Interplay between BRCA1 and GADD45A and Its Potential for Nucleotide Excision Repair in Breast Cancer Pathogenesis

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
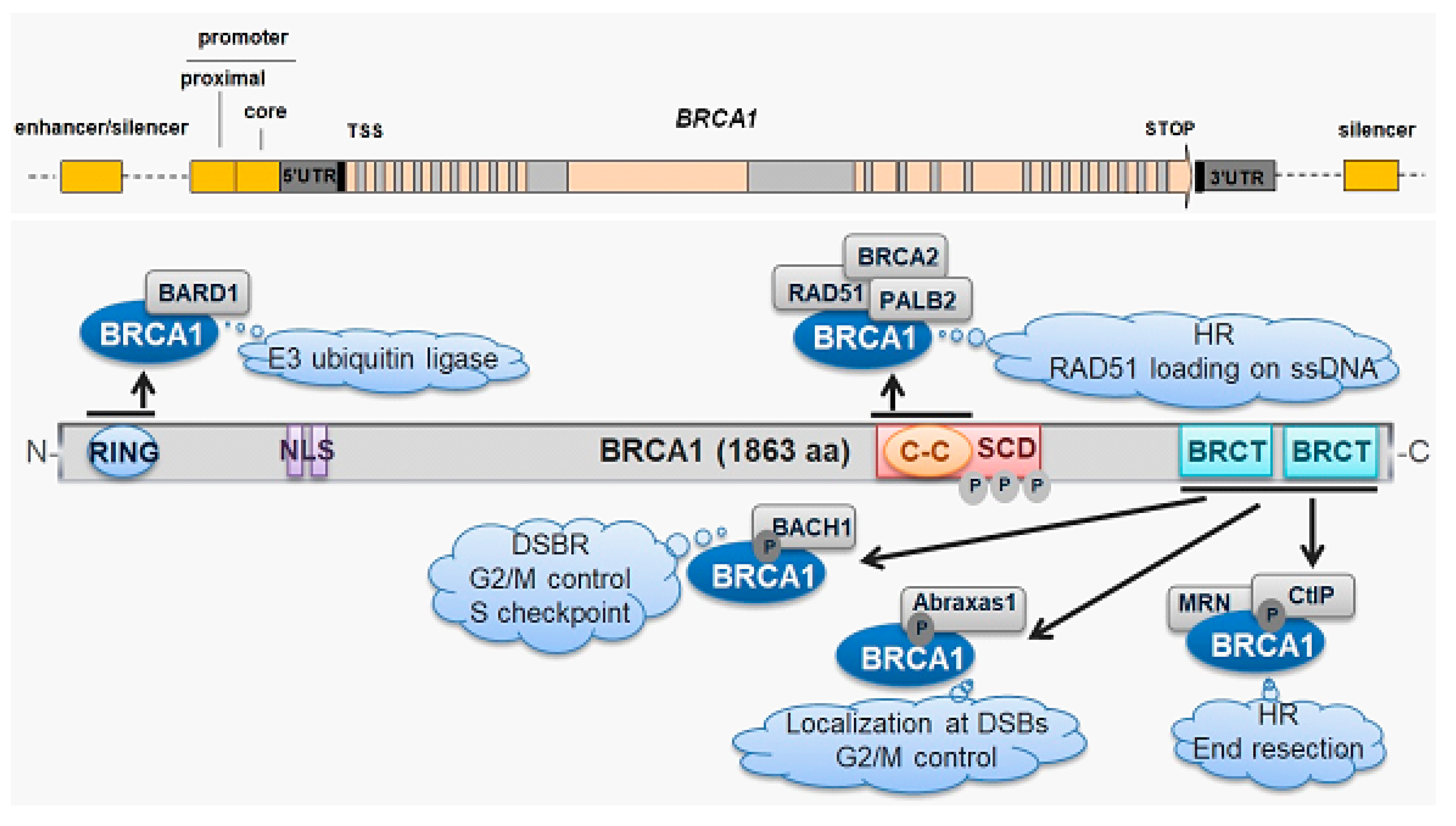
2. BRCA1—A Protein of DNA Damage Response and A Tumor Suppressor
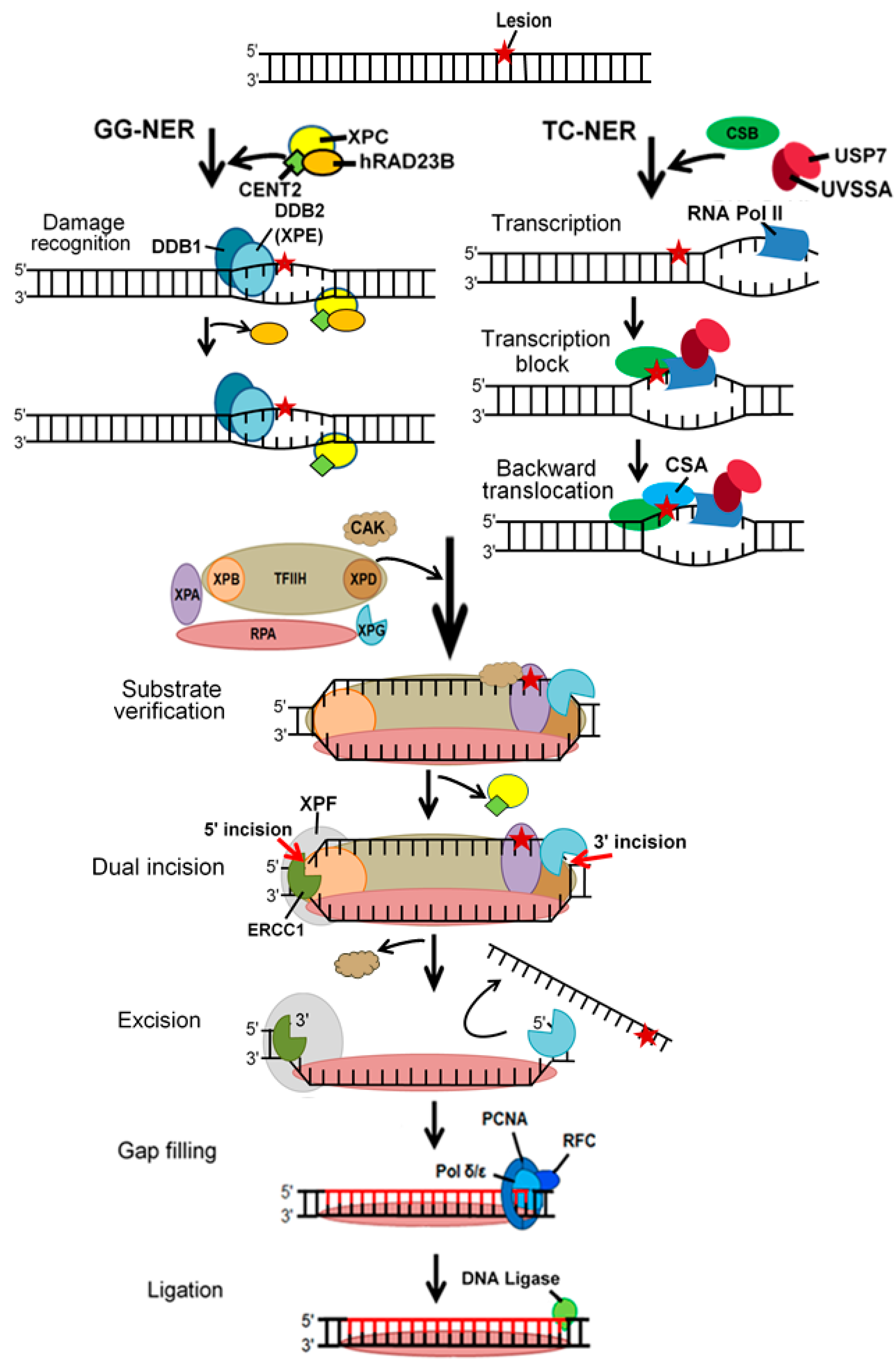
3. Nucleotide Excision Repair
4. Nucleotide Excision Repair in Breast Cancer
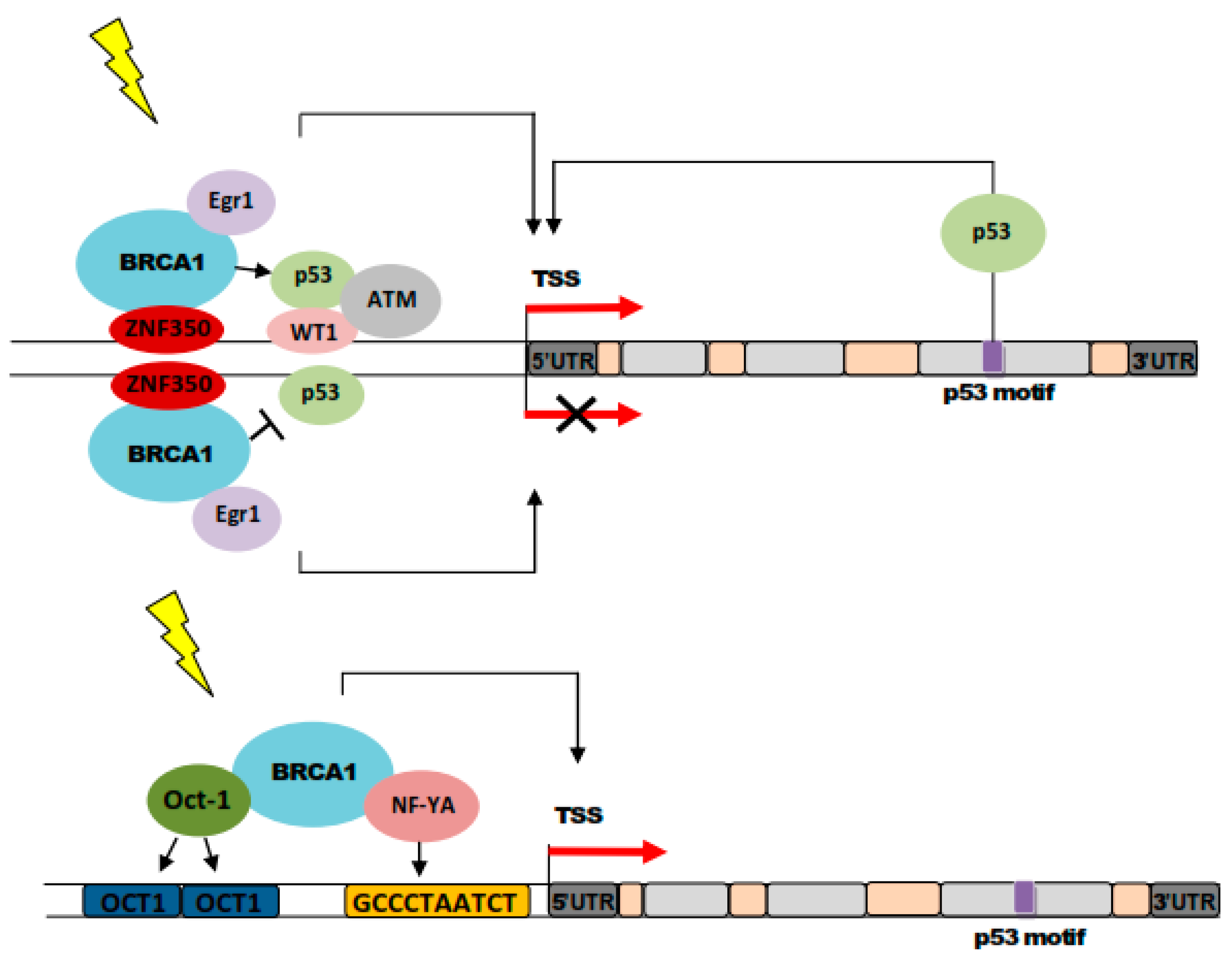
5. BRCA1 in Nucleotide Excision Repair in Breast Cancer
6. DNA Methylation in Breast Cancer
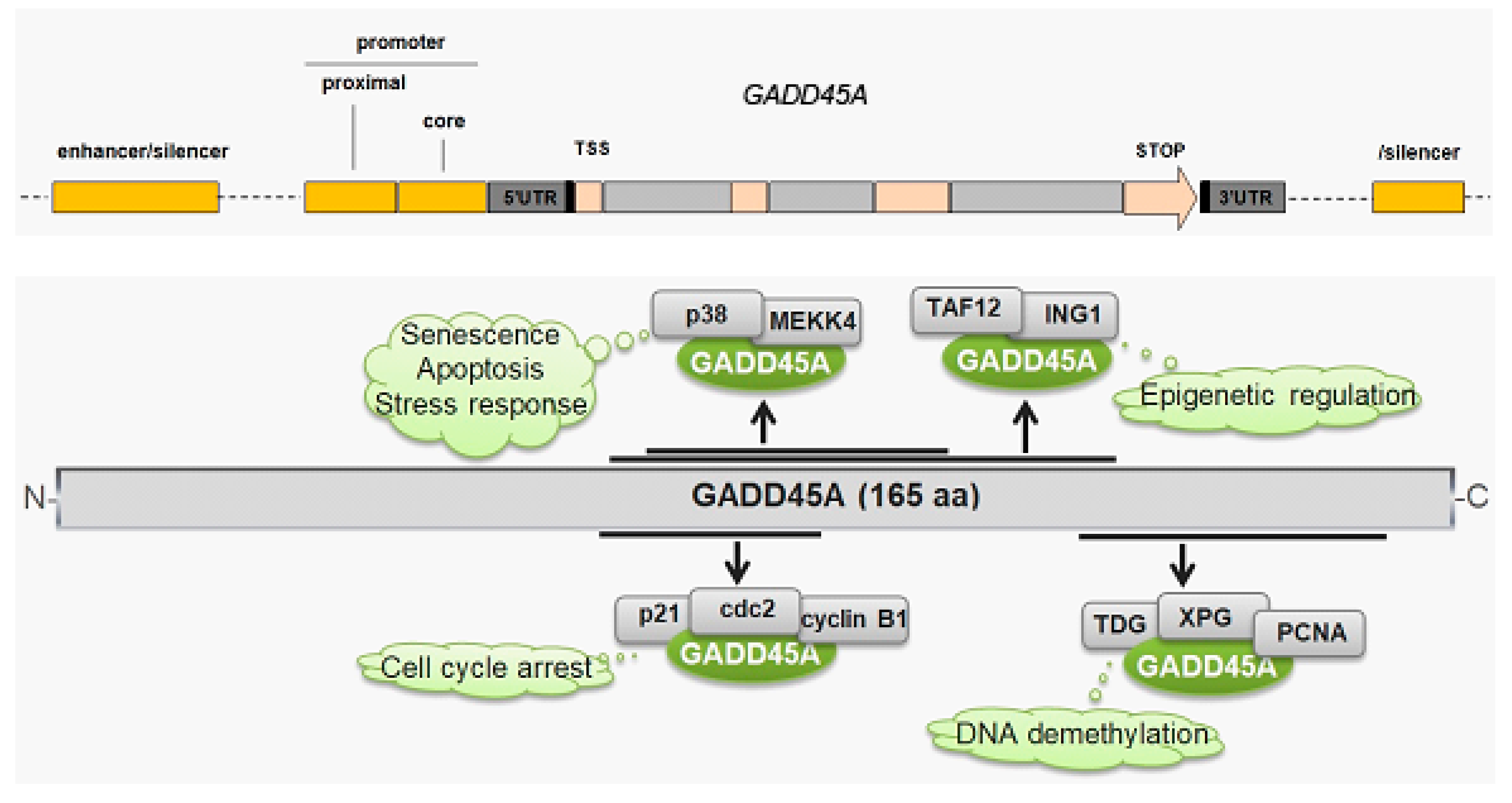
7. GADD45A and Its Role in Breast Cancer
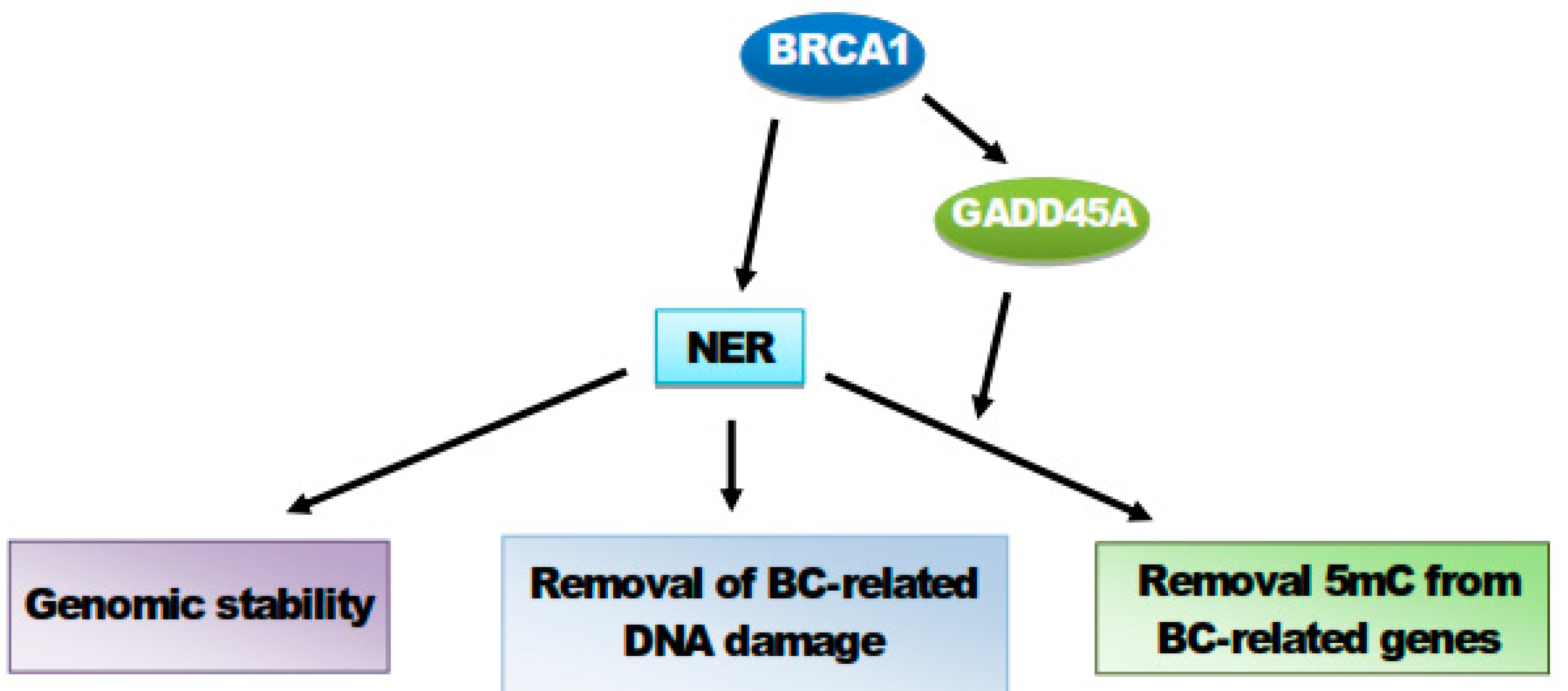
8. BRCA1 Stimulates GADD45A-Mediated NER and Active DNA Demethylation
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
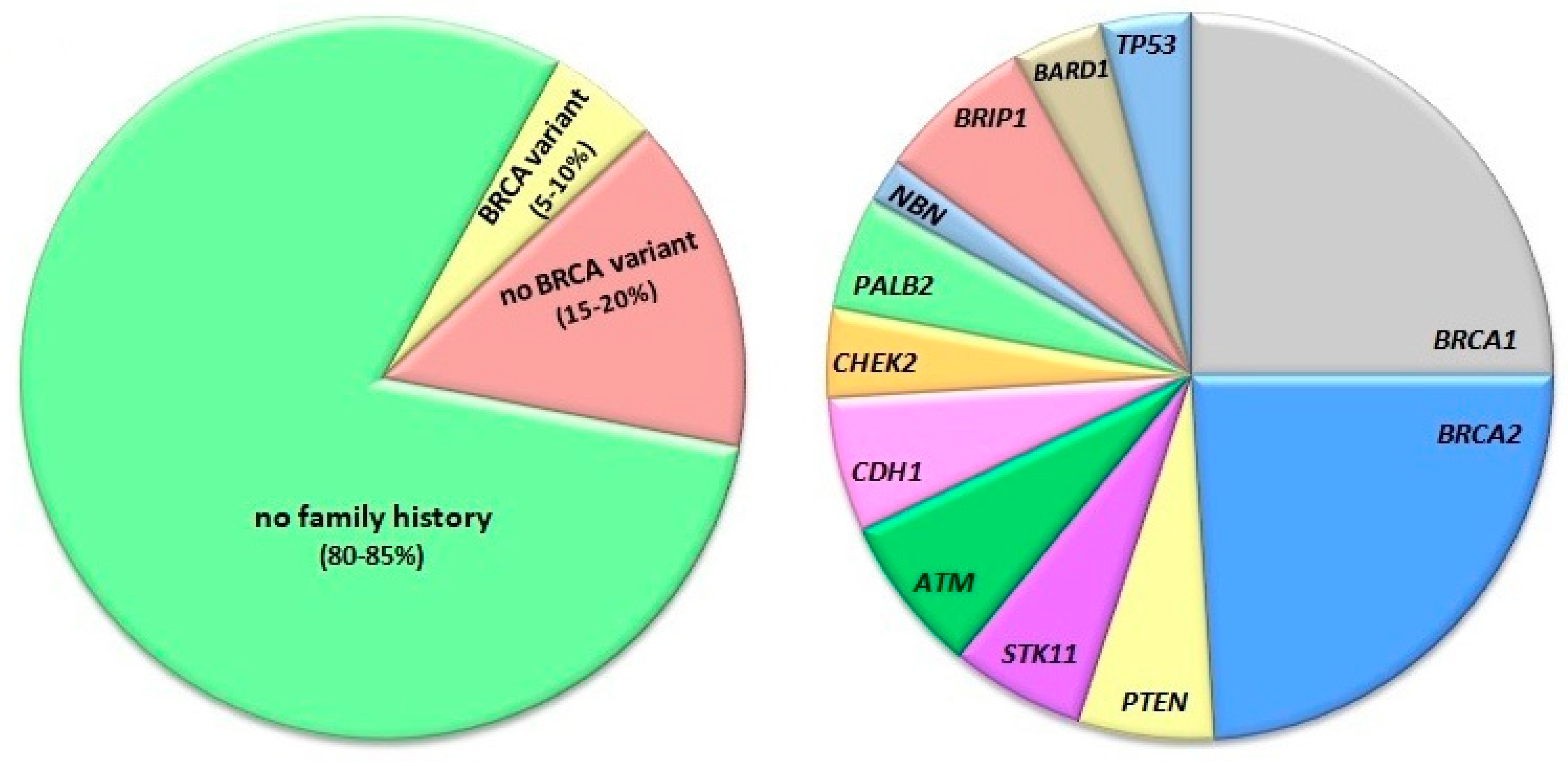
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.J. DNA double strand break repair and its association with inherited predispositions to breast cancer. Hered. Cancer Clin. Pract. 2004, 2, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Sishc, B.C.; Davis, A.J. The role of non-homologous end joining factors in carcinogenesis and cancer. Cancers (Basel) 2017, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Bogdanova, N.; Helbig, S.; Dork, T. Hereditary breast cancer: Ever more pieces to the polygenic puzzle. Hered. Cancer Clin. Pract. 2013, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Gosein, M.A.; Narinesingh, D.; Nixon, C.A.; Goli, S.R.; Maharaj, P.; Sinanan, A. Multi-organ benign and malignant tumors: Recognizing Cowden syndrome: A case report and review of the literature. BMC Res. Notes 2016, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Khincha, P.P.; Best, A.F.; Fraumeni, J.F., Jr.; Loud, J.T.; Savage, S.A.; Achatz, M.I. Reproductive factors associated with breast cancer risk in Li-Fraumeni syndrome. Eur. J. Cancer 2019, 116, 199–206. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Guilford, P.; Caldas, C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Rousset-Jablonski, C.; Gompel, A. Screening for familial cancer risk: Focus on breast cancer. Maturitas 2017, 105, 69–77. [Google Scholar] [CrossRef]
- Takaoka, M.; Miki, Y. BRCA1 gene: Function and deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef]
- Lin, D.; Izadpanah, R.; Braun, S.E.; Alt, E. A novel model to characterize structure and function of BRCA1. Cell Biol. Int. 2018, 42, 34–44. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Meza, J.E.; King, M.C.; Klevit, R.E. BRCA1 RING domain cancer-predisposing mutations. Structural consequences and effects on protein-protein interactions. J. Biol. Chem. 2001, 276, 41399–41406. [Google Scholar] [CrossRef] [Green Version]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Tirkkonen, M.; Johannsson, O.; Agnarsson, B.A.; Olsson, H.; Ingvarsson, S.; Karhu, R.; Tanner, M.; Isola, J.; Barkardottir, R.B.; Borg, A.; et al. Distinct somatic genetic changes associated with tumor progression in carriers of BRCA1 and BRCA2 germ-line mutations. Cancer Res. 1997, 57, 1222–1227. [Google Scholar]
- Tirkkonen, M.; Kainu, T.; Loman, N.; Johannsson, O.T.; Olsson, H.; Barkardottir, R.B.; Kallioniemi, O.P.; Borg, A. Somatic genetic alterations in BRCA2-associated and sporadic male breast cancer. Genes Chromosomes Cancer 1999, 24, 56–61. [Google Scholar] [CrossRef]
- Weaver, Z.; Montagna, C.; Xu, X.; Howard, T.; Gadina, M.; Brodie, S.G.; Deng, C.X.; Ried, T. Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene 2002, 21, 5097–5107. [Google Scholar] [CrossRef]
- Xu, X.; Weaver, Z.; Linke, S.P.; Li, C.; Gotay, J.; Wang, X.W.; Harris, C.C.; Ried, T.; Deng, C.X. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 1999, 3, 389–395. [Google Scholar] [CrossRef]
- Castillo, A.; Paul, A.; Sun, B.; Huang, T.H.; Wang, Y.; Yazinski, S.A.; Tyler, J.; Li, L.; You, M.J.; Zou, L.; et al. The BRCA1-interacting protein Abraxas is required for genomic stability and tumor suppression. Cell Rep. 2014, 8, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Son, M.Y.; Hasty, P. Homologous recombination defects and how they affect replication fork maintenance. Aims Genet. 2018, 5, 192–211. [Google Scholar] [CrossRef]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 Is Associated with a Human SWI/SNF-Related Complex: Linking Chromatin Remodeling to Breast Cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Saha, J.; Davis, A.J. Unsolved mystery: The role of BRCA1 in DNA end-joining. J. Radiat. Res. 2016, 57, i18–i24. [Google Scholar] [CrossRef] [Green Version]
- Saha, T.; Rih, J.K.; Roy, R.; Ballal, R.; Rosen, E.M. Transcriptional regulation of the base excision repair pathway by BRCA1. J. Biol. Chem. 2010, 285, 19092–19105. [Google Scholar] [CrossRef] [Green Version]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi Anemia Signaling and Cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465. [Google Scholar] [CrossRef]
- Datta, A.; Bagchi, S.; Nag, A.; Shiyanov, P.; Adami, G.R.; Yoon, T.; Raychaudhuri, P. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat. Res. 2001, 486, 89–97. [Google Scholar] [CrossRef]
- Friboulet, L.; Postel-Vinay, S.; Sourisseau, T.; Adam, J.; Stoclin, A.; Ponsonnailles, F.; Dorvault, N.; Commo, F.; Saulnier, P.; Salome-Desmoulez, S.; et al. ERCC1 function in nuclear excision and interstrand crosslink repair pathways is mediated exclusively by the ERCC1-202 isoform. Cell Cycle 2013, 12, 3298–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubner, R.A.; Riley, R.D.; Billingham, L.J.; Popat, S. Excision repair cross-complementation group 1 (ERCC1) status and lung cancer outcomes: A meta-analysis of published studies and recommendations. PLoS ONE 2011, 6, e25164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, S.; Ishii, G.; Goto, K.; Kubota, K.; Kim, Y.H.; Kojika, M.; Murata, Y.; Yamazaki, M.; Nishiwaki, Y.; Eguchi, K.; et al. Immunohistochemical expression of BCRP and ERCC1 in biopsy specimen predicts survival in advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy. Lung Cancer 2009, 64, 98–104. [Google Scholar] [CrossRef]
- Sodja, E.; Knez, L.; Kern, I.; Ovcaricek, T.; Sadikov, A.; Cufer, T. Impact of ERCC1 expression on treatment outcome in small-cell lung cancer patients treated with platinum-based chemotherapy. Eur. J. Cancer 2012, 48, 3378–3385. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Olaussen, K.A.; Pignon, J.P.; Shepherd, F.A.; Tsao, M.S.; Graziano, S.; Kratzke, R.; Douillard, J.Y.; Seymour, L.; Pirker, R.; et al. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. New Engl. J. Med. 2013, 368, 1101–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhu, X.; Zhang, H.; Fan, X.; Xue, X.; Chen, Y.; Ding, C.; Zhao, J.; Wu, G. ERCC1_202 Is A Prognostic Biomarker in Advanced Stage Non-Small Cell Lung Cancer Patients Treated with Platinum-Based Chemotherapy. J. Cancer 2017, 8, 2846–2853. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Santoro, J.; Kapetanaki, M.G.; Hsieh, C.L.; Gorbachinsky, I.; Levine, A.S.; Rapic-Otrin, V. The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res. 2008, 68, 5014–5022. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; Lindh, M.; Acs, K.; Vrouwe, M.G.; Pines, A.; van Attikum, H.; Mullenders, L.H.; Dantuma, N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol. 2012, 197, 267–281. [Google Scholar] [CrossRef] [Green Version]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Lake, R.J.; Geyko, A.; Hemashettar, G.; Zhao, Y.; Fan, H.Y. UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol. Cell 2010, 37, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016, 38, 9. [Google Scholar] [CrossRef] [Green Version]
- Black, J.O. Xeroderma Pigmentosum. Head Neck Pathol. 2016, 10, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, E.; Stucki, D.; Weber, W.; Muller, H. Impaired DNA-repair synthesis in lymphocytes of breast cancer patients. Eur. J. Cancer Clin. Oncol. 1986, 22, 863–869. [Google Scholar] [CrossRef]
- Kovacs, E.; Almendral, A. Reduced DNA repair synthesis in healthy women having first degree relatives with breast cancer. Eur. J. Cancer Clin. Oncol. 1987, 23, 1051–1057. [Google Scholar] [CrossRef]
- Latimer, J.J.; Johnson, J.M.; Kelly, C.M.; Miles, T.D.; Beaudry-Rodgers, K.A.; Lalanne, N.A.; Vogel, V.G.; Kanbour-Shakir, A.; Kelley, J.L.; Johnson, R.R.; et al. Nucleotide excision repair deficiency is intrinsic in sporadic stage I breast cancer. Proc. Natl. Acad. Sci. 2010, 107, 21725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Liu, H.; Qian, D.; Wang, X.; Moorman, P.G.; Luo, S.; Hwang, S.; Wei, Q. Genetic variants of genes in the NER pathway associated with risk of breast cancer: A large-scale analysis of 14 published GWAS datasets in the DRIVE study. Int. J. Cancer 2019, 145, 1270–1279. [Google Scholar] [CrossRef]
- He, B.S.; Xu, T.; Pan, Y.Q.; Wang, H.J.; Cho, W.C.; Lin, K.; Sun, H.L.; Gao, T.Y.; Wang, S.K. Nucleotide excision repair pathway gene polymorphisms are linked to breast cancer risk in a Chinese population. Oncotarget 2016, 7, 84872–84882. [Google Scholar] [CrossRef] [Green Version]
- Shakil Malik, S.; Mubarik, S.; Baig, M.; Masood, N.; Chaudhry, N. Genetic polymorphism in ERCC5 and breast cancer risk. Mol. Biol. Res. Commun. 2019, 8, 27–31. [Google Scholar] [CrossRef]
- Smolarz, B.; Makowska, M.; Samulak, D.; Michalska, M.M.; Mojs, E.; Wilczak, M.; Romanowicz, H. Single nucleotide polymorphisms (SNPs) of ERCC2, hOGG1, and XRCC1 DNA repair genes and the risk of triple-negative breast cancer in Polish women. Tumour Biol. 2014, 35, 3495–3502. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.M.; Xie, L.C.; Yuan, L.L.; Hu, X.L.; Jin, J.Q.; Niu, Y.M. Association of xeroderma pigmentosum complementation group G Asp1104His polymorphism with breast cancer risk: A cumulative meta-analysis. Mol. Clin. Oncol. 2014, 2, 1177–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crew, K.D.; Gammon, M.D.; Terry, M.B.; Zhang, F.F.; Zablotska, L.B.; Agrawal, M.; Shen, J.; Long, C.M.; Eng, S.M.; Sagiv, S.K.; et al. Polymorphisms in nucleotide excision repair genes, polycyclic aromatic hydrocarbon-DNA adducts, and breast cancer risk. Cancer Epidemiol. Prev. Biomark. 2007, 16, 2033–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Hoglund, L.; Zhao, C.; Forsti, A.; Snellman, E.; Hemminki, K. Single nucleotide polymorphisms in the XPG gene: Determination of role in DNA repair and breast cancer risk. Int. J. Cancer 2003, 103, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Singh, S.S.; Rama, R.; Sridevi, V.; Rajkumar, T. XPD Lys751Gln increases the risk of breast cancer. Oncol. Lett. 2011, 2, 155–159. [Google Scholar] [CrossRef]
- Tang, D.; Cho, S.; Rundle, A.; Chen, S.; Phillips, D.; Zhou, J.; Hsu, Y.; Schnabel, F.; Estabrook, A.; Perera, F.P. Polymorphisms in the DNA repair enzyme XPD are associated with increased levels of PAH-DNA adducts in a case-control study of breast cancer. Breast Cancer Res. Treat. 2002, 75, 159–166. [Google Scholar] [CrossRef]
- Terry, M.B.; Gammon, M.D.; Zhang, F.F.; Eng, S.M.; Sagiv, S.K.; Paykin, A.B.; Wang, Q.; Hayes, S.; Teitelbaum, S.L.; Neugut, A.I.; et al. Polymorphism in the DNA repair gene XPD, polycyclic aromatic hydrocarbon-DNA adducts, cigarette smoking, and breast cancer risk. Cancer Epidemiol. Prev. Biomark. 2004, 13, 2053–2058. [Google Scholar]
- Cosman, M.; Hingerty, B.E.; Luneva, N.; Amin, S.; Geacintov, N.E.; Broyde, S.; Patel, D.J. Solution conformation of the (-)-cis-anti-benzo[a]pyrenyl-dG adduct opposite dC in a DNA duplex: Intercalation of the covalently attached BP ring into the helix with base displacement of the modified deoxyguanosine into the major groove. Biochemistry 1996, 35, 9850–9863. [Google Scholar] [CrossRef]
- Boysen, G.; Hecht, S.S. Analysis of DNA and protein adducts of benzo[a]pyrene in human tissues using structure-specific methods. Mutat. Res. 2003, 543, 17–30. [Google Scholar] [CrossRef]
- Goldvaser, H.; Gal, O.; Rizel, S.; Hendler, D.; Neiman, V.; Shochat, T.; Sulkes, A.; Brenner, B.; Yerushalmi, R. The association between smoking and breast cancer characteristics and outcome. BMC Cancer 2017, 17, 624. [Google Scholar] [CrossRef] [Green Version]
- Xiong, P.; Bondy, M.L.; Li, D.; Shen, H.; Wang, L.E.; Singletary, S.E.; Spitz, M.R.; Wei, Q. Sensitivity to benzo(a)pyrene diol-epoxide associated with risk of breast cancer in young women and modulation by glutathione S-transferase polymorphisms: A case-control study. Cancer Res. 2001, 61, 8465–8469. [Google Scholar]
- Motykiewicz, G.; Faraglia, B.; Wang, L.W.; Terry, M.B.; Senie, R.T.; Santella, R.M. Removal of benzo(a)pyrene diol epoxide (BPDE)-DNA adducts as a measure of DNA repair capacity in lymphoblastoid cell lines from sisters discordant for breast cancer. Environ. Mol. Mutagenesis 2002, 40, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.O.; Agrawal, M.; Shen, J.; Terry, M.B.; Zhang, F.F.; Senie, R.T.; Motykiewicz, G.; Santella, R.M. DNA repair capacity of lymphoblastoid cell lines from sisters discordant for breast cancer. J. Natl. Cancer Inst. 2005, 97, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, Y.; Ji, W.; Song, L.; Dai, C.; Zhan, L. Effects of exposure to benzo[a]pyrene on metastasis of breast cancer are mediated through ROS-ERK-MMP9 axis signaling. Toxicol. Lett. 2015, 234, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Olivares-Illana, V.; Wang, L.; Karakostis, K.; Chen, S.; Fåhraeus, R. The p53 mRNA: An integral part of the cellular stress response. Nucleic Acids Res. 2019, 47, 3257–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Ford, J.M.; Hanawalt, P.C. Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc. Natl. Acad. Sci. USA 1995, 92, 8876–8880. [Google Scholar] [CrossRef] [Green Version]
- Ford, J.M. Regulation of DNA damage recognition and nucleotide excision repair: Another role for p53. Mutat. Res. 2005, 577, 195–202. [Google Scholar] [CrossRef]
- Fan, S.; Smith, M.L.; Rivet, D.J., 2nd; Duba, D.; Zhan, Q.; Kohn, K.W.; Fornace, A.J., Jr.; O’Connor, P.M. Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995, 55, 1649–1654. [Google Scholar]
- Hartman, A.R.; Ford, J.M. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat. Genet. 2002, 32, 180–184. [Google Scholar] [CrossRef]
- Bowman, K.K.; Sicard, D.M.; Ford, J.M.; Hanawalt, P.C. Reduced global genomic repair of ultraviolet light-induced cyclobutane pyrimidine dimers in simian virus 40-transformed human cells. Mol. Carcinog. 2000, 29, 17–24. [Google Scholar] [CrossRef]
- Ford, J.M.; Baron, E.L.; Hanawalt, P.C. Human fibroblasts expressing the human papillomavirus E6 gene are deficient in global genomic nucleotide excision repair and sensitive to ultraviolet irradiation. Cancer Res. 1998, 58, 599–603. [Google Scholar] [PubMed]
- Ford, J.M.; Hanawalt, P.C. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J. Biol. Chem. 1997, 272, 28073–28080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamer, M.; Darbre, P.D. Effects of exposure to six chemical ultraviolet filters commonly used in personal care products on motility of MCF-7 and MDA-MB-231 human breast cancer cells in vitro. J. Appl. Toxicol. 2018, 38, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Barr, L.; Alamer, M.; Darbre, P.D. Measurement of concentrations of four chemical ultraviolet filters in human breast tissue at serial locations across the breast. J. Appl. Toxicol. 2018, 38, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Murria, R.; Palanca, S.; de Juan, I.; Alenda, C.; Egoavil, C.; Segui, F.J.; Garcia-Casado, Z.; Juan, M.J.; Sanchez, A.B.; Segura, A.; et al. Immunohistochemical, genetic and epigenetic profiles of hereditary and triple negative breast cancers. Relevance in personalized medicine. Am. J. Cancer Res. 2015, 5, 2330–2343. [Google Scholar]
- McFarlane, R.J.; Feichtinger, J.; Larcombe, L. Cancer germline gene activation: Friend or foe? Cell Cycle 2014, 13, 2151–2152. [Google Scholar] [CrossRef] [Green Version]
- Van Tongelen, A.; Loriot, A.; De Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett. 2017, 396, 130–137. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Ali, M.M. Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, A.; Ji, W.; Zhang, X.Y.; Marrogi, A.; Graff, J.R.; Baylin, S.B.; Ehrlich, M. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int. J. Cancer 1998, 77, 833–838. [Google Scholar] [CrossRef]
- Bodelon, C.; Ambatipudi, S.; Dugue, P.A.; Johansson, A.; Sampson, J.N.; Hicks, B.; Karlins, E.; Hutchinson, A.; Cuenin, C.; Chajes, V.; et al. Blood DNA methylation and breast cancer risk: A meta-analysis of four prospective cohort studies. Breast Cancer Res. 2019, 21, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Cheng, J.; Cao, X.; Surowy, H.; Burwinkel, B. Blood-based DNA methylation as biomarker for breast cancer: A systematic review. Clin. Epigenetics 2016, 8, 115. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Huang, Y.; Liu, Z.; Zhao, R.; Liu, Q.; Wei, L.; Yu, X.; Li, B.; Qin, Y. Hypomethylation of BORIS is a promising prognostic biomarker in hepatocellular carcinoma. Gene 2017, 629, 29–34. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Du, P.; Zhang, X.; Li, X.; Cheng, G. Hypomethylation of the lncRNA SOX21-AS1 has clinical prognostic value in cervical cancer. Life Sci. 2019, 233, 116708. [Google Scholar] [CrossRef]
- Wolf, C.; Garding, A.; Filarsky, K.; Bahlo, J.; Robrecht, S.; Becker, N.; Zucknick, M.; Rouhi, A.; Weigel, A.; Claus, R.; et al. NFATC1 activation by DNA hypomethylation in chronic lymphocytic leukemia correlates with clinical staging and can be inhibited by ibrutinib. Int. J. Cancer 2018, 142, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Zelic, R.; Fiano, V.; Grasso, C.; Zugna, D.; Pettersson, A.; Gillio-Tos, A.; Merletti, F.; Richiardi, L. Global DNA hypomethylation in prostate cancer development and progression: A systematic review. Prostate Cancer Prostatic Dis. 2015, 18, 1–12. [Google Scholar] [CrossRef]
- Kushwaha, G.; Dozmorov, M.; Wren, J.D.; Qiu, J.; Shi, H.; Xu, D. Hypomethylation coordinates antagonistically with hypermethylation in cancer development: A case study of leukemia. Hum. Genom. 2016, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Hakkarainen, M.; Wahlfors, J.; Myohanen, S.; Hiltunen, M.O.; Eskelinen, M.; Johansson, R.; Janne, J. Hypermethylation of calcitonin gene regulatory sequences in human breast cancer as revealed by genomic sequencing. Int. J. Cancer 1996, 69, 471–474. [Google Scholar] [CrossRef]
- Bernardino, J.; Roux, C.; Almeida, A.; Vogt, N.; Gibaud, A.; Gerbault-Seureau, M.; Magdelenat, H.; Bourgeois, C.A.; Malfoy, B.; Dutrillaux, B. DNA hypomethylation in breast cancer: An independent parameter of tumor progression? Cancer Genet. Cytogenet. 1997, 97, 83–89. [Google Scholar] [CrossRef]
- Soares, J.; Pinto, A.E.; Cunha, C.V.; Andre, S.; Barao, I.; Sousa, J.M.; Cravo, M. Global DNA hypomethylation in breast carcinoma: Correlation with prognostic factors and tumor progression. Cancer 1999, 85, 112–118. [Google Scholar] [CrossRef]
- Ghecham, A.; Senator, A.; Pawlowska, E.; Bouafia, W.; Blasiak, J. Epigenetic modifiers 5-aza-2’-deoxycytidine and valproic acid differentially change viability, DNA damage and gene expression in metastatic and non-metastatic colon cancer cell lines. Acta Biochim. Pol. 2019, 66, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Kirn, V.; Strake, L.; Thangarajah, F.; Richters, L.; Eischeid, H.; Koitzsch, U.; Odenthal, M.; Fries, J. ESR1-promoter-methylation status in primary breast cancer and its corresponding metastases. Clin. Exp. Metastasis 2018, 35, 707–712. [Google Scholar] [CrossRef]
- Zhang, Y.; Bhatia, D.; Xia, H.; Castranova, V.; Shi, X.; Chen, F. Nucleolin links to arsenic-induced stabilization of GADD45alpha mRNA. Nucleic Acids Res. 2006, 34, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Kearsey, J.M.; Coates, P.J.; Prescott, A.R.; Warbrick, E.; Hall, P.A. Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene 1995, 11, 1675–1683. [Google Scholar]
- Vairapandi, M.; Balliet, A.G.; Fornace, A.J., Jr.; Hoffman, B.; Liebermann, D.A. The differentiation primary response gene MyD118, related to GADD45, encodes for a nuclear protein which interacts with PCNA and p21WAF1/CIP1. Oncogene 1996, 12, 2579–2594. [Google Scholar]
- Leung, C.H.; Lam, W.; Zhuang, W.J.; Wong, N.S.; Yang, M.S.; Fong, W.F. PKCdelta-dependent deubiquitination and stabilization of Gadd45 in A431 cells overexposed to EGF. Biochem. Biophys. Res. Commun. 2001, 285, 283–288. [Google Scholar] [CrossRef]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J., Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar] [CrossRef]
- Hildesheim, J.; Bulavin, D.V.; Anver, M.R.; Alvord, W.G.; Hollander, M.C.; Vardanian, L.; Fornace, A.J., Jr. Gadd45a protects against UV irradiation-induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res. 2002, 62, 7305–7315. [Google Scholar]
- Hollander, M.C.; Kovalsky, O.; Salvador, J.M.; Kim, K.E.; Patterson, A.D.; Haines, D.C.; Fornace, A.J., Jr. Dimethylbenzanthracene carcinogenesis in Gadd45a-null mice is associated with decreased DNA repair and increased mutation frequency. Cancer Res. 2001, 61, 2487–2491. [Google Scholar]
- Hollander, M.C.; Sheikh, M.S.; Bulavin, D.V.; Lundgren, K.; Augeri-Henmueller, L.; Shehee, R.; Molinaro, T.A.; Kim, K.E.; Tolosa, E.; Ashwell, J.D.; et al. Genomic instability in Gadd45a-deficient mice. Nat. Genet. 1999, 23, 176–184. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Long, F.; Yan, F.; Wang, N.; Wang, Y. The expression and clinical significance of GADD45A in breast cancer patients. PeerJ 2018, 6, e5344. [Google Scholar] [CrossRef]
- Tront, J.S.; Hoffman, B.; Liebermann, D.A. Gadd45a suppresses Ras-driven mammary tumorigenesis by activation of c-Jun NH2-terminal kinase and p38 stress signaling resulting in apoptosis and senescence. Cancer Res. 2006, 66, 8448–8454. [Google Scholar] [CrossRef] [Green Version]
- Tront, J.S.; Huang, Y.; Fornace, A.J., Jr.; Hoffman, B.; Liebermann, D.A. Gadd45a functions as a promoter or suppressor of breast cancer dependent on the oncogenic stress. Cancer Res. 2010, 70, 9671–9681. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.D.; Di, G.H.; Li, W.F.; Rao, N.Y.; Fan, L.; Yuan, W.T.; Hu, Z.; Wu, J.; Shen, Z.Z.; Huang, W.; et al. Genetic contribution of GADD45A to susceptibility to sporadic and non-BRCA1/2 familial breast cancers: A systematic evaluation in Chinese populations. Breast Cancer Res. Treat. 2010, 121, 157–167. [Google Scholar] [CrossRef]
- Desjardins, S.; Ouellette, G.; Labrie, Y.; Simard, J.; Durocher, F. Analysis of GADD45A sequence variations in French Canadian families with high risk of breast cancer. J. Hum. Genet. 2008, 53, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Tront, J.S.; Willis, A.; Huang, Y.; Hoffman, B.; Liebermann, D.A. Gadd45a levels in human breast cancer are hormone receptor dependent. J. Transl. Med. 2013, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Fabbro, M.; Henderson, B.R. BARD1 regulates BRCA1-mediated transactivation of the p21WAF1/CIP1 and Gadd45 promoters. Cancer Lett. 2008, 263, 189–196. [Google Scholar] [CrossRef]
- Wang, W.; Huper, G.; Guo, Y.; Murphy, S.K.; Olson, J.A., Jr.; Marks, J.R. Analysis of methylation-sensitive transcriptome identifies GADD45a as a frequently methylated gene in breast cancer. Oncogene 2005, 24, 2705–2714. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat. Res. 2005, 569, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, R.H.; Li, W.; Xu, X.; Hollander, M.C.; Fornace, A.J., Jr.; Deng, C.X. Genetic interactions between Brca1 and Gadd45a in centrosome duplication, genetic stability, and neural tube closure. J. Biol. Chem. 2004, 279, 29606–29614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkin, D.P.; Bean, J.M.; Miklos, D.; Song, Y.H.; Truong, V.B.; Englert, C.; Christians, F.C.; Ellisen, L.W.; Maheswaran, S.; Oliner, J.D.; et al. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell 1999, 97, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Zhao, H.; Fan, F.; Blanck, P.; Fan, W.; Colchagie, A.B.; Fornace, A.J., Jr.; Zhan, Q. BRCA1 activation of the GADD45 promoter. Oncogene 2000, 19, 4050–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Pan, H.; Li, S.; Flesken-Nikitin, A.; Chen, P.L.; Boyer, T.G.; Lee, W.H. Sequence-specific transcriptional corepressor function for BRCA1 through a novel zinc finger protein, ZBRK1. Mol. Cell 2000, 6, 757–768. [Google Scholar] [CrossRef]
- Zhan, Q.; Bae, I.; Kastan, M.B.; Fornace, A.J., Jr. The p53-dependent gamma-ray response of GADD45. Cancer Res. 1994, 54, 2755–2760. [Google Scholar] [PubMed]
- Amundson, S.A.; Lee, R.A.; Koch-Paiz, C.A.; Bittner, M.L.; Meltzer, P.; Trent, J.M.; Fornace, A.J., Jr. Differential responses of stress genes to low dose-rate gamma irradiation. Mol. Cancer Res. 2003, 1, 445–452. [Google Scholar]
- Bishop, A.J.; Hollander, M.C.; Kosaras, B.; Sidman, R.L.; Fornace, A.J., Jr.; Schiestl, R.H. Atm-, p53-, and Gadd45a-deficient mice show an increased frequency of homologous recombination at different stages during development. Cancer Res. 2003, 63, 5335–5343. [Google Scholar]
- Chen, Y.; Yang, R.; Guo, P.; Ju, Z. Gadd45a deletion aggravates hematopoietic stem cell dysfunction in ATM-deficient mice. Protein Cell 2014, 5, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.L.; Chen, I.T.; Zhan, Q.; Bae, I.; Chen, C.Y.; Gilmer, T.M.; Kastan, M.B.; O’Connor, P.M.; Fornace, A.J., Jr. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 1994, 266, 1376–1380. [Google Scholar] [CrossRef] [Green Version]
- Kazantsev, A.; Sancar, A. Does the p53 up-regulated Gadd45 protein have a role in excision repair? Science 1995, 270, 1003–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearsey, J.M.; Shivji, M.K.; Hall, P.A.; Wood, R.D. Does the p53 up-regulated Gadd45 protein have a role in excision repair? Science 1995, 270, 1004–1005. [Google Scholar] [PubMed]
- Carrier, F.; Georgel, P.T.; Pourquier, P.; Blake, M.; Kontny, H.U.; Antinore, M.J.; Gariboldi, M.; Myers, T.G.; Weinstein, J.N.; Pommier, Y.; et al. Gadd45, a p53-responsive stress protein, modifies DNA accessibility on damaged chromatin. Mol. Cell. Biol. 1999, 19, 1673–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.L.; Ford, J.M.; Hollander, M.C.; Bortnick, R.A.; Amundson, S.A.; Seo, Y.R.; Deng, C.X.; Hanawalt, P.C.; Fornace, A.J., Jr. p53-mediated DNA repair responses to UV radiation: Studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol. Cell. Biol. 2000, 20, 3705–3714. [Google Scholar] [CrossRef]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Amente, S.; Zhang, J.; Lavadera, M.L.; Lania, L.; Avvedimento, E.V.; Majello, B. Myc and PI3K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 2011, 39, 9498–9507. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Espino, R.A.; Chomey, E.G.; Luong, L.; Bano, A.; Meakins, D.; Tron, V.A. Loss of p21WAF1/Cip1 in Gadd45-deficient keratinocytes restores DNA repair capacity. Carcinogenesis 2005, 26, 1804–1810. [Google Scholar] [CrossRef] [Green Version]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Stoyanova, T.; Yoon, T.; Kopanja, D.; Mokyr, M.B.; Raychaudhuri, P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol. Cell. Biol. 2008, 28, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Waga, S.; Stillman, B. Cyclin-dependent kinase inhibitor p21 modulates the DNA primer-template recognition complex. Mol. Cell. Biol. 1998, 18, 4177–4187. [Google Scholar] [CrossRef] [Green Version]
- Soria, G.; Speroni, J.; Podhajcer, O.L.; Prives, C.; Gottifredi, V. p21 differentially regulates DNA replication and DNA-repair-associated processes after UV irradiation. J. Cell Sci. 2008, 121, 3271–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stivala, L.A.; Riva, F.; Cazzalini, O.; Savio, M.; Prosperi, E. p21(waf1/cip1)-null human fibroblasts are deficient in nucleotide excision repair downstream the recruitment of PCNA to DNA repair sites. Oncogene 2001, 20, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzalini, O.; Perucca, P.; Savio, M.; Necchi, D.; Bianchi, L.; Stivala, L.A.; Ducommun, B.; Scovassi, A.I.; Prosperi, E. Interaction of p21 CDKN1A with PCNA regulates the histone acetyltransferase activity of p300 in nucleotide excision repair. Nucleic Acids Res. 2008, 36, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Tillhon, M.; Cazzalini, O.; Nardo, T.; Necchi, D.; Sommatis, S.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair 2012, 11, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A. Gadd45 proteins: Key players of repair-mediated DNA demethylation. Adv. Exp. Med. Biol. 2013, 793, 35–50. [Google Scholar] [CrossRef]
- Barreto, G.; Schafer, A.; Marhold, J.; Stach, D.; Swaminathan, S.K.; Handa, V.; Doderlein, G.; Maltry, N.; Wu, W.; Lyko, F.; et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007, 445, 671–675. [Google Scholar] [CrossRef]
- Schuermann, D.; Weber, A.R.; Schar, P. Active DNA demethylation by DNA repair: Facts and uncertainties. DNA Repair 2016, 44, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C.; Schafer, A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 2012, 22, 220–227. [Google Scholar] [CrossRef]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [Green Version]
- Hartman, A.R.; Ford, J.M. BRCA1 and p53: Compensatory roles in DNA repair. J. Mol. Med. 2003, 81, 700–707. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrasik, S.; Zajac, G.; Morawiec, J.; Soszynski, M.; Fila, M.; Blasiak, J. Interplay between BRCA1 and GADD45A and Its Potential for Nucleotide Excision Repair in Breast Cancer Pathogenesis. Int. J. Mol. Sci. 2020, 21, 870. https://doi.org/10.3390/ijms21030870
Pietrasik S, Zajac G, Morawiec J, Soszynski M, Fila M, Blasiak J. Interplay between BRCA1 and GADD45A and Its Potential for Nucleotide Excision Repair in Breast Cancer Pathogenesis. International Journal of Molecular Sciences. 2020; 21(3):870. https://doi.org/10.3390/ijms21030870
Chicago/Turabian StylePietrasik, Sylwia, Gabriela Zajac, Jan Morawiec, Miroslaw Soszynski, Michal Fila, and Janusz Blasiak. 2020. "Interplay between BRCA1 and GADD45A and Its Potential for Nucleotide Excision Repair in Breast Cancer Pathogenesis" International Journal of Molecular Sciences 21, no. 3: 870. https://doi.org/10.3390/ijms21030870