Mass Spectrometry-Based Metabolomics Analysis of Obese Patients’ Blood Plasma

 ,
, 
 ,
,  ,
,
Abstract
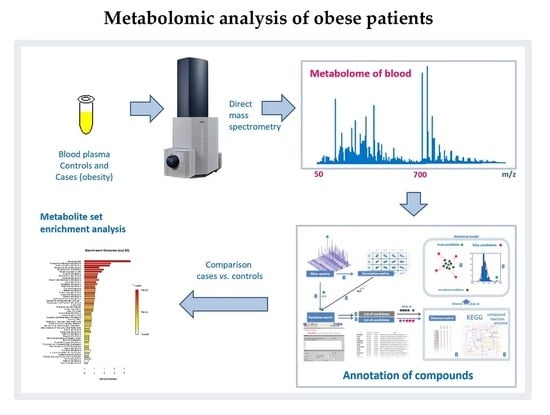
:
1. Introduction
2. Results
2.1. Mass Spectrometry Measurement of Compounds in Blood
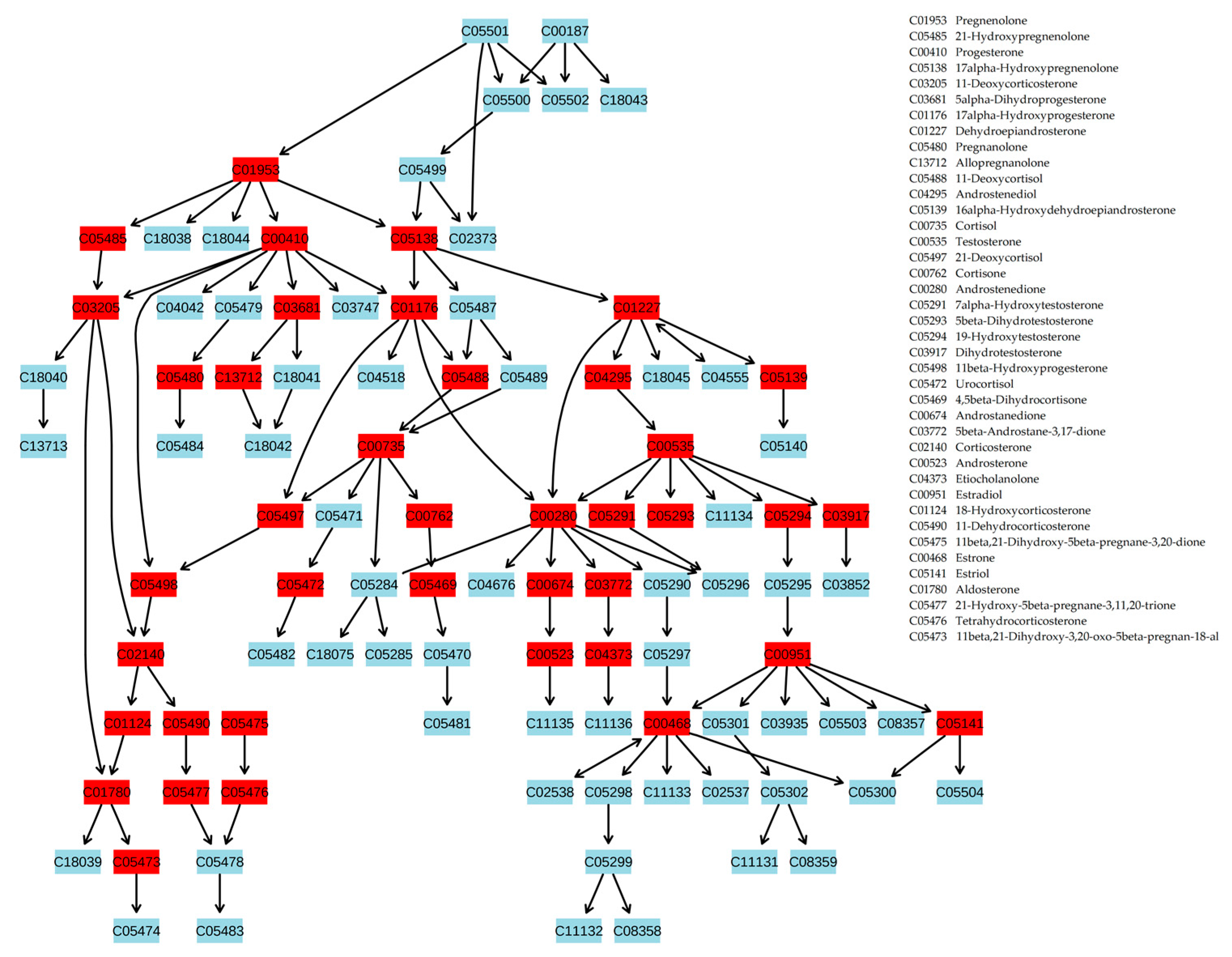
2.2. Compound Annotation Algorithm
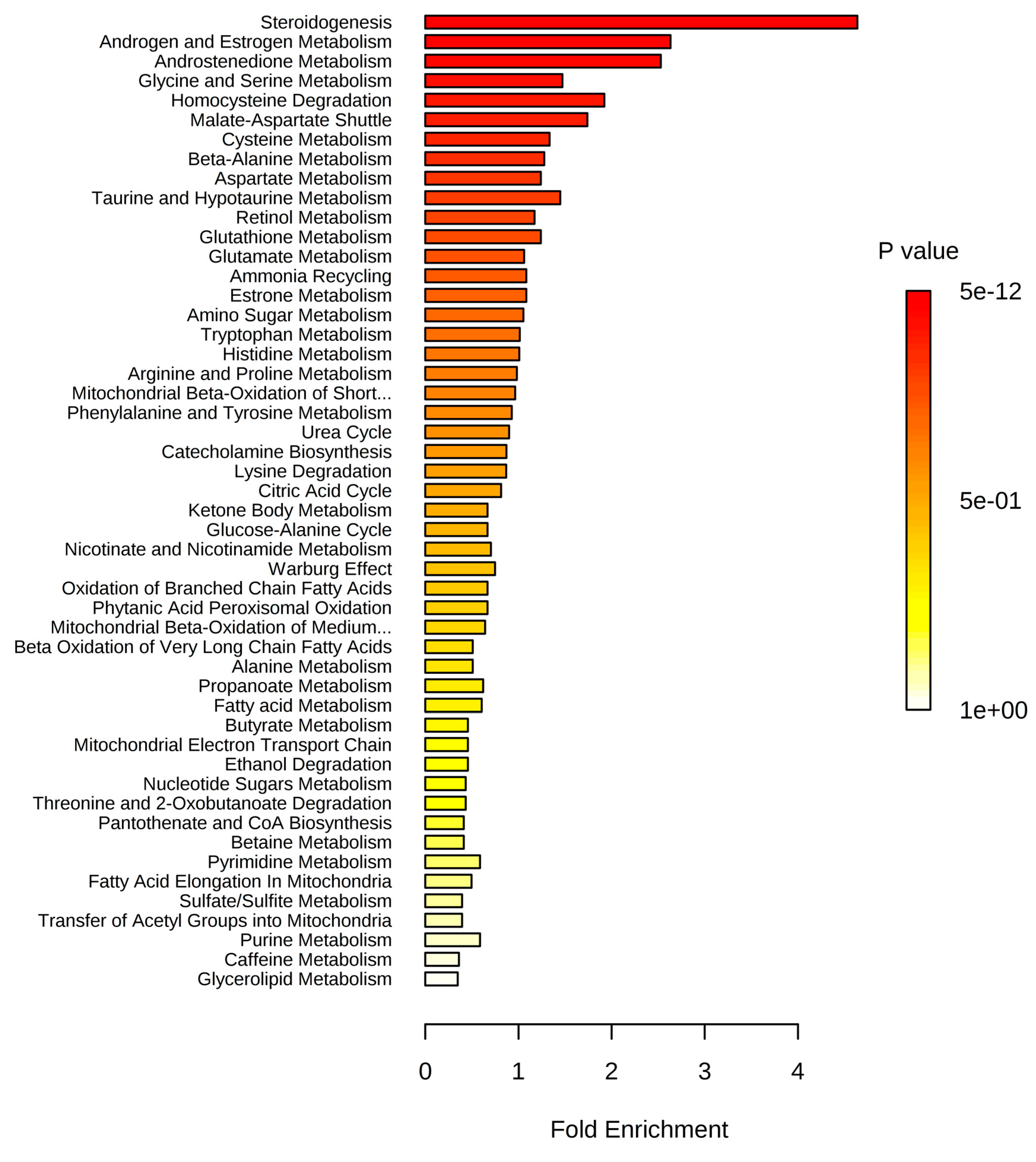
2.3. Metabolite Set Enrichment Analysis (MSEA)
3. Discussion
4. Materials and Methods
4.1. Blood Samples
4.2. Mass Spectrometry
4.3. Mass Spectra Preprocessing
4.4. Compound Annotation Algorithm
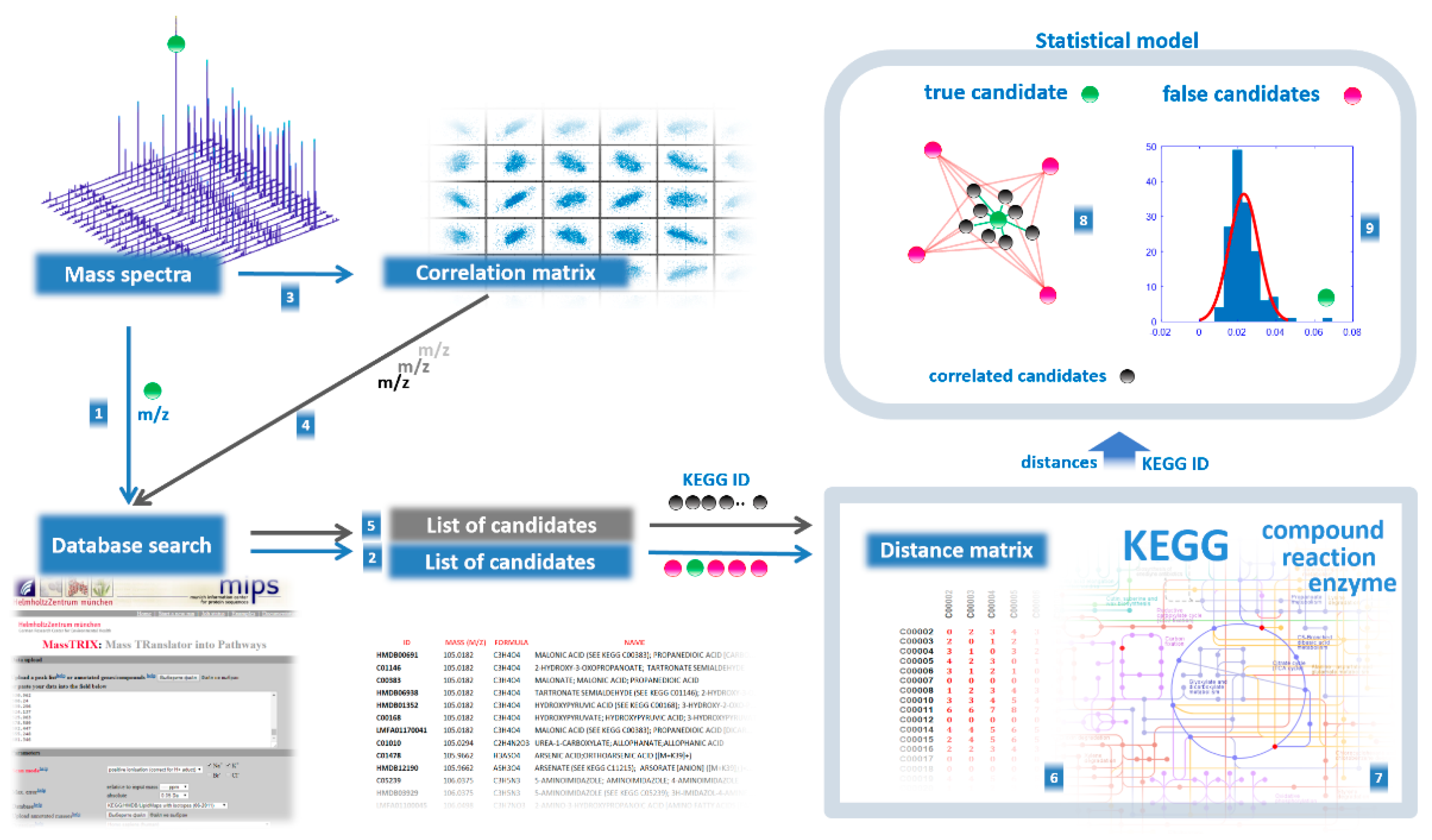
- Coefficients for Pearson correlation between aligned mass peak intensities were calculated using the corr function in MATLAB (MathWorks, Natick, MA, USA). As a result, the correlation matrix was built (columns correspond to m/z values of peaks for which correlation was calculated, rows and columns correspond to other m/z values of peaks taken to calculate pairwise correlation, in matrix cells correlation coefficients (r) are presented). Correlation values were calculated only if at least nine nonzero values were available to calculate correlation. Zero values in the matrix cells were not used in the calculation of the correlation. To find correlations, the mass spectra of all samples involved in the study were used.
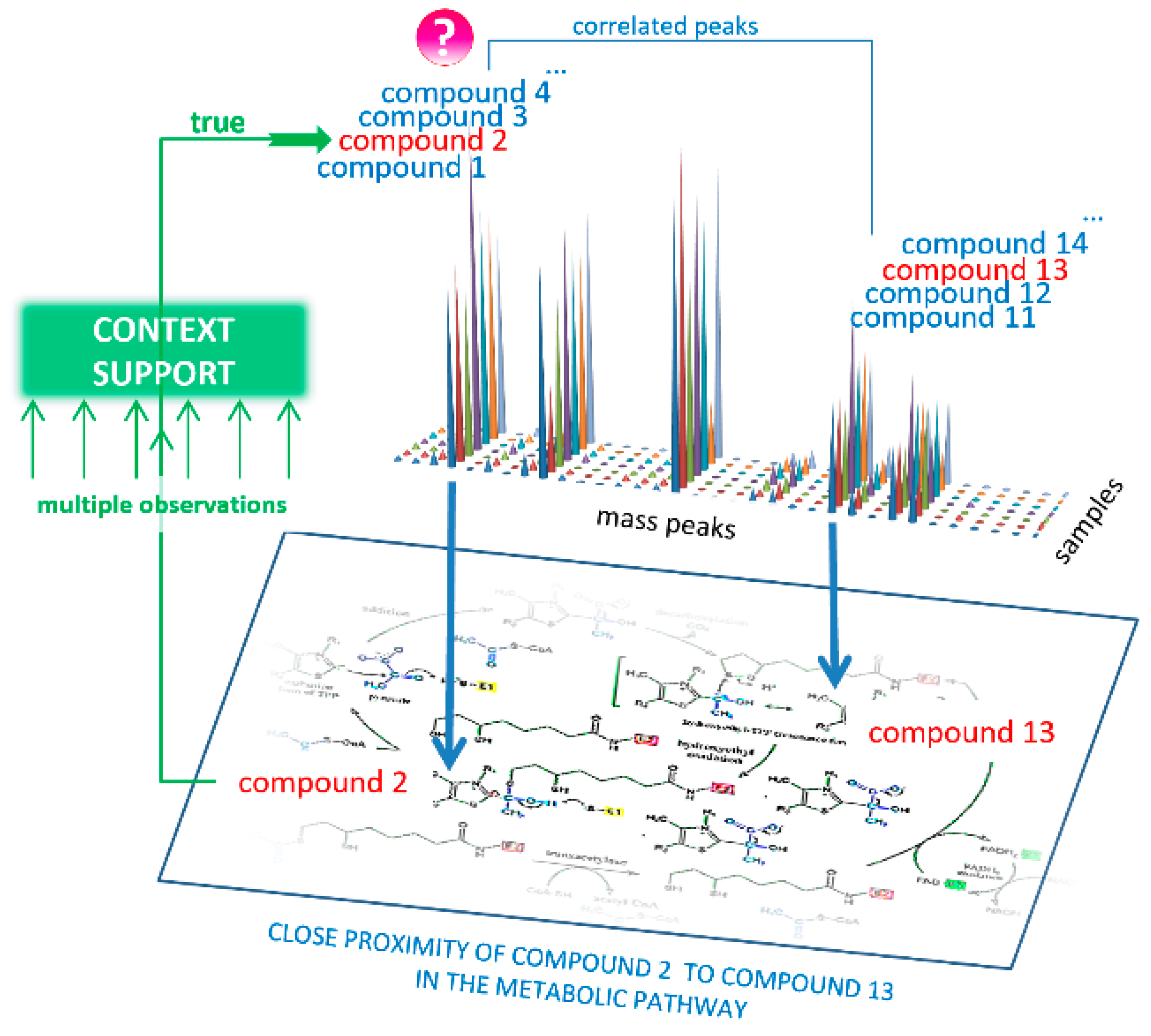
- Each mass spectrometry peak was tested for support from context data (Figure 4). Using the correlation matrix, the top 30 (empirically established as optimal) highly correlated (positive or negative, with r < 0.85) mass peaks were also associated with KEGG IDs proposed by MassTRIX, which were considered CS for the tested peak. Highly correlated peaks from CS with r > 0.85 were annotated as the derivatives of the tested peaks (fragment, adduct, or multi-ion of the compound).
- To reveal the true compound name, the distance between each candidate name of the compound and the compound name in its CS was calculated. To do this, the distance matrix between compounds in the human metabolic pathways was generated using Metabonetworks toolbox [32]. A distance equal to N means that the distance from the testing annotation to a particular annotation from the CS lies through the N-1 intermediate compounds (i.e., shortest way through the N biochemical reactions). The retrieved values for the N = 1, 2, 3, and 4 positions were tested against random cause. CS was randomly generated 300 times, and the retrieved data for the N = 1, 2, 3, and 4 positions were used to calculate the p-value for candidates using the cumulative function for generalized extreme value distribution (1-cdf function with gev option in MATLAB).
- A probability of p > 0.95 for any N 1–4 position statistically confirms that the tested annotation is verified by CS, i.e., it is a true annotation.
- Retrieved true annotations were submitted to the compound annotation algorithm again, and this process was repeated six times. Each time, the CS became more competent. Finally, annotations with a Z score<-1.64 (correspond to probability of p > 0.95) were considered the output of the annotation algorithm.
4.5. Statistical Analysis
4.6. Metabolite Set Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cacciatore, S.; Loda, M. Innovation in metabolomics to improve personalized healthcare. Ann. N. Y. Acad. Sci. 2015, 1346, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viant, M.R.; Kurland, I.J.; Jones, M.R.; Dunn, W.B. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2017, 36, 64–69. [Google Scholar] [CrossRef] [PubMed]
- de Jong, F.; Beecher, C.; Raskind, A.; Chamberlain, C.; Guingab, J.; Garrett, T. MetaboNews. Available online: http://www.metabonews.ca/Aug2017/MetaboNews_Aug2017.htm (accessed on 1 August 2017).
- Rogers, S.; Scheltema, R.A.; Girolami, M.; Breitling, R. Probabilistic assignment of formulas to mass peaks in metabolomics experiments. Bioinformatics 2009, 25, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, R.R.; Jourdan, F.; Salvanha, D.M.; Letisse, F.; Jamin, E.L.; Guidetti-Gonzalez, S.; Labate, C.A.; Vêncio, R.Z.N. ProbMetab: An R package for Bayesian probabilistic annotation of LC-MS-based metabolomics. Bioinformatics 2014, 30, 1336–1337. [Google Scholar] [CrossRef] [Green Version]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Tchernof, A.; Després, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef]
- Fritscher, L.G.; Mottin, C.C.; Canani, S.; Chatkin, J.M. Obesity and obstructive sleep apnea-hypopnea syndrome: the impact of bariatric surgery. Obes. Surg. 2007, 17, 95–99. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Dossus, L.; Marant-Micallef, C.; His, M. Obesity and Cancer. Bull. Cancer 2019, 106, 635–646. [Google Scholar] [CrossRef]
- Steuer, R.; Kurths, J.; Fiehn, O.; Weckwerth, W. Observing and interpreting correlations in metabolomic networks. Bioinformatics 2003, 19, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, A.; Klaasen, C.D.; Watkins, J.B. Biotransformation of xenobiotics. Casarett Doull’s Essent. Toxicol. 2001, 133–144. [Google Scholar]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.D.; Wittmann, B.M.; Evans, A.M.; Miller, L.A.D.; Toal, D.R.; Lonergan, S.; Elsea, S.H.; Pappan, K.L. Metabolomics in the clinic: A review of the shared and unique features of untargeted metabolomics for clinical research and clinical testing. J. Mass Spectrom. 2018, 53, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.K.; Kumar, P.; Mahalingam, K. Molecular genetics of human obesity: A comprehensive review. Comptes Rendus Biol. 2017, 340, 87–108. [Google Scholar] [CrossRef]
- Hall, J.E.; Do Carmo, J.M.; Da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-Induced Hypertension: Interaction of Neurohumoral and Renal Mechanisms. Circ. Res. 2015, 616, 991–1006. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, C.; Collison, M. Sex steroids and insulin resistance. Clin. Sci. 2002, 102, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Quarta, C.; Clemmensen, C.; Zhu, Z.; Yang, B.; Joseph, S.S.; Lutter, D.; Yi, C.X.; Graf, E.; García-Cáceres, C.; Legutko, B.; et al. Molecular integration of incretin and glucocorticoid action reverses immunometabolic dysfunction and obesity. Cell Metab. 2017, 26, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Rossouw, J.E. Hormones for coronary disease - Full circle. Lancet 2002, 360, 1996–1997. [Google Scholar] [CrossRef]
- Bhasin, S.; Calof, O.M.; Storer, T.W.; Lee, M.L.; Mazer, N.A.; Jasuja, R.; Montori, V.M.; Gao, W.; Dalton, J.T. Drug Insight: Testosterone and selective androgen receptor modulators as anabolic therapies for chronic illness and aging. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 146–159. [Google Scholar] [CrossRef]
- Di Cesare, M.; Bentham, J.; Stevens, G.A.; Zhou, B.; Danaei, G.; Lu, Y.; Bixby, H.; Cowan, M.J.; Riley, L.M.; Hajifathalian, K.; et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377. [Google Scholar]
- Cai, L.; He, J.; Song, Y.; Zhao, K.; Cui, W. Association of obesity with socio-economic factors and obesity-related chronic diseases in rural southwest China. Public Health 2012, 127, 247. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Panagiotou, O.A.; Graubard, B.I. Estimating population attributable fractions to quantify the health burden of obesity. Ann. Epidemiol. 2015, 25, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korhonen, P.E.; Seppälä, T.; Järvenpää, S.; Kautiainen, H. Body mass index and health-related quality of life in apparently healthy individuals. Qual. Life Res. 2014, 23, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Renzaho, A.; Wooden, M.; Houng, B. Associations between body mass index and health-related quality of life among Australian adults. Qual. Life Res. 2010, 19, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Guyatt, G.H.; Feeny, D.H.; Patrick, D.L. Measuring health-related quality of life. Ann. Intern. Med. 1993, 38, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Brazier, J. Health, Health-Related Quality of Life, and Quality of Life: What is the Difference? Pharmacoeconomics 2016, 34, 645–649. [Google Scholar] [CrossRef]
- Søltoft, F.; Hammer, M.; Kragh, N. The association of body mass index and health-related quality of life in the general population: Data from the 2003 Health Survey of England. Qual. Life Res. 2009, 18, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xu, L.; Li, J.; Sun, L.; Qin, W.; Ding, G.; Wang, Q.; Zhu, J.; Yu, Z.; Xie, S.; et al. Gender differences in the association between body mass index and health-related quality of life among adults:a cross-sectional study in Shandong, China. BMC Public Health 2019, 19, 1021. [Google Scholar] [CrossRef] [Green Version]
- BD Vacutainer® Evacuated Blood Collection System For In Vitro Diagnostic Use. Becton, Dickinson and Company. 2019. Available online: www.bd.com/vacutainer/referencematerial (accessed on 1 January 2011).
- Lokhov, P.G.; Kharybin, O.N.; Archakov, A.I. Diagnosis of lung cancer based on direct-infusion electrospray mass spectrometry of blood plasma metabolites. Int. J. Mass Spectrom. 2011, 309, 200–205. [Google Scholar] [CrossRef]
- Posma, J.M.; Robinette, S.L.; Holmes, E.; Nicholson, J.K. MetaboNetworks, an interactive Matlab-based toolbox for creating, customizing and exploring sub-networks from KEGG. Bioinformatics 2014, 30, 893–895. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wishart, D.S. Using metaboanalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.9. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Detection mass range of compounds (m/z) | 45–900 |
| Number of detected compound mass peaks | 9333 ± 416 1 |
| Number of masses submitted to MassTrix | 14,738 |
| Number of mass peaks/compound candidate submitted to the annotation algorithm | 26,430 |
| Number of mass peaks with putatively annotated compound(s) by the annotation algorithm | 2015 |
| Number of unique compound names retrieved by the annotation algorithm | 386 |
| Metabolite Set | p Value 1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ♂♀ vs. ♂♀ | ♂ vs. ♂ | ♀ vs. ♀ | ||||||||||
| Normal vs. Overweight | Normal vs. Stage 1 Obesity | Normal vs. Stage 2 Obesity | Normal vs. Stage 3 Obesity | Normal vs. Overweight | Normal vs. Stage 1 Obesity | Normal vs. Stage 2 Obesity | Normal vs. Stage 3 Obesity | Normal vs. Overweight | Normal vs. Stage 1 Obesity | Normal vs. Stage 2 Obesity | Normal vs. Stage 3 Obesity | |
| Steroidogenesis | 3 × 10−6 | 2 × 10−5 | 5 × 10−8 | 5 × 10−12 | 0.477 | - | 0.004 | 0.002 | 0.094 | 6 × 10−7 | 1 × 10−6 | 1 × 10−10 |
| Androstenedione metabolism | 4 × 10−4 | 0.013 | 0.017 | 0.014 | - | 0.173 | 0.113 | 0.045 | 0.258 | 0.011 | 0.065 | 6 × 10−4 |
| Androgen and estrogen metabolism | 9 × 10−4 | 0.011 | 0.011 | 0.002 | - | - | 0.076 | 0.023 | 0.042 | 0.002 | 0.004 | 2 × 10−5 |
| Aspartate metabolism | 0.062 | 0.047 | 0.047 | 0.378 | 0.409 | 0.244 | 0.251 | 0.338 | 0.426 | 0.015 | 0.191 | 0.198 |
| Urea cycle | 0.085 | 0.041 | 0.120 | 0.669 | 0.352 | 0.019 | 0.429 | 0.261 | 0.027 | 0.035 | 0.294 | 0.302 |
| Ammonia recycling | 0.119 | 0.154 | 0.171 | 0.514 | 0.381 | 0.225 | 0.21 | 0.3 | 0.744 | 0.061 | 0.633 | 0.358 |
| Estrone metabolism | 0.131 | 0.449 | 0.56 | 0.536 | - | - | 0.711 | 0.57 | 0.258 | 0.426 | 0.204 | 0.210 |
| Beta-alanine metabolism | 0.144 | 0.733 | 0.381 | 0.353 | - | 0.237 | 0.829 | 0.699 | 0.765 | 0.322 | 0.387 | 0.674 |
| Malate-aspartate shuttle | 0.199 | 0.158 | 0.106 | 0.323 | 0.138 | 0.075 | 0.401 | 0.295 | 0.343 | 0.148 | 0.135 | 0.139 |
| Glucose-alanine cycle | 0.294 | 0.276 | 0.471 | 0.798 | - | 0.097 | 0.487 | 0.365 | 0.422 | 0.553 | 0.587 | 0.593 |
| Glycine and serine metabolism | 0.364 | 0.005 | 0.038 | 0.130 | - | 0.379 | 0.338 | 0.137 | 0.219 | 0.010 | 0.55 | 0.191 |
| Cysteine metabolism | 0.368 | 0.515 | 0.376 | 0.351 | - | 0.187 | 0.375 | 0.600 | - | 0.491 | 0.517 | 0.836 |
| Retinol metabolism | 0.369 | 0.794 | 0.662 | 0.426 | - | - | 0.279 | 0.73 | 0.794 | 0.774 | 0.219 | 0.227 |
| Tryptophan metabolism | 0.38 | 0.772 | 0.662 | 0.55 | 0.598 | 0.384 | 0.586 | 0.337 | 0.228 | 0.744 | 0.985 | 0.778 |
| Glutamate metabolism | 0.38 | 0.034 | 0.662 | 0.505 | 0.523 | 0.325 | 0.715 | 0.826 | 0.609 | 0.0617 | 0.636 | 0.646 |
| Phenylalanine and tyrosine metabolism | 0.415 | 0.783 | 0.433 | 0.645 | - | 0.0182 | 0.411 | 0.248 | 0.0245 | 0.177 | 0.275 | 0.283 |
| Alanine metabolism | 0.419 | 0.211 | 0.33 | 0.877 | - | 0.384 | 0.583 | 0.449 | 0.512 | 0.422 | 0.686 | 0.692 |
| Lysine degradation | 0.461 | 0.822 | 0.715 | 0.692 | - | 0.212 | 0.447 | 0.653 | - | 0.611 | 0.873 | 0.605 |
| Histidine metabolism | 0.486 | 0.585 | 0.407 | 0.692 | - | 0.291 | 0.363 | 0.438 | 0.841 | 0.379 | 0.544 | 0.795 |
| Citric acid cycle | 0.505 | 0.483 | 0.542 | 0.734 | - | - | 0.81 | 0.677 | 0.744 | 0.271 | 0.350 | 0.641 |
| Group | Body Height (cm) | Body Weight (kg) | Age (Years) | Body Mass Index | Gender (Male/Female) |
|---|---|---|---|---|---|
| Normal | 173.5 ± 8.2 1 | 66.9 ± 9.4 | 31.3 ± 5.5 | 22.1 ± 1.9 | 10/10 |
| Overweight | 172.1 ± 12.2 | 82.0 ± 13.1 | 32.9 ± 6.7 | 27.5 ± 1.3 | 10/10 |
| Stage 1 obesity | 170.6 ± 11.7 | 95.1 ± 13.7 | 29.7 ± 8.0 | 32.5 ± 11.7 | 10/10 |
| Stage 2 obesity | 171.5 ± 9.4 | 109.1 ± 13.8 | 32.8 ± 8.1 | 36.9 ± 1.3 | 10/10 |
| Stage 3 obesity | 172.3 ± 9.9 | 141.0 ± 27.4 | 34.5 ± 6.5 | 47.3 ± 6.1 | 10/10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lokhov, P.G.; Balashova, E.E.; Trifonova, O.P.; Maslov, D.L.; Ponomarenko, E.A.; Archakov, A.I. Mass Spectrometry-Based Metabolomics Analysis of Obese Patients’ Blood Plasma. Int. J. Mol. Sci. 2020, 21, 568. https://doi.org/10.3390/ijms21020568
Lokhov PG, Balashova EE, Trifonova OP, Maslov DL, Ponomarenko EA, Archakov AI. Mass Spectrometry-Based Metabolomics Analysis of Obese Patients’ Blood Plasma. International Journal of Molecular Sciences. 2020; 21(2):568. https://doi.org/10.3390/ijms21020568
Chicago/Turabian StyleLokhov, Petr G., Elena E. Balashova, Oxana P. Trifonova, Dmitry L. Maslov, Elena A. Ponomarenko, and Alexander I. Archakov. 2020. "Mass Spectrometry-Based Metabolomics Analysis of Obese Patients’ Blood Plasma" International Journal of Molecular Sciences 21, no. 2: 568. https://doi.org/10.3390/ijms21020568