Loss of p53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation

 , ,
, ,
Abstract
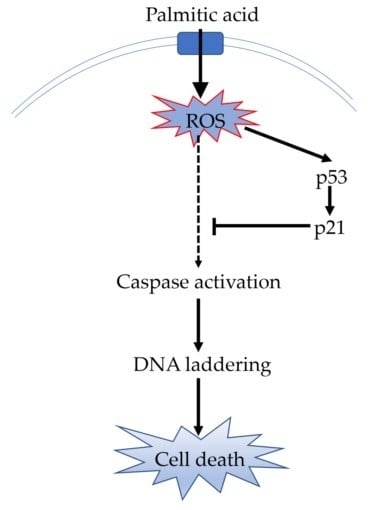
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
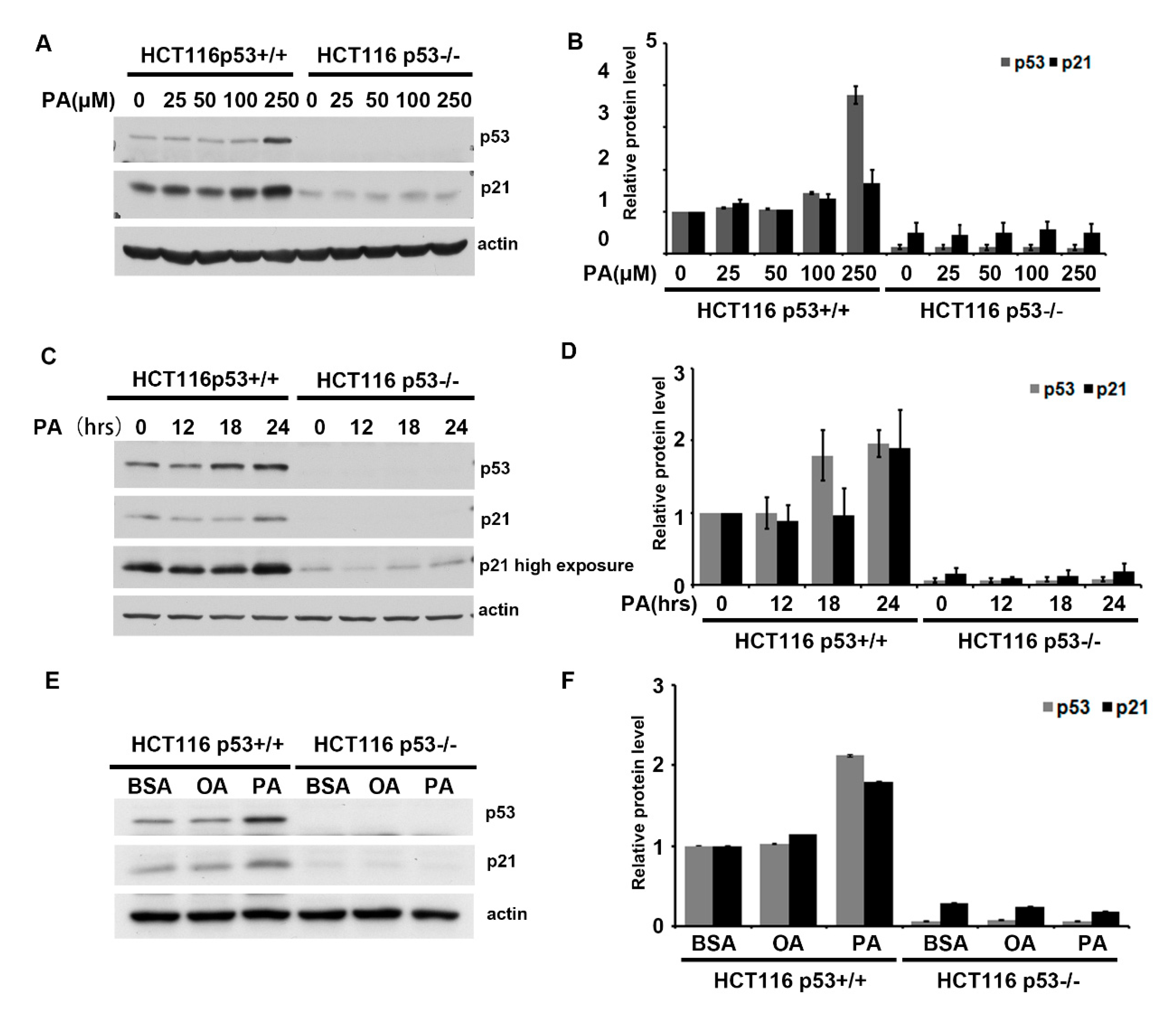
2.1. Palmitic Acid Activates p53 in a Dose-and Time-Dependent Manner
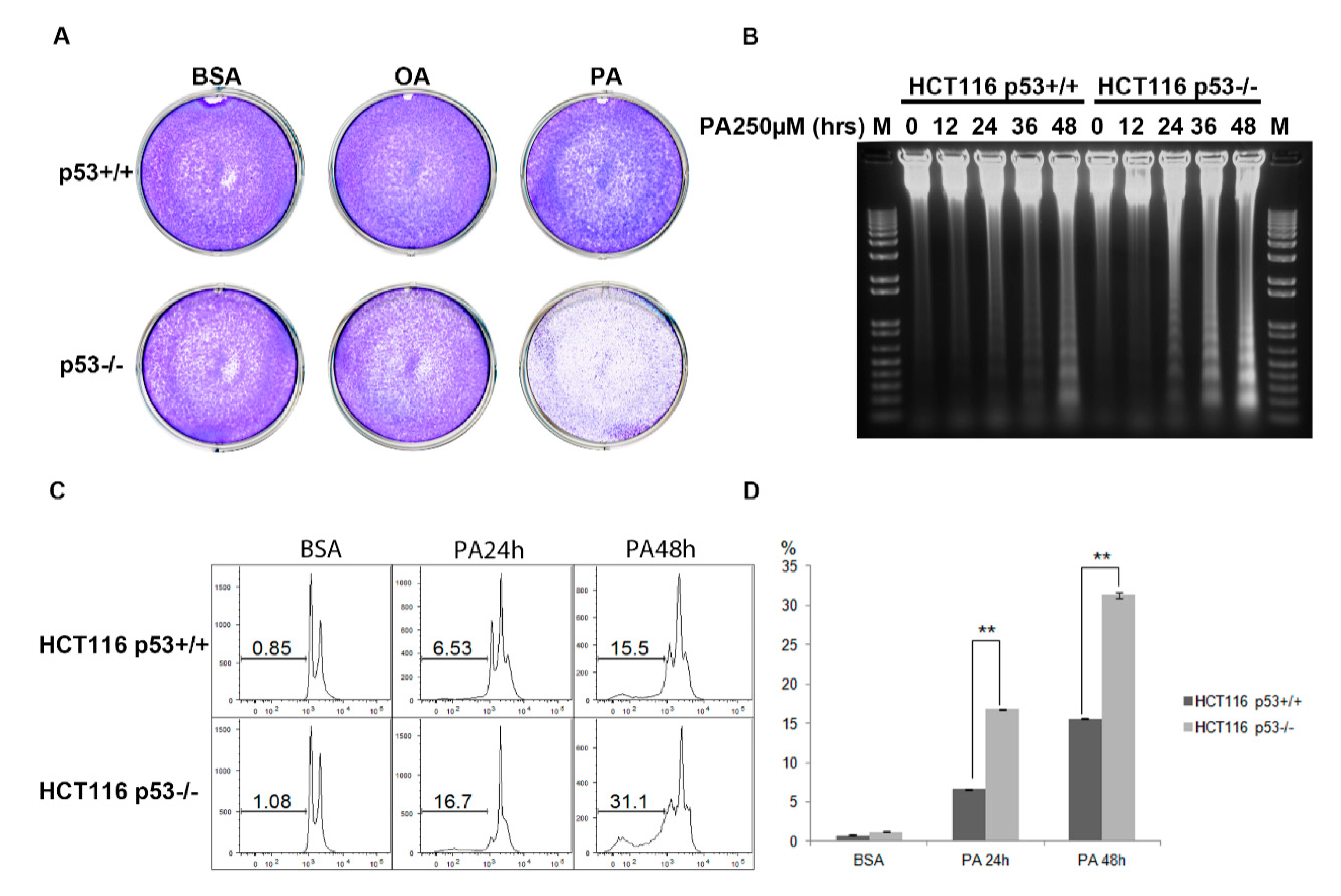
2.2. Cells are more Sensitive to PA in HCT116 p53−/− Cell Line Compared to HCT116 p53+/+ Cell Line
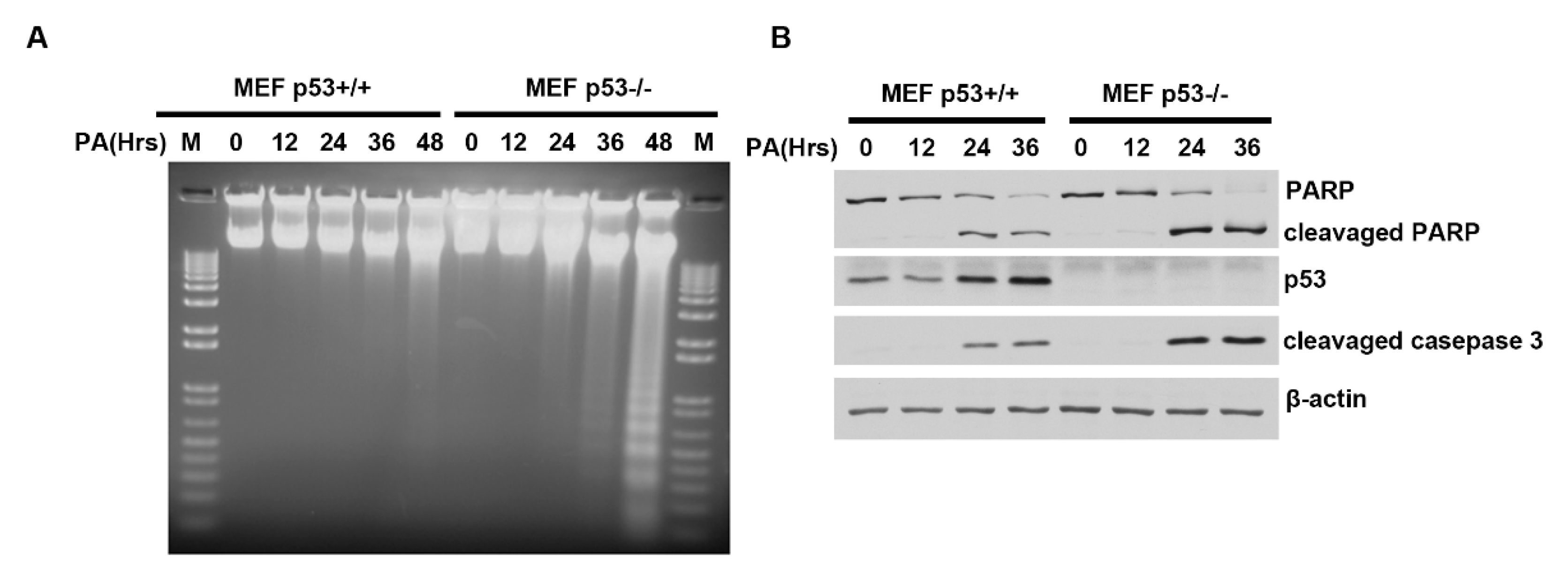
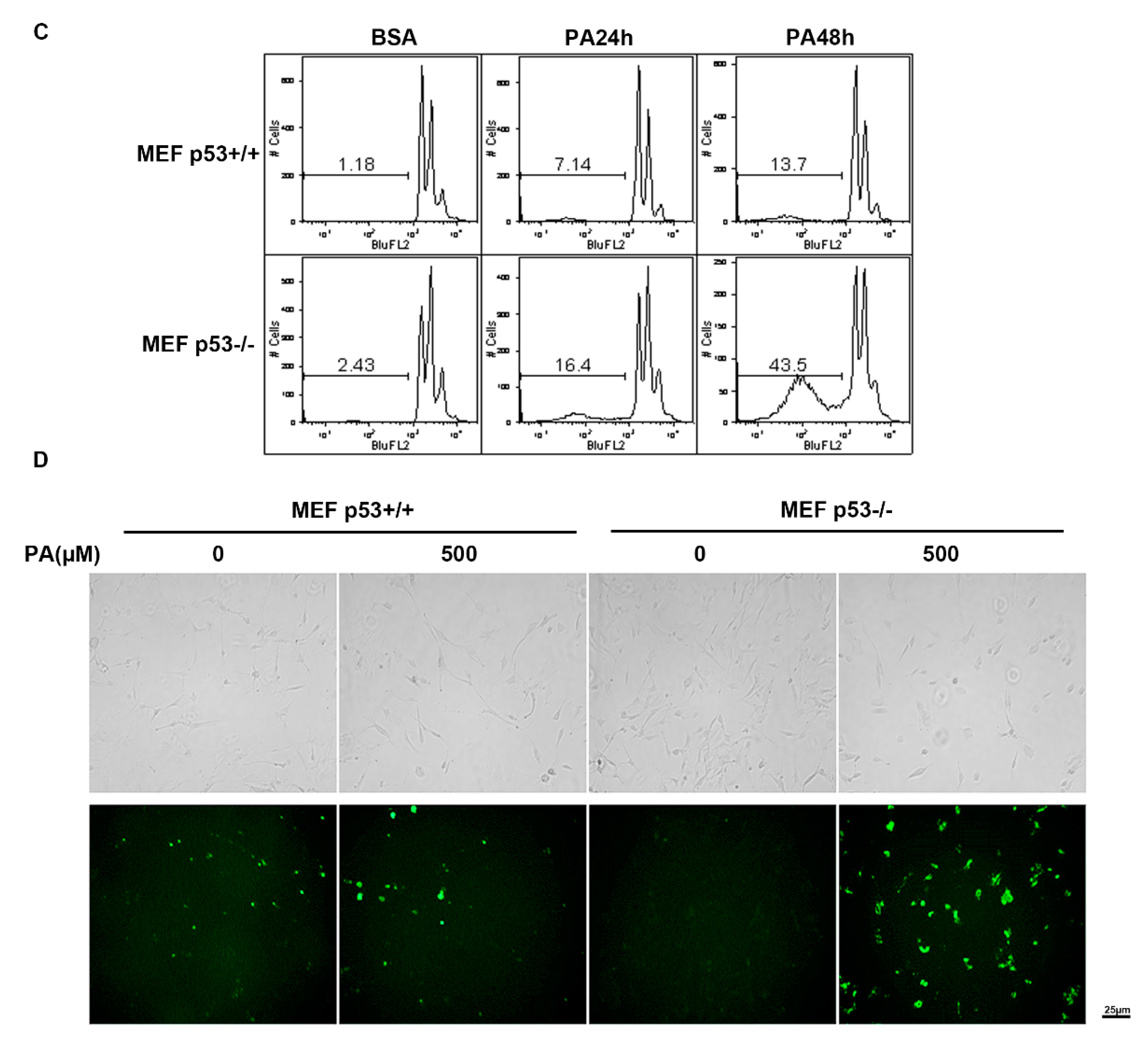
2.3. Distinct PA-Induced Apoptosis Effects in p53+/+ and p53−/− Primary Mouse Embryo Fibroblast Cell Lines
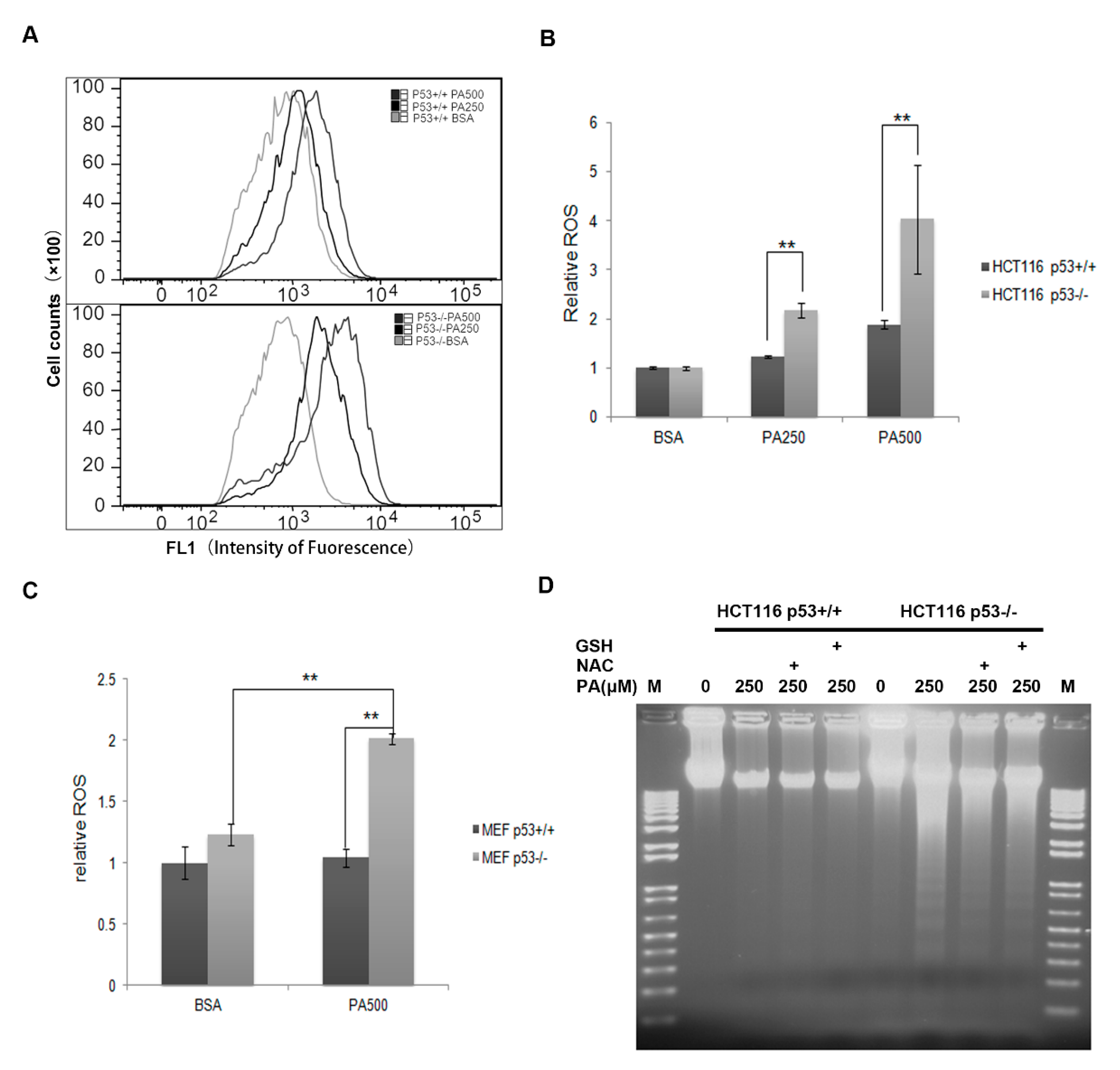
2.4. ROS Contributes at Least Partly to the Cell Apoptosis Induced by PA
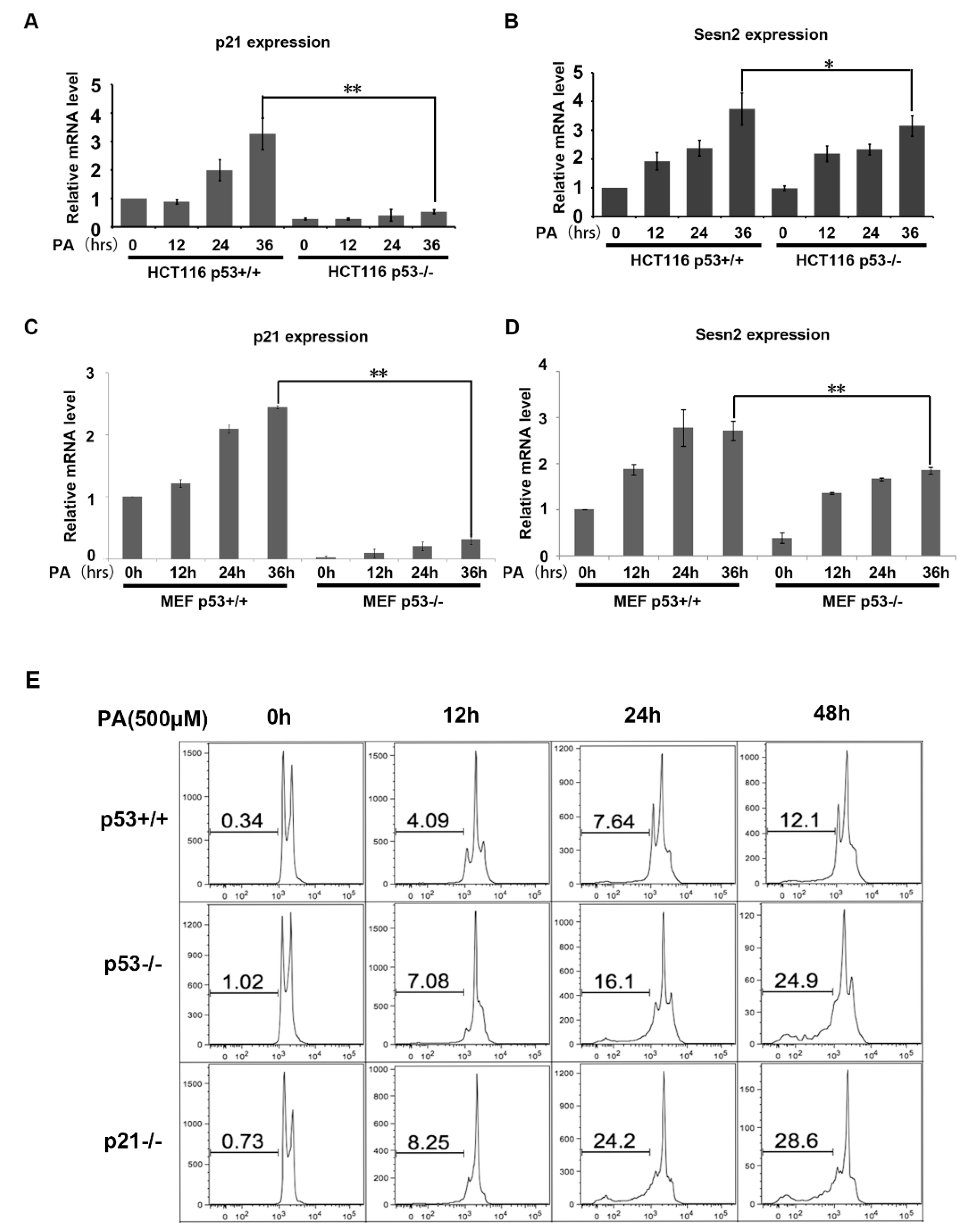
2.5. Gene Expression Induced by p53 Under PA Stress in HCT116 and MEF Cell Lines.
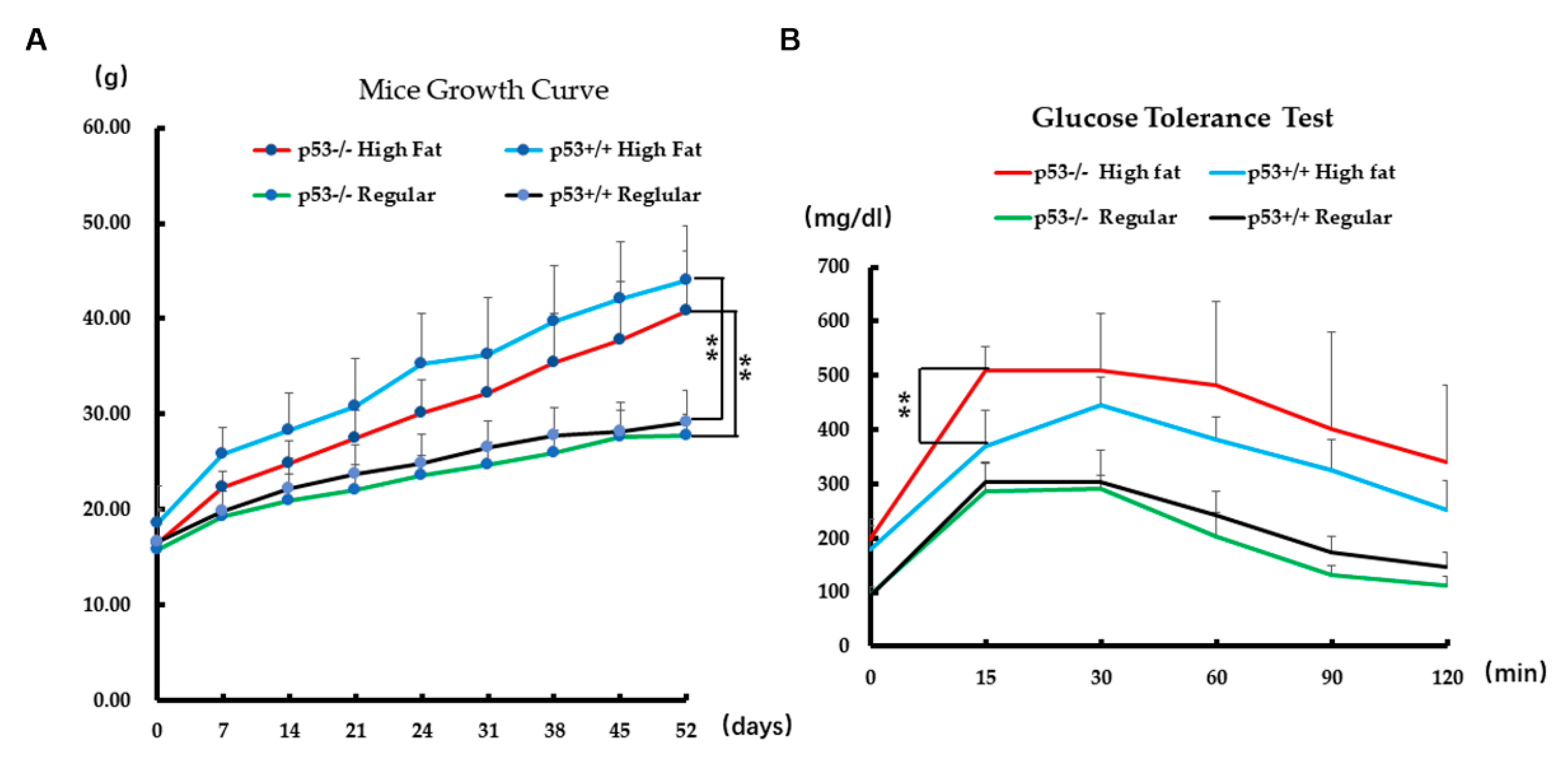
2.6. Biological Effect on Mice in PA Stress
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of Albumin-Bound Fatty Acid
4.3. Cell Culture and Isolation of Mouse Embryonic Fibroblasts
4.4. SDS-PAGE and Immunoblotting
4.5. Cell Viability Analysis and ANNEXIN V-FITC Staining
4.6. Propidium Iodide (PI) Staining and Flow Cytometry.
4.7. ROS Measurement and DNA Ladder Assay
4.8. Construction of a Model of Mice Obesity and Glucose Resistance Assay
4.9. Real Time PCR and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | bovine serum albumin |
| MEF | mouse embryo fibroblasts |
| OA | oleic acid |
| PI | propidium iodide |
| ROS | reactive oxygen species |
| HCT116 | human colon carcinoma cells |
| NAC | N-Acetyl cysteine |
| PA | palmitic acid |
| PLG | phase lock gel |
References
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, A.; Imperlini, E.; Nigro, E.; Montagnese, C.; Daniele, A.; Orru, S.; Buono, P. Biological and Nutritional Properties of Palm Oil and Palmitic Acid: Effects on Health. Molecules 2015, 20, 17339–17361. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Banerjee, H.; Dutta, S.; Hazra, A.K.; Majumdar, K. Macronutrients influence yield and oil quality of hybrid maize (Zea mays L.). PLoS ONE 2019, 14, e0216939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Geng, H.; Tang, H.; Wang, L.; Yu, D.; Wang, J.; Song, Y. Enzyme-assisted Aqueous Extraction of Oil from Rice Germ and its Physicochemical Properties and Antioxidant Activity. J. Oleo Sci. 2019, 69, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amalfitano, C.; Golubkina, N.A.; Del Vacchio, L.; Russo, G.; Cannoniero, M.; Somma, S.; Morano, G.; Cuciniello, A.; Caruso, G. Yield, Antioxidant Components, Oil Content, and Composition of Onion Seeds Are Influenced by Planting Time and Density. Plants (Basel) 2019, 8, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marafie, S.K.; Al-Shawaf, E.M.; Abubaker, J.; Arefanian, H. Palmitic acid-induced lipotoxicity promotes a novel interplay between Akt-mTOR, IRS-1, and FFAR1 signaling in pancreatic beta-cells. Biol. Res. 2019, 52, 44. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, X.; Sun, Y.; Du, J.; Li, X.; Xun, Z.; Li, Y.C. High fat diet promotes experimental colitis by inducing oxidative stress in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G453–G462. [Google Scholar] [CrossRef]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Hannaert, J.C.; Hoorens, A.; Eizirik, D.L.; Pipeleers, D.G. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 2001, 50, 1771–1777. [Google Scholar] [CrossRef] [Green Version]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.; Chen, H.; Zhang, H.; Wan, X.; Su, Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine 2012, 42, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Langelier, Y.; Prentki, M. Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res. 2000, 60, 6353–6358. [Google Scholar] [PubMed]
- de Vries, J.E.; Vork, M.M.; Roemen, T.H.; de Jong, Y.F.; Cleutjens, J.P.; van der Vusse, G.J.; van Bilsen, M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J. Lipid Res. 1997, 38, 1384–1394. [Google Scholar] [PubMed]
- Zhang, Y.; Xia, G.; Zhang, Y.; Liu, J.; Liu, X.; Li, W.; Lv, Y.; Wei, S.; Liu, J.; Quan, J. Palmitate induces VSMC apoptosis via toll like receptor (TLR)4/ROS/p53 pathway. Atherosclerosis 2017, 263, 74–81. [Google Scholar] [CrossRef]
- Shan, X.; Miao, Y.; Fan, R.; Song, C.; Wu, G.; Wan, Z.; Zhu, J.; Sun, G.; Zha, W.; Mu, X.; et al. Suppression of Grb2 expression improved hepatic steatosis, oxidative stress, and apoptosis induced by palmitic acid in vitro partly through insulin signaling alteration. In Vitro Cell Dev. Biol. Anim. 2013, 49, 576–582. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhao, S.; Wang, F.; Zhang, H.; Chen, Z.J.; Wang, J.; Wang, Z.; Du, Z.; Ling, E.A.; Liu, Q.; et al. Palmitic acid increases apoptosis of neural stem cells via activating c-Jun N-terminal kinase. Stem Cell Res. 2013, 10, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro, M.; Koyama, K.; Lee, Y.; Unger, R.H. Leptin- or troglitazone-induced lipopenia protects islets from interleukin 1beta cytotoxicity. J. Clin. Investig. 1997, 100, 1750–1754. [Google Scholar] [CrossRef]
- Ostrander, D.B.; Zhang, M.; Mileykovskaya, E.; Rho, M.; Dowhan, W. Lack of mitochondrial anionic phospholipids causes an inhibition of translation of protein components of the electron transport chain. A yeast genetic model system for the study of anionic phospholipid function in mitochondria. J. Biol. Chem. 2001, 276, 25262–25272. [Google Scholar] [CrossRef] [Green Version]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [Green Version]
- Wawryk-Gawda, E.; Chylinska-Wrzos, P.; Lis-Sochocka, M.; Chlapek, K.; Bulak, K.; Jedrych, M.; Jodlowska-Jedrych, B. P53 protein in proliferation, repair and apoptosis of cells. Protoplasma 2014, 251, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic functions of the p53 tumor suppressor: Regulation of energy metabolism and antioxidant defense. Semin. Cancer Biol. 2009, 19, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.F.; Szalay, M.G.; Paskar, L.; Sahai, B.M.; Boyer, M.; Singh, B.; Green, D.R. Activation-induced cell death in T cell hybridomas is due to apoptosis. Morphologic aspects and DNA fragmentation. J. Immunol. 1990, 144, 3326–3333. [Google Scholar]
- Desoize, B.; Sen, S. Apoptosis or programmed cell death: Concepts, mechanisms and contribution in oncology. Bull. Cancer 1992, 79, 413–425. [Google Scholar]
- Schmid, I.; Uittenbogaart, C.H.; Giorgi, J.V. Sensitive method for measuring apoptosis and cell surface phenotype in human thymocytes by flow cytometry. Cytometry 1994, 15, 12–20. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, J.; Liu, W.; Yao, G.; Xu, F.; Hayashi, T.; Onodera, S.; Ikejima, T. Attenuating effect of silibinin on palmitic acid-induced apoptosis and mitochondrial dysfunction in pancreatic beta-cells is mediated by estrogen receptor alpha. Mol. Cell Biochem. 2019, 460, 81–92. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Chen, M.Z.; Olefsky, J.M.; Marth, J.D. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat. Med. 2011, 17, 1067–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Guan, G.; Lei, L.; Liu, J.; Cao, L.; Wang, X. Oxidative and endoplasmic reticulum stresses are involved in palmitic acid-induced H9c2 cell apoptosis. Biosci. Rep. 2019, 39, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkel, O.; Taylor, N.; Prutsch, N.; Staber, P.B.; Moriggl, R.; Turner, S.D.; Kenner, L. When the guardian sleeps: Reactivation of the p53 pathway in cancer. Mutat. Res. 2017, 773, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Green, D.R. Detection of apoptosis by annexin V labeling. Methods Enzymol. 2000, 322, 15–18. [Google Scholar] [PubMed]
- van Engeland, M.; Nieland, L.J.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Le, B.H.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [PubMed]
- Guri, A.J.; Hontecillas, R.; Bassaganya-Riera, J. Abscisic acid synergizes with rosiglitazone to improve glucose tolerance and down-modulate macrophage accumulation in adipose tissue: Possible action of the cAMP/PKA/PPAR gamma axis. Clin. Nutr. 2010, 29, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Heijden, R.A.; Sheedfar, F.; Morrison, M.C.; Hommelberg, P.P.; Kor, D.; Kloosterhuis, N.J.; Gruben, N.; Youssef, S.A.; de Bruin, A.; Hofker, M.H.; et al. High-fat diet induced obesity primes inflammation in adipose tissue prior to liver in C57BL/6j mice. Aging (Albany NY) 2015, 7, 256–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 5. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Impact of Premature Senescence on Radiosensitivity Measured by High Throughput Cell-Based Assays. Int. J. Mol. Sci. 2017, 18, 7. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Weiss, R.H.; Murray, D. Spontaneous gammaH2AX Foci in Human Solid Tumor-Derived Cell Lines in Relation to p21WAF1 and WIP1 Expression. Int. J. Mol. Sci. 2015, 16, 11609–11628. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- de la Torre, A.J.; Rogoff, D.; White, P.C. P53 and cellular glucose uptake. Endocr. Res. 2013, 38, 32–39. [Google Scholar] [CrossRef]
- Mai, W.X.; Gosa, L.; Daniels, V.W.; Ta, L.; Tsang, J.E.; Higgins, B.; Gilmore, W.B.; Bayley, N.A.; Harati, M.D.; Lee, J.T.; et al. Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat. Med. 2017, 23, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.Y.; Kim, H.B.; Piao, C.; Lee, K.H.; Hyun, J.W.; Chang, I.Y.; You, H.J. The critical role of catalase in prooxidant and antioxidant function of p53. Cell Death Differ. 2013, 20, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Popowich, D.A.; Vavra, A.K.; Walsh, C.P.; Bhikhapurwala, H.A.; Rossi, N.B.; Jiang, Q.; Aalami, O.O.; Kibbe, M.R. Regulation of reactive oxygen species by p53: Implications for nitric oxide-mediated apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2192–H2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.M.; Yu, Z.X.; Ferrans, V.J.; Lowenstein, R.A.; Finkel, T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 11848–11852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef]
- Hollander, M.C.; Alamo, I.; Jackman, J.; Wang, M.G.; McBride, O.W.; Fornace, A.J., Jr. Analysis of the mammalian gadd45 gene and its response to DNA damage. J. Biol. Chem. 1993, 268, 24385–24393. [Google Scholar]
- Das, S.K.; Mondal, A.K.; Elbein, S.C. Distinct gene expression profiles characterize cellular responses to palmitate and oleate. J. Lipid Res. 2010, 51, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Viability Assessment Following Anticancer Treatment Requires Single-Cell Visualization. Cancers (Basel) 2018, 10, 255. [Google Scholar] [CrossRef] [Green Version]
- Spector, A.A. Structure and lipid binding properties of serum albumin. Methods Enzymol. 1986, 128, 320–339. [Google Scholar]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, G.; Luo, H.; Zhang, N.; Wang, Y.; Li, Y.; Huang, H.; Liu, Y.; Hu, Y.; Liu, H.; Zhang, J.; et al. Loss of p53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation. Int. J. Mol. Sci. 2019, 20, 6268. https://doi.org/10.3390/ijms20246268
Yu G, Luo H, Zhang N, Wang Y, Li Y, Huang H, Liu Y, Hu Y, Liu H, Zhang J, et al. Loss of p53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation. International Journal of Molecular Sciences. 2019; 20(24):6268. https://doi.org/10.3390/ijms20246268
Chicago/Turabian StyleYu, Guowu, Hongwei Luo, Na Zhang, Yongbin Wang, Yangping Li, Huanhuan Huang, Yinghong Liu, Yufeng Hu, Hanmei Liu, Junjie Zhang, and et al. 2019. "Loss of p53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation" International Journal of Molecular Sciences 20, no. 24: 6268. https://doi.org/10.3390/ijms20246268