Acadesine Circumvents Azacitidine Resistance in Myelodysplastic Syndrome and Acute Myeloid Leukemia

 , add
Show full author list
, add
Show full author list
Abstract
:1. Introduction
2. Results
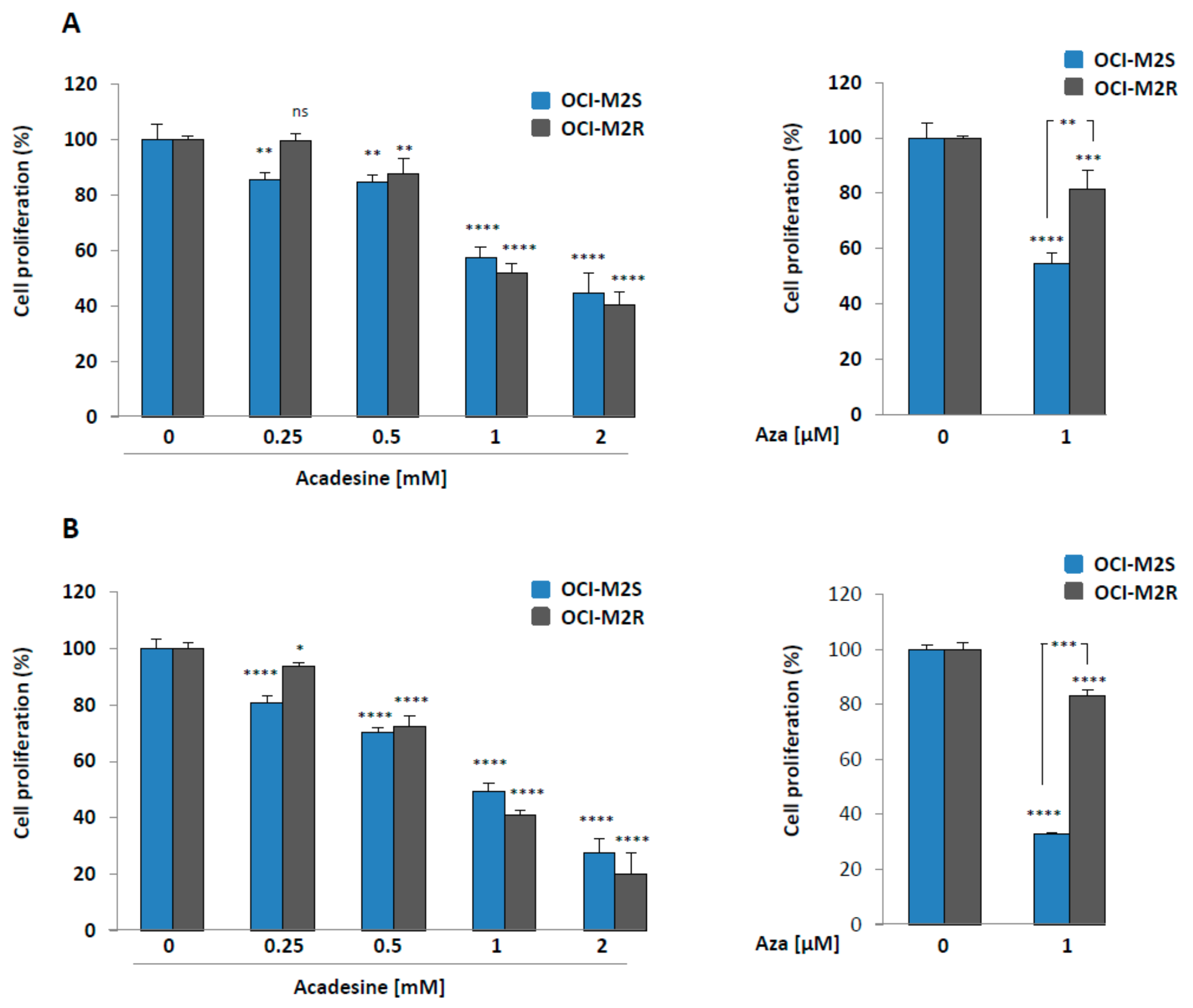
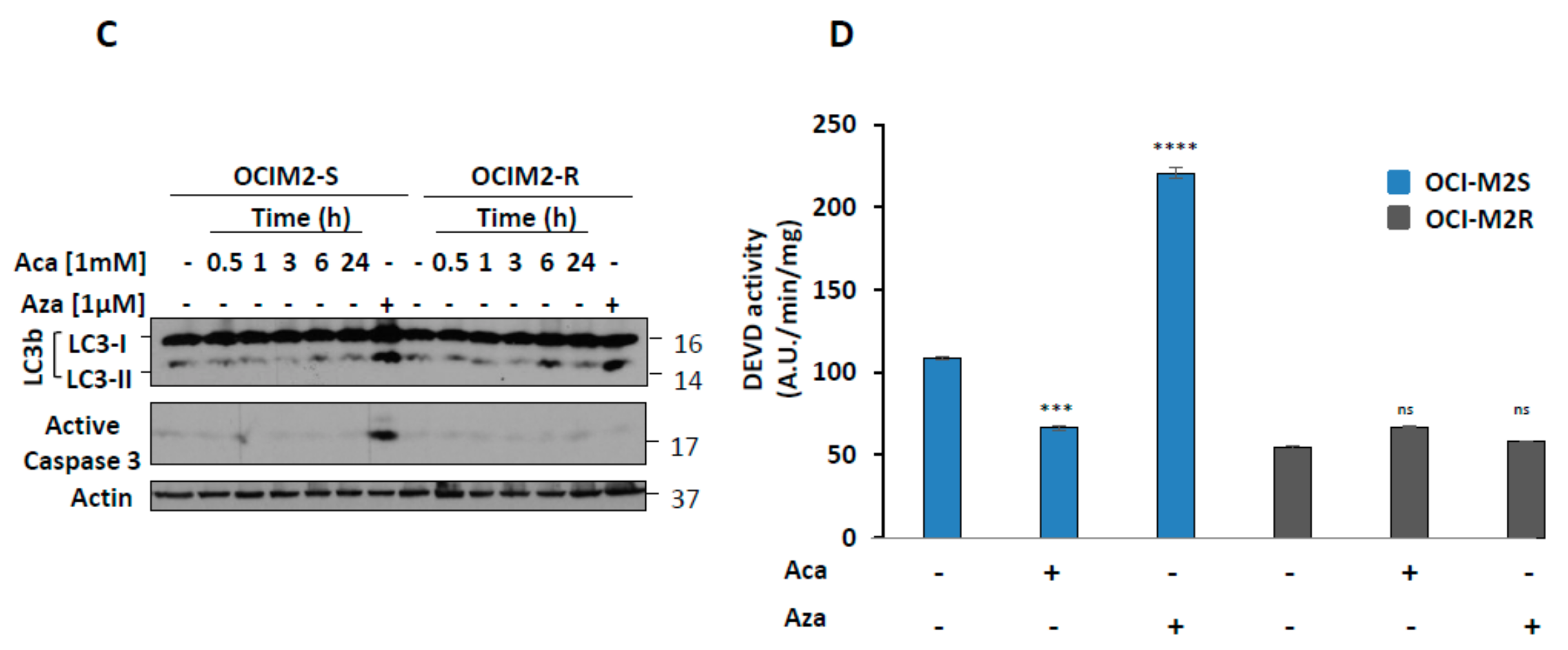
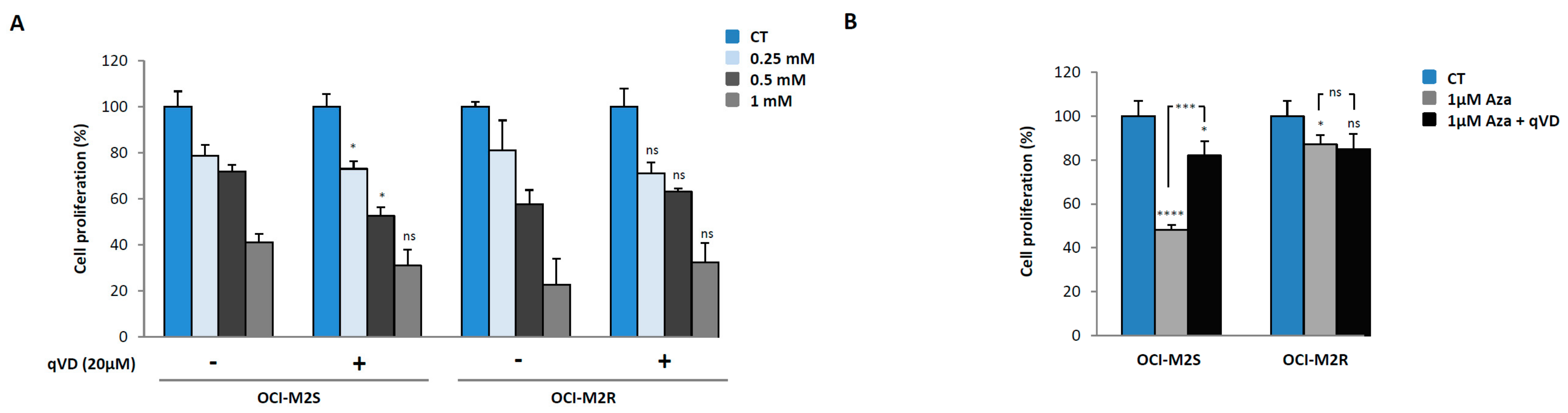
2.1. Aca Induces Cell Death in an Apoptosis-Independent Manner in MDS Cell Lines
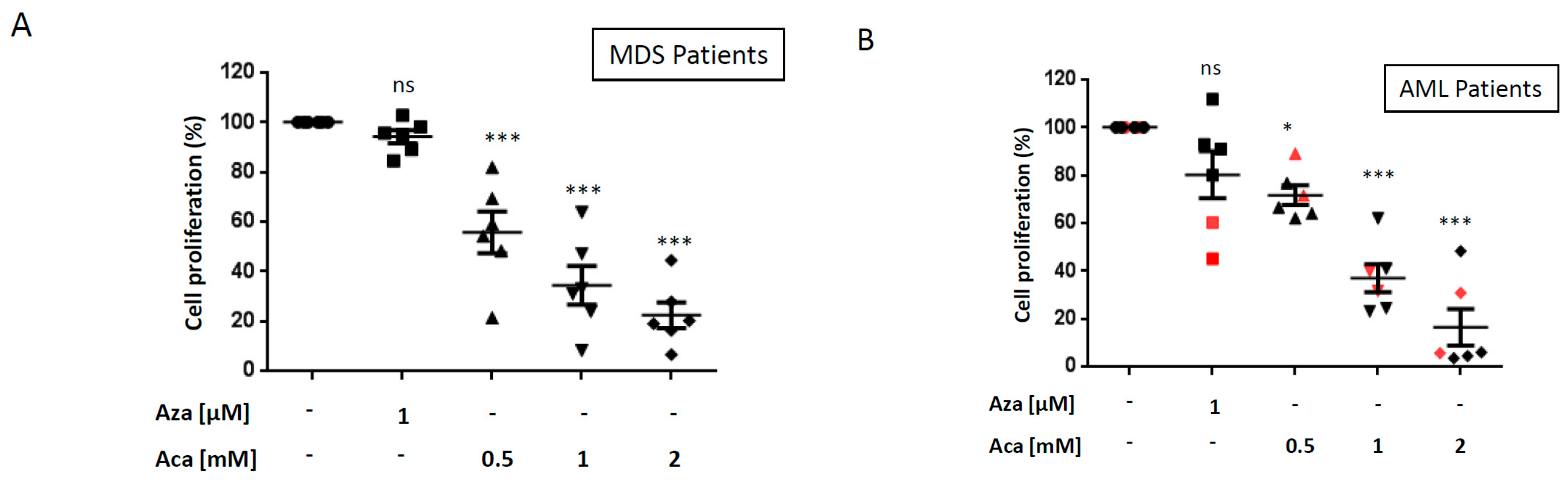
2.2. Aca Efficiently Kills Primary Cell from Aza-Resistant MDS and AML Patient ex vivo.
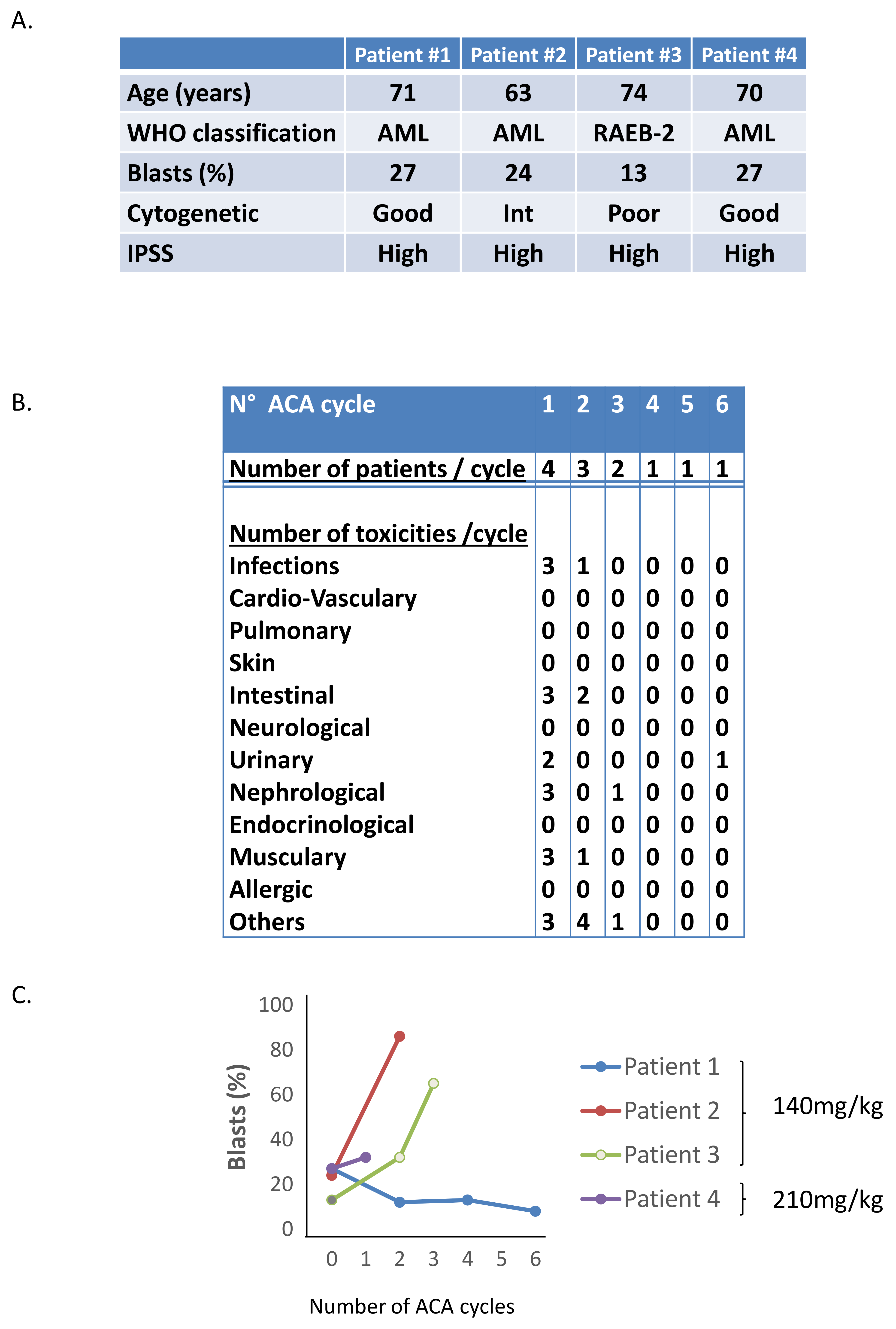
2.3. Phase I/II Clinical Trial of Aca in MDS/AML Patients
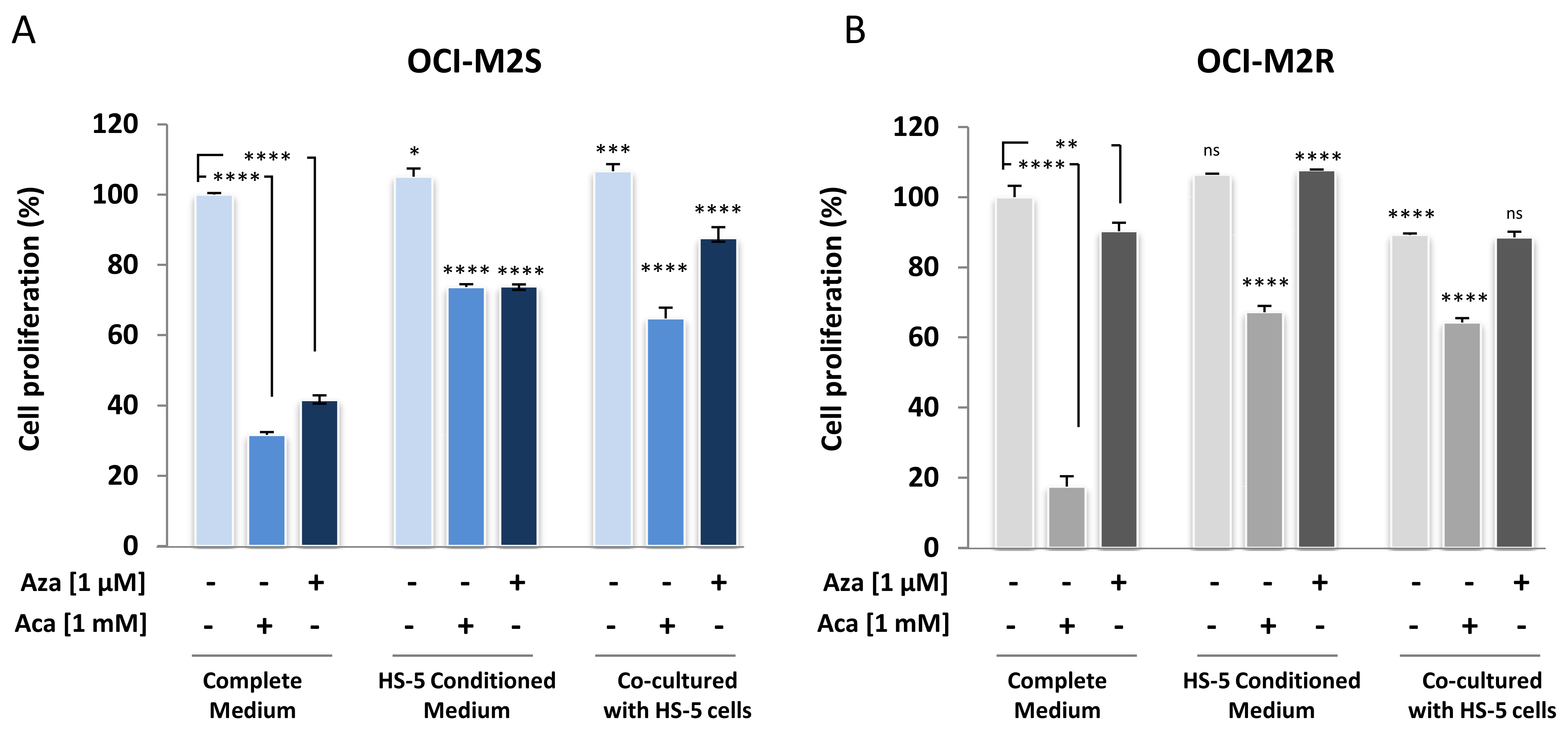
2.4. Medullary Stromal Cells Inhibit Aca-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines
4.3. Bone Marrow Samples
4.4. Assessment of Cell Proliferation:
4.5. Western Blot Analysis
4.6. Caspase 3 Activity Assay
4.7. Phase I-II Clinical Trial of Aca in MDS/AML Patients
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prebet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Ades, L.; et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluzeau, T.; Robert, G.; Mounier, N.; Karsenti, J.M.; Dufies, M.; Puissant, A.; Jacquel, A.; Renneville, A.; Preudhomme, C.; Cassuto, J.P.; et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget 2012, 3, 490–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluzeau, T.; Robert, G.; Puissant, A.; Jean-Michel, K.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Azacitidine-resistant SKM1 myeloid cells are defective for AZA-induced mitochondrial apoptosis and autophagy. Cell Cycle 2011, 10, 2339–2343. [Google Scholar] [CrossRef] [Green Version]
- Campas, C.; Lopez, J.M.; Santidrian, A.F.; Barragan, M.; Bellosillo, B.; Colomer, D.; Gil, J. Acadesine activates AMPK and induces apoptosis in B-cell chronic lymphocytic leukemia cells but not in T lymphocytes. Blood 2003, 101, 3674–3680. [Google Scholar] [CrossRef]
- Campas, C.; Santidrian, A.F.; Domingo, A.; Gil, J. Acadesine induces apoptosis in B cells from mantle cell lymphoma and splenic marginal zone lymphoma. Leukemia 2005, 19, 292–294. [Google Scholar] [CrossRef] [Green Version]
- Coll-Mulet, L.; Iglesias-Serret, D.; Santidrian, A.F.; Cosialls, A.M.; de Frias, M.; Castano, E.; Campas, C.; Barragan, M.; de Sevilla, A.F.; Domingo, A.; et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood 2006, 107, 4109–4114. [Google Scholar] [CrossRef] [Green Version]
- Santidrian, A.F.; Gonzalez-Girones, D.M.; Iglesias-Serret, D.; Coll-Mulet, L.; Cosialls, A.M.; de Frias, M.; Campas, C.; Gonzalez-Barca, E.; Alonso, E.; Labi, V.; et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood 2010, 116, 3023–3032. [Google Scholar] [CrossRef] [Green Version]
- Robert, G.; Ben Sahra, I.; Puissant, A.; Colosetti, P.; Belhacene, N.; Gounon, P.; Hofman, P.; Bost, F.; Cassuto, J.P.; Auberger, P. Acadesine kills chronic myelogenous leukemia (CML) cells through PKC-dependent induction of autophagic cell death. PLoS ONE 2009, 4, e7889. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Robert, G.; Auberger, P. Targeting autophagy to fight hematopoietic malignancies. Cell Cycle 2010, 9, 3470–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Montraveta, A.; Xargay-Torrent, S.; Lopez-Guerra, M.; Rosich, L.; Perez-Galan, P.; Salaverria, I.; Bea, S.; Kalko, S.G.; de Frias, M.; Campas, C.; et al. Synergistic anti-tumor activity of acadesine (AICAR) in combination with the anti-CD20 monoclonal antibody rituximab in in vivo and in vitro models of mantle cell lymphoma. Oncotarget 2014, 5, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Vakana, E.; Altman, J.K.; Glaser, H.; Donato, N.J.; Platanias, L.C. Antileukemic effects of AMPK activators on BCR-ABL-expressing cells. Blood 2011, 118, 6399–6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, A.; Furstoss, N.; Calleja, A.; Zerhouni, M.; Cluzeau, T.; Savy, C.; Marchetti, S.; Hamouda, M.A.; Boulakirba, S.; Orange, F.; et al. LAMP2 expression dictates azacytidine response and prognosis in MDS/AML. Leukemia 2019, 33, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Van Den Neste, E.; Cazin, B.; Janssens, A.; Gonzalez-Barca, E.; Terol, M.J.; Levy, V.; Perez de Oteyza, J.; Zachee, P.; Saunders, A.; de Frias, M.; et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): A multicenter phase I/II study. Cancer Chemother. Pharmacol. 2013, 71, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amdouni, H.; Robert, G.; Driowya, M.; Furstoss, N.; Metier, C.; Dubois, A.; Dufies, M.; Zerhouni, M.; Orange, F.; Lacas-Gervais, S.; et al. In Vitro and in Vivo Evaluation of Fully Substituted (5-(3-Ethoxy-3-oxopropynyl)-4-(ethoxycarbonyl)-1,2,3-triazolyl-glycosides as Original Nucleoside Analogues to Circumvent Resistance in Myeloid Malignancies. J. Med. Chem. 2017, 60, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Cheson, B.D.; Greenberg, P.L.; Bennett, J.M.; Lowenberg, B.; Wijermans, P.W.; Nimer, S.D.; Pinto, A.; Beran, M.; de Witte, T.M.; Stone, R.M.; et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006, 108, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Cheson, B.D.; Bennett, J.M.; Kopecky, K.J.; Buchner, T.; Willman, C.L.; Estey, E.H.; Schiffer, C.A.; Doehner, H.; Tallman, M.S.; Lister, T.A.; et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J. Clin. Oncol. 2003, 21, 4642–4649. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | n = 12 | ||
|---|---|---|---|
| WHO Classification | |||
| RAEB-2 | 6 (50%) | ||
| AML 20–30 % | 6 (50%) | ||
| IPSS Cytogenetic Risk | |||
| Good | 3 (25%) | ||
| Intermediate | 3 (25%) | ||
| Poor | 6 (50%) | ||
| Cytogenetics Abnormalities | |||
| Normal Karyotype | 3 (25%) | ||
| +8 | 1 (8%) | ||
| 7 Abnormalities | 3 (25%) | ||
| 5 Abnormalities | 3 (25%) | ||
| Complex > 3 Abnormalities | 2 (17%) | ||
| IPSS Classification | |||
| Low | 0 | ||
| Intermediate 1 | 0 | ||
| Intermediate 2 | 2 (17%) | ||
| High | 10 (83%) | ||
| Aza Response Status | |||
| Failure | 8 (67%) | ||
| Relapse | 4 (33%) | ||
| Number of Aza Cycles before Relapse or Failure | 6 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27] | ||
| (B) | |||
| Patients | Disease | IPSS Cytogenetic Risk | Cytogenetic Abnormalities |
| #1 | RAEB-2 | Intermediate | Normal |
| #2 | RAEB-2 | Intermediate | Normal |
| #3 | RAEB-2 | Intermediate | Normal |
| #4 | RAEB-2 | Intermediate | 47,XY,+8 |
| #5 | RAEB-2 | Intermediate | Normal |
| #6 | RAEB-2 | Intermediate | 46,XY(del(9)(q12q31)/47,idem,+21 |
| #7 | AML | Poor | 45,XX,-7 |
| #8 | AML | Poor | 45,XX,t(3;11)(q22,q33),del(9)(q22,q33),-7 |
| #9 | AML | Poor | 45,XX,-7, inv(3), del11q |
| #10 | AML | Intermediate | 46,XY,inv(3)(q21q26) |
| #11 | AML | Poor | 46,XX,del(5)(q13q33)/47, idem, del(1)(p3?4),+mar/46,XX |
| #12 | AML | Good | 45,X,-Y |
| (C) | |||
| Patients | IPSS Classification | AZA Response Status | Number of AZA Cycles before Relapse or Failure |
| #1 | High | Failure | 3 |
| #2 | Intermediate 2 | Relapse | 27 |
| #3 | High | Failure | 6 |
| #4 | High | Relapse | 9 |
| #5 | Intermediate 2 | Relapse | 19 |
| #6 | High | Relapse | 26 |
| #7 | High | Failure | 6 |
| #8 | High | Failure | 4 |
| #9 | High | Failure | 6 |
| #10 | High | Failure | 1 |
| #11 | High | Failure | 6 |
| #12 | High | Failure | 6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cluzeau, T.; Furstoss, N.; Savy, C.; El Manaa, W.; Zerhouni, M.; Blot, L.; Calleja, A.; Dufies, M.; Dubois, A.; Ginet, C.; et al. Acadesine Circumvents Azacitidine Resistance in Myelodysplastic Syndrome and Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 164. https://doi.org/10.3390/ijms21010164
Cluzeau T, Furstoss N, Savy C, El Manaa W, Zerhouni M, Blot L, Calleja A, Dufies M, Dubois A, Ginet C, et al. Acadesine Circumvents Azacitidine Resistance in Myelodysplastic Syndrome and Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2020; 21(1):164. https://doi.org/10.3390/ijms21010164
Chicago/Turabian StyleCluzeau, Thomas, Nathan Furstoss, Coline Savy, Wejdane El Manaa, Marwa Zerhouni, Lauriane Blot, Anne Calleja, Maeva Dufies, Alix Dubois, Clemence Ginet, and et al. 2020. "Acadesine Circumvents Azacitidine Resistance in Myelodysplastic Syndrome and Acute Myeloid Leukemia" International Journal of Molecular Sciences 21, no. 1: 164. https://doi.org/10.3390/ijms21010164