 | LASS2, TRH3, TMSG1 | ENSG00000143418 | 1q21.3 | 14/9 | CERS | 40 | 3 |
| CRX | Cone-rod homeobox | CORD2, OTX3, LCA7 | ENSG00000105392 | 19q13.33 | 7/4 | PRD | 43 | 18 |
| HESX1 | Homeobox gene expressed in embryonic stem cells 1 | ANF, CPHD5, RPX | ENSG00000163666 | 3p14.3 | 4/4 | PRD | 4 | – |
| HMX1 | H6 family homeobox 1 | Nkx5-3, H6 | ENSG00000215612 | 4p16.1 | 2/2 | ANTP | 13 | 0 |
| LMX1B | LIM homeobox transcription factor 1 beta | NPS1, LMX-1.2 | ENSG00000136944 | 9q33.3 | 4/4 | LIM | 3 | 0 |
| MEIS1 | Myeloid ecotropic insertion site homeobox 1 | – | ENSG00000143995 | 2p14 | 17/6 | TALE | 6 | 2 |
| MSX2 | Muscle segment homeobox 2 | MSH, HOX8 | ENSG00000120149 | 5q35.2 | 2/2 | ANTP | 4 | 0 |
| OTX2 | Orthodenticle homeobox | – | ENSG00000165588 | 14q22.3 | 11/11 | PRD | 4 | 12 |
| PAX2 | Paired box 2 | – | ENSG00000075891 | 10q24.31 | 9/6 | PRD | 3 | 0 |
| PAX6 | Paired box 6 | – | ENSG00000007372 | 11p13 | 82/57 | PRD | 75 | 5 |
| RAX | Retina and anterior neural fold homeobox | RX | ENSG00000134438 | 18q21.32 | 4/3 | PRD | 4 | 0.52 |
| RAX2 | Retina and anterior neural fold homeobox 2 | RAXL1, QRX | ENSG00000173976 | 19p13.3 | 2/2 | PRD | 13 | 5 |
| VAX1 | Ventral anterior homeobox 1 | – | ENSG00000148704 | 10q25.3 | 2/2 | ANTP | – | – |
| VAX2 | Ventral anterior homeobox 2 | – | ENSG00000116035 | 2p13.3 | 3/3 | ANTP | 3 | – |
| VSX1 | Visual system homeobox 1 | RINX, KTCN | ENSG00000100987 | 20p11.21 | 7/6 | PRD | 3 | – |
| VSX2 | Visual system homeobox 2 | CHX10, HOX10, RET1 | ENSG00000119614 | 14q24.3 | 1/1 | PRD | 4 | 4 |
Ensembl ID: data from Ensembl, version 98, September 2019, for human genes; RNA expression: microarray data from BioGPS; Protein expression: estimated protein expression log10 (ppm) (according to ProteomicsDB). “–“ synonyms are unknown. Homeobox classes: ANTP, named for the Antennapedia (Antp) gene of Drosophila; CERS, CERamide Synthase; LIM, named by the initials of the three homeodomain proteins Lin11, Isl-1 and Mec-3; PRD, PaiReD; TALE, Three Amino acid Loop Extension; ZF, Zinc Finger.
Table 2.
Cell-type specificity and function of homeobox genes expressed in neural retina and associated with inherited eye diseases in humans.
Table 2.
Cell-type specificity and function of homeobox genes expressed in neural retina and associated with inherited eye diseases in humans.
| Gene | Full Name | Expression in the Retina/RPE | Cell Functions | Disease or Syndrome | ||
|---|---|---|---|---|---|---|
| Single-Cell RNAseq (mouse) | RT-PCR, IHC | Disease/Syndrome (OMIM#) | Ocular Manifestations in Humans | |||
| ADNP (611386) | Activity-dependent neuroprotector homeobox | BC, rods | INP, OPL | Neuroprotection, promotion of neuronal growth and differentiation, autophagy | Helsmoortel–van der Aa syndrome (615873) | Macular laminations, foveal hypoplasia, cone degeneration |
| ALX1 (601527) | Aristaless-like homeobox 1 | Early RPC (E14) | Retinal margin | Cranial neural crest migration and differentiation, retinogenesis | Frontonasal dysplasia, type 3 (613456) | Anophthalmia, microphthalmia, optic nerve hypoplasia, coloboma |
| CERS2 (606920) | Ceramide synthase 2 | AC | All neurons, MG | Apoptosis, inflammation, signal transduction, lipid metabolism | Rhegmatogenous retinal detachment (Stickler syndrome) (PS108300) AMD | Retinal detachment, photoreceptor degradation, lipofuscin granules, RPE atrophy |
| CRX (602225) | Cone-rod homeobox | BC, cones, rods | RPC | Development and maintenance of Phr, renewal of Phr disks | Cone-rod retinal dystrophy type 2 (120970) Leber congenital amaurosis type 7 (613829) Dominant retinitis pigmentosa (268000) | Cone-rod retinal dystrophy, widespread retinal pigmentation, chorioretinal atrophy, attenuated retinal vessels, cystoid macular edema |
| HESX1 (601802) | Homeobox gene expressed in embryonic stem cells 1 | – | Early RPC, AC | Maintenance of stemness, neural cell determination | Septo-optic dysplasia (182230) Ocular coloboma (120200) | Optic nerve hypoplasia, hypoplastic optic discs, microphthalmia |
| HMX1 (142992) | H6 family homeobox 1 | AC, cones, RGC | Optic vesicle, early RPC, nasal RGC | Neurogenesis, nasotemporal patterning of retina | Oculoauricular syndrome (612109) | Microphthalmia, ocular coloboma, RPE anomalies, rod-cone dystrophy, macular hypoplasia |
| LMX1B (602575) | LIM homeobox transcription factor 1 beta | – | Periocular mesenchyme | Optic cup morphogenesis, nasotemporal patterning of retina | Nail-patella syndrome (161200) Primary open-angle glaucoma (137760) | Isolated optic disk excavation, ocular hypertension |
| MEIS1 (601739) | Myeloid ecotropic insertion site homeobox 1 | RPC, AC, cones, PhR precursors | – | Control of cell cycle in RPE, dorsoventral and nasotemporal patterning of retina | Microphthalmia (?) | Microphthalmia |
| MSX2 (123101) | Muscle segment homeobox 2 | AC, cones | – | EMT, suppression of transcription, apoptosis, RGC commitment and differentiation | Craniosynostosis type 2 (Boston-type) (604757) | Chorioretinal coloboma |
| OTX2 (600037) | Orthodenticle homeobox 2 | Late RPC, AC, rods, cones, BC, RGC (E14-P0) | RPC, RPE, RGC, PhR, BC | RPE specification, differentiation of photoreceptors and bipolar cells | Microphthalmia, syndromic type 5 (610125) Early-onset retinal dystrophy (610125) | Microphthalmia or anophthalmia, ocular coloboma, retinal dystrophy, optic nerve dysplasia |
| PAX2 (167409) | Paired box 2 | Rods, cones (E18) | Ventral optic vesicle, optic fissure, optic stalk, astrocytes | Inhibition of neurogenesis, induction of gliogenesis, axon guidance | Papillorenal syndrome (120330) | Retinal and optic nerve colobomas, optic disc dysplasia or hyperplasia, microphthalmia, gliosis of optic nerve, abnormal retinal vessels, chorioretinal degeneration |
| PAX6 (607108) | Paired box 6 | Early and late RPC, AC, cones, rods, RGC, MG | RPC, AC, RGC, HC | Maintenance of RPC multipotency, proliferation and differentiation of RGC and RPE, differentiation of HC | Foveal hypoplasia 1 (136520) Optic nerve hypoplasia (165550) Coloboma of optic nerve (120430) | Optic nerve hypoplasia, coloboma of optic nerve, microphthalmia |
| RAX (601881) | Retina and anterior neural fold homeobox | Early and late RPC, AC, rods, cones, RGC | RPC, rods, cones, MG | Proliferation of RPC, differentiation of PhR | Microphthalmia, isolated, type 3 (611038) | Microphthalmia (retinal size reduction) |
| RAX2 | Retina and anterior neural fold homeobox 2 | – | RPC, cones | Differentiation of cones | Cone-rod dystrophy type 11 (610381); AMD type 6 (613757) | Progressive macular atrophy, pigment granularity of peripheral retina, mixed rod and cone dysfunction on electroretinography, atrophy of macular RPE, progressive attenuation of retinal vessels, macular degeneration |
| VAX1 (604294) | Ventral anterior homeobox 1 | Late RPC | Glia of the optic nerve, ventral optic stalk | Repression of retinogenesis, RGC axon growth and guidance, dorsoventral patterning of retina | Microphthalmia, syndromic type 11 (614402) | Bilateral severe microphthalmia, small optic nerve |
| VAX2 (604295) | Ventral anterior homeobox 2 | AC | Cones, RGC | Repression of retinogenesis, dorsoventral patterning of retina, differentiation of cones, organization of nerve fiber layer within retina | Cone dystrophy, coloboma | Macular and cone degeneration |
| VSX1 (605020) | Visual system homeobox 1 | BC, cones | Cone BC (cBC), RGC | cBC differentiation | Craniofacial anomalies and anterior segment dysgenesis syndrome (614195) | Macular degeneration, BC dysfunction |
| VSX2 (142993) | Visual system homeobox 2 | Early and late RPC, BC, cones | RPC, all BC, MG | RPC proliferation, neuroretina specification | Microphthalmia, isolated type 2 (610093) Microphthalmia with coloboma type 3 (610092) | Microphthalmia, coloboma |
AC, amacrine cells; BC, bipolar cells; HC, horizontal cells; MG, Muller glia; PhR, photoreceptor; RGC, retinal ganglion cells; RPC, retinal progenitory cells; RPE, retinal pigment epithelium; OPL, outer plexiform layer; IPL, inner plexiform layer; IHC, immunohistochemistry. Single-cell RNAseq (mouse) – data from https://eyeintegration.nei.nih.gov [77] showing cells that expressed high levels of transcripts (decile of mean gene expression = 8–10) in prenatal and postnatal mouse retina. “–“ data are not found.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Markitantova, Y.; Simirskii, V. Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes. Int. J. Mol. Sci. 2020, 21, 1602. https://doi.org/10.3390/ijms21051602
AMA Style
Markitantova Y, Simirskii V. Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes. International Journal of Molecular Sciences. 2020; 21(5):1602. https://doi.org/10.3390/ijms21051602
Chicago/Turabian StyleMarkitantova, Yuliya, and Vladimir Simirskii. 2020. "Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes" International Journal of Molecular Sciences 21, no. 5: 1602. https://doi.org/10.3390/ijms21051602
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.