Dysregulated Choline, Methionine, and Aromatic Amino Acid Metabolism in Patients with Wilson Disease: Exploratory Metabolomic Profiling and Implications for Hepatic and Neurologic Phenotypes


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
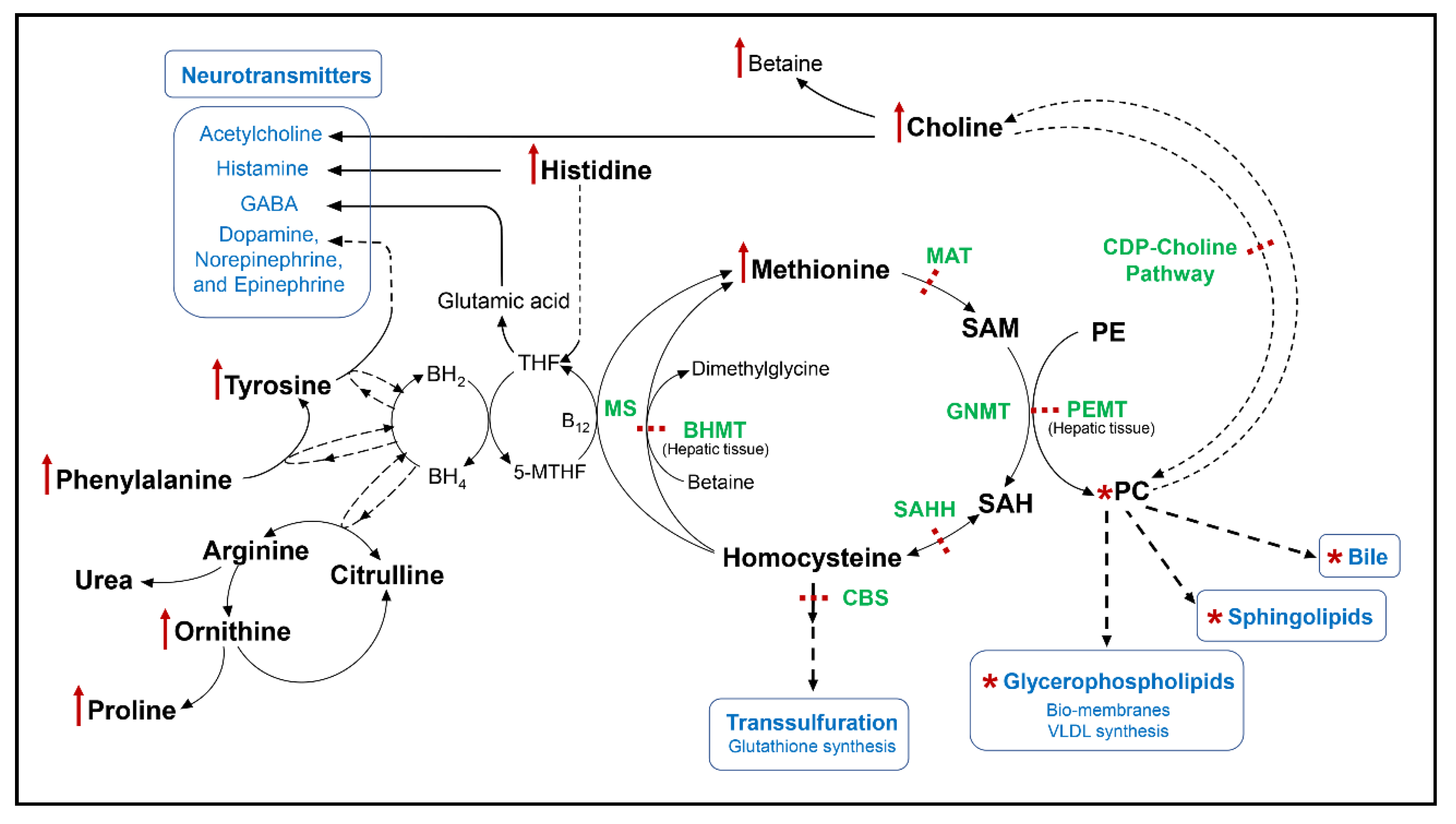
Abstract
:1. Introduction
2. Results
2.1. Natural Clustering
2.2. Identification of Distinctive Metabolites in Serum of WD Subjects Compared to HC
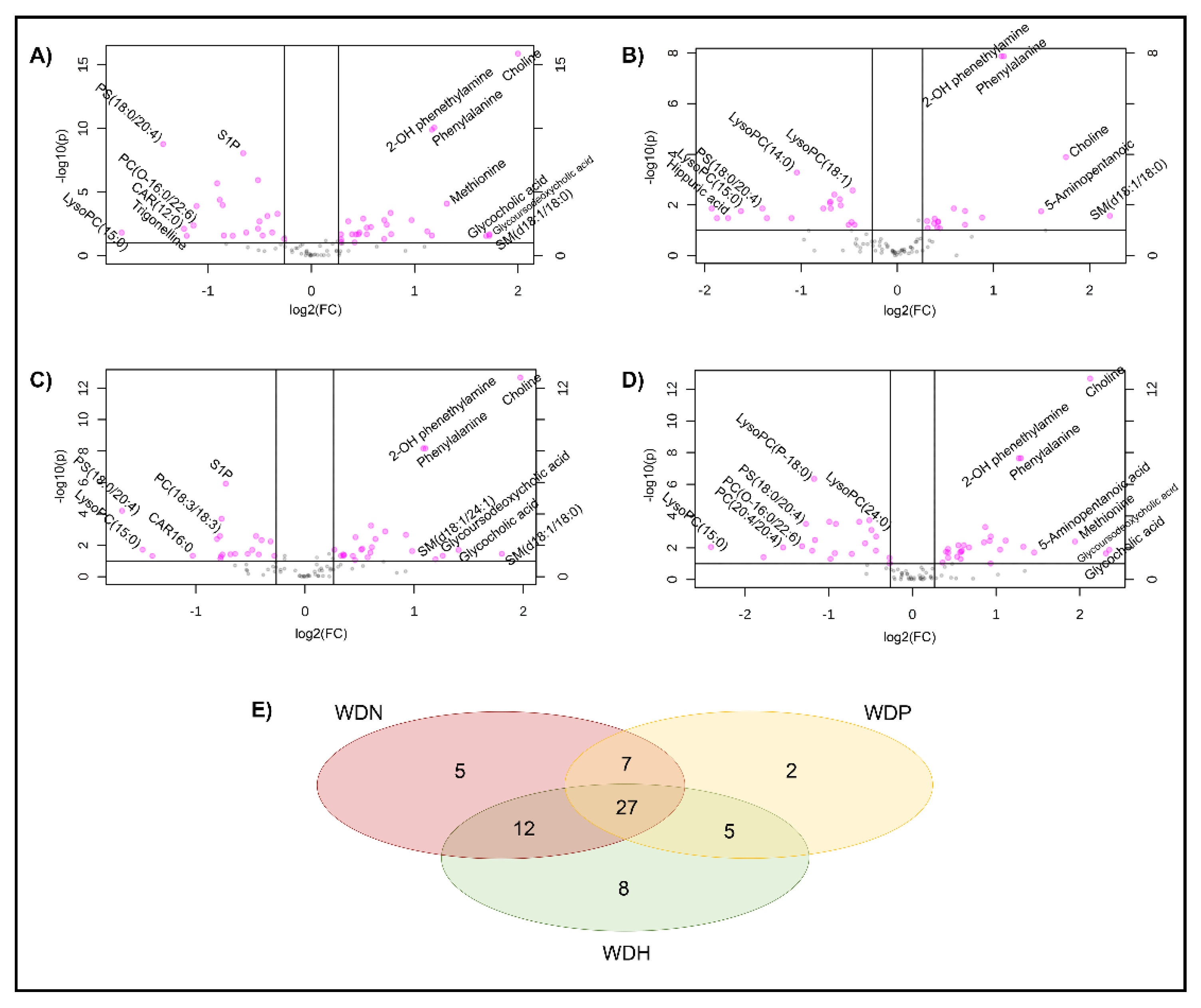
2.3. Identification of Serum Metabolites Distinguishing between WD Phenotypes
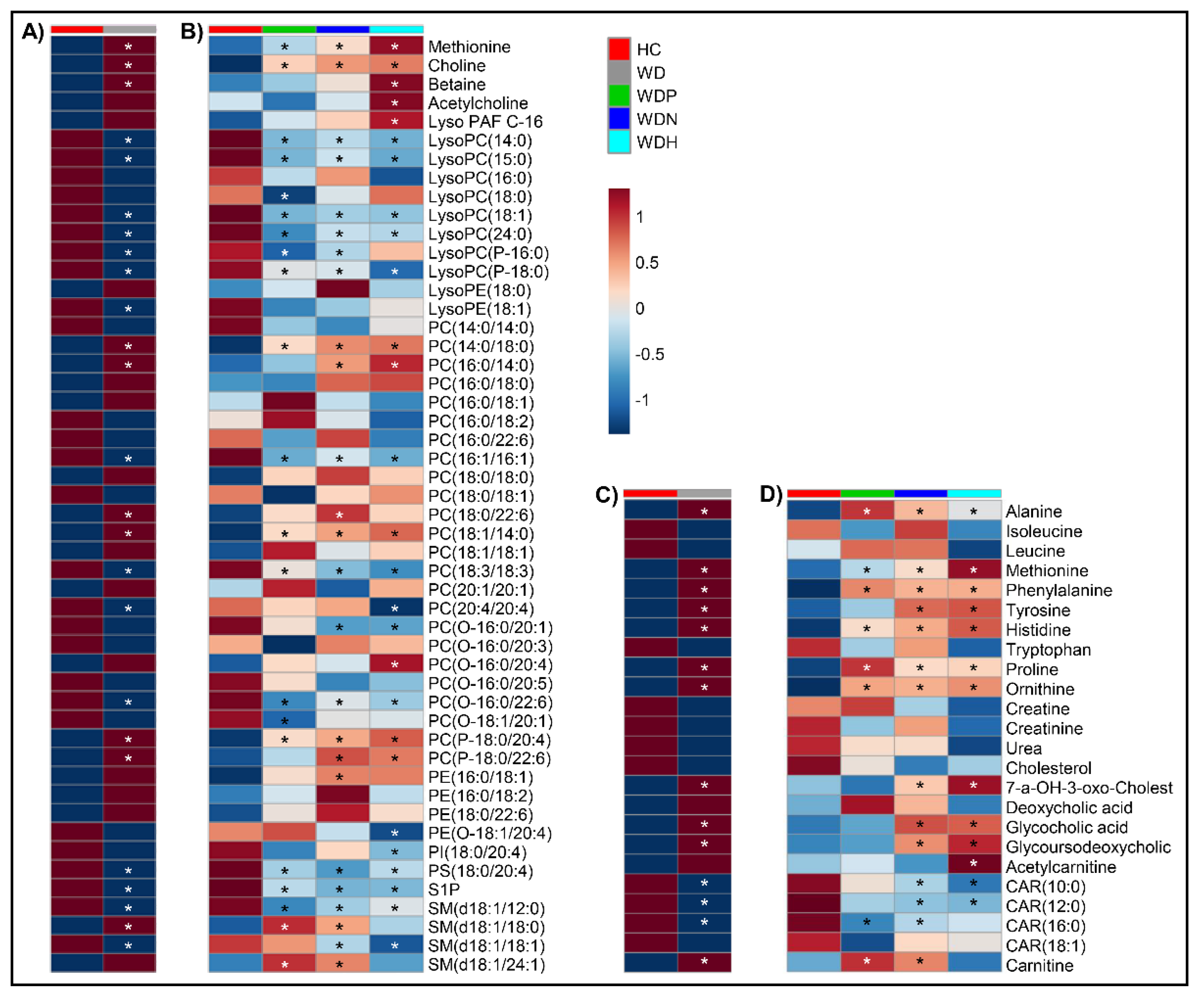
2.4. Choline Is a Hallmark Alteration in Patients with WD
2.5. Patients with WD Present Elevation in Some Amino Acid Levels
2.6. Altered Circulating Phospholipids in WD
2.7. Secondary Bile Acids Are Increased in WD
2.8. Decreased Serum Levels of Long-Chain Acylcarnitines in WD
2.9. No Sex-Specific Differences between WD and HC
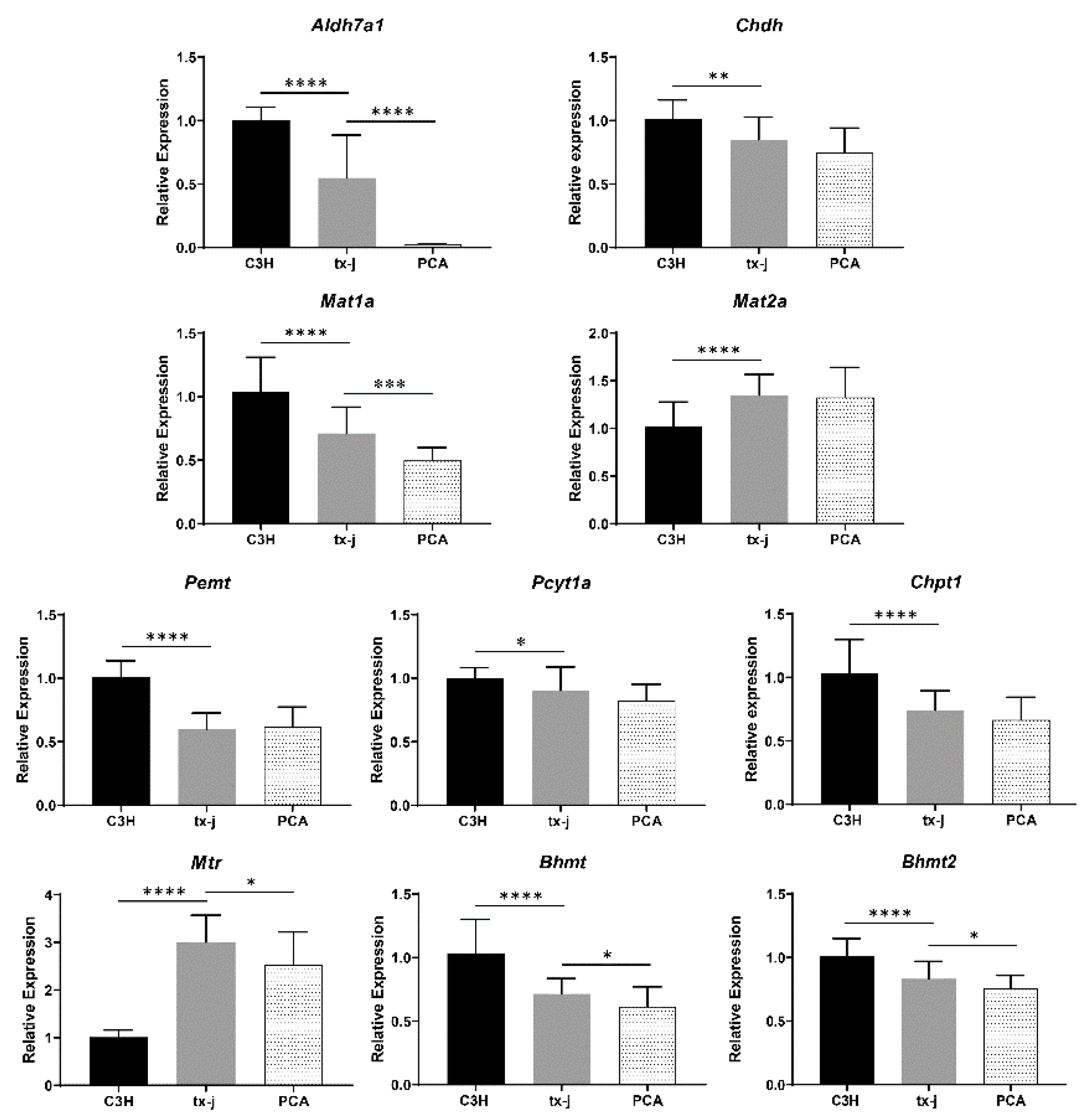
2.10. Gene Expression Analysis in tx-j Mice and Response to Copper Chelation
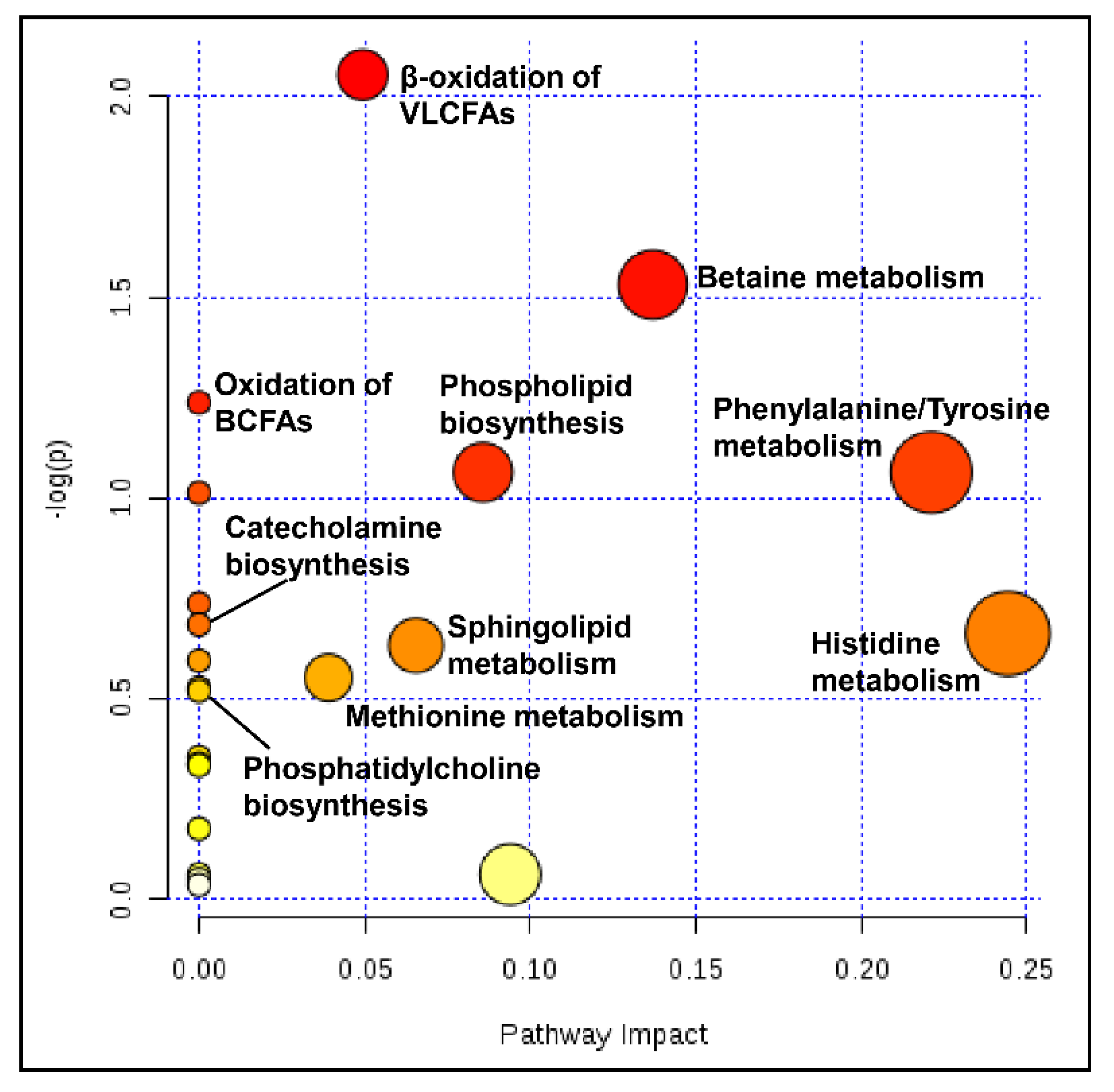
2.11. Pathway Analysis
3. Discussion
4. Methods
4.1. Subject Recruitment and Features
4.2. Metabolomic Analysis
4.3. Mouse Models and Diets
4.4. RNA Isolation and qPCR
4.5. Statistical Analysis
4.6. Metabolite Enrichment and Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aldh7a1 | Aldehyde dehydrogenase family 7, member A1 |
| BHMT | Betaine-homocysteine S-methyltransferase |
| C3H | C3HeB/FeJ |
| CAR | Acylcarnitine |
| CDP-choline | Cytidine-diphosphate-choline |
| Chdh | Choline dehydrogenase |
| Chpt1 | Cholinephosphotransferase 1 |
| FAC | Fatty acyl chain |
| FDR | False discovery rate |
| HC | Healthy control |
| LysoPC | Lysophosphatidylcholine |
| Mat1a | Methionine adenosyltransferase I alpha |
| Mat2a | Methionine adenosyltransferase II, alpha |
| Mtr | Methionine synthase |
| NASH | Non-alcoholic steatohepatitis |
| PC | Phosphatidylcholine |
| PCA | Penicillamine |
| Pcyt1a | Phosphate cytidylyltransferase 1, choline, alpha isoform |
| PEMT | Phosphatidylethanolamine N-methyltransferase |
| PL | Phospholipid |
| PUFA | Polyunsaturated fatty acyl |
| ROS | Reactive oxygen species |
| S1P | Sphingosine-1-phosphate |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosylmethionine |
| SL | Sphingolipid |
| SM | Sphingomyelin |
| SPHK | Sphingosine kinase |
| tx-j | C3He-Atp7btx-J/J |
| VLDL | Very low-density lipoprotein |
| WD | Wilson disease |
| WDH | Wilson disease hepatic |
| WDN | Wilson disease neurologic |
| WDP | Wilson disease pre-clinical |
References
- Riordan, S.M.; Williams, R. The Wilson’s disease gene and phenotypic diversity. J. Hepatol. 2001, 34, 165–171. [Google Scholar] [CrossRef]
- Lutsenko, S. Modifying factors and phenotypic diversity in Wilson’s disease. Ann. New York Acad. Sci. 2014, 1315, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, J.; Pu, C.; Qiao, L.; Jiang, C. Wilson’s disease: A comprehensive review of the molecular mechanisms. Int. J. Mol. Sci. 2015, 16, 6419–6431. [Google Scholar] [CrossRef] [PubMed]
- Boga, S.; Ala, A.; Schilsky, M.L. Hepatic features of Wilson disease. In Handbook of clinical neurology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 142, pp. 91–99. [Google Scholar]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef]
- Sokol, R.J.; Twedt, D.; McKim, J.M., Jr.; Devereaux, M.W.; Karrer, F.M.; Kam, I.; Von Steigman, G.; Narkewicz, M.R.; Bacon, B.R.; Britton, R.S. Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. Gastroenterology 1994, 107, 1788–1798. [Google Scholar] [CrossRef]
- Zischka, H.; Lichtmannegger, J. Pathological mitochondrial copper overload in livers of Wilson’s disease patients and related animal models. Ann. New York Acad. Sci. 2014, 1315, 6–15. [Google Scholar] [CrossRef]
- Park, E.I.; Garrow, T.A. Interaction between dietary methionine and methyl donor intake on rat liver betaine-homocysteine methyltransferase gene expression and organization of the human gene. J. Biol. Chem. 1999, 274, 7816–7824. [Google Scholar] [CrossRef]
- Medici, V.; Shibata, N.M.; Kharbanda, K.K.; LaSalle, J.M.; Woods, R.; Liu, S.; Engelberg, J.A.; Devaraj, S.; Török, N.J.; Jiang, J.X. Wilson’s disease: Changes in methionine metabolism and inflammation affect global DNA methylation in early liver disease. Hepatology 2013, 57, 555–565. [Google Scholar] [CrossRef]
- Delgado, M.; Pérez-Miguelsanz, J.; Garrido, F.; Rodríguez-Tarduchy, G.; Pérez-Sala, D.; Pajares, M.A. Early effects of copper accumulation on methionine metabolism. Cell. Mol. Life Sci. 2008, 65, 2080–2090. [Google Scholar] [CrossRef]
- Le, A.; Shibata, N.M.; French, S.W.; Kim, K.; Kharbanda, K.K.; Islam, M.S.; LaSalle, J.M.; Halsted, C.H.; Keen, C.L.; Medici, V. Characterization of timed changes in hepatic copper concentrations, methionine metabolism, gene expression, and global DNA methylation in the Jackson toxic milk mouse model of Wilson disease. Int. J. Mol. Sci. 2014, 15, 8004–8023. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The Critical Role of Phosphatidylcholine and Phosphatidylethanolamine Metabolism in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef] [PubMed]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into non-alcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 2012, 28, 159. [Google Scholar] [CrossRef] [PubMed]
- Vance, D.E.; Walkey, C.J.; Cui, Z. Phosphatidylethanolamine N-methyltransferase from liver. Biochim. Et Biophys. 1997, 134, 142–150. [Google Scholar] [CrossRef]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef]
- Ma, D.; Arendt, B.; Hillyer, L.; Fung, S.; McGilvray, I.; Guindi, M.; Allard, J. Plasma phospholipids and fatty acid composition differ between liver biopsy-proven nonalcoholic fatty liver disease and healthy subjects. Nutr. Diabetes 2016, 6, e220. [Google Scholar] [CrossRef]
- Arendt, B.M.; Ma, D.W.; Simons, B.; Noureldin, S.A.; Therapondos, G.; Guindi, M.; Sherman, M.; Allard, J.P. Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl. Physiol. Nutr. Metab. 2012, 38, 334–340. [Google Scholar] [CrossRef]
- Whiley, L.; Sen, A.; Heaton, J.; Proitsi, P.; García-Gómez, D.; Leung, R.; Smith, N.; Thambisetty, M.; Kloszewska, I.; Mecocci, P. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 271–278. [Google Scholar] [CrossRef]
- Demirkan, A.; Isaacs, A.; Ugocsai, P.; Liebisch, G.; Struchalin, M.; Rudan, I.; Wilson, J.F.; Pramstaller, P.P.; Gyllensten, U.; Campbell, H. Plasma phosphatidylcholine and sphingomyelin concentrations are associated with depression and anxiety symptoms in a Dutch family-based lipidomics study. J. Psychiatr. Res. 2013, 47, 357–362. [Google Scholar] [CrossRef]
- Cole, L.K.; Vance, J.E.; Vance, D.E. Phosphatidylcholine biosynthesis and lipoprotein metabolism. Biochim. Et Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 2012, 1821, 754–761. [Google Scholar] [CrossRef]
- Slotte, J.P. Biological functions of sphingomyelins. Prog. Lipid Res. 2013, 52, 424–437. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Functions of the multifaceted family of sphingosine kinases and some close relatives. J. Biol. Chem. 2007, 282, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397. [Google Scholar] [CrossRef] [PubMed]
- Lebman, D.A.; Spiegel, S. Thematic review series: Sphingolipids. Cross-talk at the crossroads of sphingosine-1-phosphate, growth factors, and cytokine signaling. J. Lipid Res. 2008, 49, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S. Catecholamines in the periphery. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 1997; Volume 42, pp. 529–539. [Google Scholar]
- Kurian, M.A.; Gissen, P.; Smith, M.; Heales, S.J.; Clayton, P.T. The monoamine neurotransmitter disorders: An expanding range of neurological syndromes. Lancet Neurol. 2011, 10, 721–733. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.; Fitzpatrick, D. (Eds.) The Biogenic Amines. In Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK11035 (accessed on 26 November 2019).
- Parthasarathy, A.; Cross, P.J.; Dobson, R.C.; Adams, L.E.; Savka, M.A.; Hudson, A.O. A three-ring circus: Metabolism of the three proteogenic aromatic amino acids and their role in the health of plants and animals. Front. Mol. Biosci. 2018, 5, 29. [Google Scholar] [CrossRef]
- Snyder, S.H.; Innis, R.B. Peptide neurotransmitters. Annu. Rev. Biochem. 1979, 48, 755–782. [Google Scholar] [CrossRef]
- Papakostas, G.I.; Alpert, J.E.; Fava, M. S-adenosyl-methionine in depression: A comprehensive review of the literature. Curr. Psychiatry Rep. 2003, 5, 460–466. [Google Scholar] [CrossRef]
- Wilmarth, P.A.; Short, K.K.; Fiehn, O.; Lutsenko, S.; David, L.L.; Burkhead, J.L. A systems approach implicates nuclear receptor targeting in the Atp7b−/− mouse model of Wilson’s disease. Metallomics 2012, 4, 660–668. [Google Scholar] [CrossRef]
- Santos, E.M.; Ball, J.S.; Williams, T.D.; Wu, H.; Ortega, F.; Van Aerle, R.; Katsiadaki, I.; Falciani, F.; Viant, M.R.; Chipman, J.K. Identifying health impacts of exposure to copper using transcriptomics and metabolomics in a fish model. Environ. Sci. Technol. 2009, 44, 820–826. [Google Scholar] [CrossRef]
- Huster, D.; Purnat, T.D.; Burkhead, J.L.; Ralle, M.; Fiehn, O.; Stuckert, F.; Olson, N.E.; Teupser, D.; Lutsenko, S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 2007, 282, 8343–8355. [Google Scholar] [CrossRef]
- Sarode, G.V.; Kim, K.; Kieffer, D.A.; Shibata, N.M.; Litwin, T.; Czlonkowska, A.; Medici, V. Metabolomics profiles of patients with Wilson disease reveal a distinct metabolic signature. Metabolomics 2019, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- DeLong, C.J.; Shen, Y.-J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekas, G.; Patton, G.M.; Lincoln, E.C.; Robins, S.J. Origin of plasma lysophosphatidylcholine: Evidence for direct hepatic secretion in the rat. J. Lab. Clin. Med. 1985, 105, 185–189. [Google Scholar]
- Jonas, A. Lecithin cholesterol acyltransferase. Biochim. Et Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 2000, 1529, 245–256. [Google Scholar] [CrossRef]
- Wu, P.; Dong, J.; Cheng, N.; Yang, R.; Han, Y.; Han, Y. Inflammatory cytokines expression in Wilson’s disease. Neurol. Sci. 2019, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Law, S.-H.; Chan, M.-L.; Marathe, G.K.; Parveen, F.; Chen, C.-H.; Ke, L.-Y. An updated review of lysophosphatidylcholine metabolism in human diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, A.B.; Iaciura, B.M.F.; Nohara, L.L.; Lopes, C.D.; Veas, E.M.C.; Mariano, V.S.; Bozza, P.T.; Lopes, U.G.; Atella, G.C.; Almeida, I.C. Lysophosphatidylcholine triggers TLR2-and TLR4-mediated signaling pathways but counteracts LPS-induced NO synthesis in peritoneal macrophages by inhibiting NF-κB translocation and MAPK/ERK phosphorylation. PLoS ONE 2013, 8, e76233. [Google Scholar] [CrossRef] [Green Version]
- Radu, C.G.; Yang, L.V.; Riedinger, M.; Au, M.; Witte, O.N. T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Grzelczyk, A.; Gendaszewska-Darmach, E. Novel bioactive glycerol-based lysophospholipids: New data–new insight into their function. Biochimie 2013, 95, 667–679. [Google Scholar] [CrossRef]
- Hung, N.D.; Sok, D.-E.; Kim, M.R. Prevention of 1-palmitoyl lysophosphatidylcholine-induced inflammation by polyunsaturated acyl lysophosphatidylcholine. Inflamm. Res. 2012, 61, 473–483. [Google Scholar] [CrossRef]
- Hung, N.D.; Kim, M.R.; Sok, D.E. Mechanisms for anti-inflammatory effects of 1-[15 (S)-hydroxyeicosapentaenoyl] lysophosphatidylcholine, administered intraperitoneally, in zymosan A-induced peritonitis. Br. J. Pharmacol. 2011, 162, 1119–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalita, J.; Kumar, V.; Misra, U.K.; Ranjan, A.; Khan, H.; Konwar, R. A study of oxidative stress, cytokines and glutamate in Wilson disease and their asymptomatic siblings. J. Neuroimmunol. 2014, 274, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.N.; Risis, S.; Yang, C.; Meikle, P.J.; Staples, M.; Febbraio, M.A.; Bruce, C.R. Plasma lysophosphatidylcholine levels are reduced in obesity and type 2 diabetes. PLoS ONE 2012, 7, e41456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimerl, S.; Fischer, M.; Baessler, A.; Liebisch, G.; Sigruener, A.; Wallner, S.; Schmitz, G. Alterations of plasma lysophosphatidylcholine species in obesity and weight loss. PLoS ONE 2014, 9, e111348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, J.; Liu, Y.; Wu, R.; Yang, H.; Hu, X.; Ling, X. Studies on diagnostic biomarkers and therapeutic mechanism of Alzheimer’s disease through metabolomics and hippocampal proteomics. Eur. J. Pharm. Sci. 2017, 105, 119–126. [Google Scholar] [CrossRef]
- Drobnik, W.; Liebisch, G.; Audebert, F.-X.; Fröhlich, D.; Glück, T.; Vogel, P.; Rothe, G.; Schmitz, G. Plasma ceramide and lysophosphatidylcholine inversely correlate with mortality in sepsis patients. J. Lipid Res. 2003, 44, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Jacobs, R.L.; Watts, J.L.; Rottiers, V.; Jiang, K.; Finnegan, D.M.; Shioda, T.; Hansen, M.; Yang, F.; Niebergall, L.J. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011, 147, 840–852. [Google Scholar] [CrossRef] [Green Version]
- Pyszko, J.; Strosznajder, J.B. Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.-X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Katsel, P.; Li, C.; Haroutunian, V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: A shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Bandaru, V.V.; Bae, M.; Mattson, M.P. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim. Et Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 2010, 1801, 878–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grin’kina, N.M.; Karnabi, E.E.; Damania, D.; Wadgaonkar, S.; Muslimov, I.A.; Wadgaonkar, R. Sphingosine kinase 1 deficiency exacerbates LPS-induced neuroinflammation. PLoS ONE 2012, 7, e36475. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Seppänen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepää, A.L.; Yki-Järvinen, H.; Orešič, M. Comparison of lipid and fatty acid composition of the liver, subcutaneous and intra-abdominal adipose tissue, and serum. Obesity 2010, 18, 937–944. [Google Scholar] [CrossRef]
- Kolak, M.; Westerbacka, J.; Velagapudi, V.R.; Wågsäter, D.; Yetukuri, L.; Makkonen, J.; Rissanen, A.; Häkkinen, A.-M.; Lindell, M.; Bergholm, R. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes 2007, 56, 1960–1968. [Google Scholar] [CrossRef] [Green Version]
- Olivera, A.; Spiegel, S. Sphingosine kinase: A mediator of vital cellular functions. Prostaglandins Other Lipid Mediat. 2001, 64, 123–134. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120. [Google Scholar] [CrossRef]
- Kalita, J.; Kumar, V.; Misra, U. A study on apoptosis and anti-apoptotic status in Wilson disease. Mol. Neurobiol. 2016, 53, 6659–6667. [Google Scholar] [CrossRef]
- Strand, S.; Hofmann, W.J.; Grambihler, A.; Hug, H.; Volkmann, M.; Otto, G.; Wesch, H.; Mariani, S.M.; Hack, V.; Stremmel, W. Hepatic failure and liver cell damage in acute Wilson’s disease involve CD95 (APO-1/Fas) mediated apoptosis. Nat. Med. 1998, 4, 588. [Google Scholar] [CrossRef]
- Barth, B.M.; Gustafson, S.J.; Kuhn, T.B. Neutral sphingomyelinase activation precedes NADPH oxidase-dependent damage in neurons exposed to the proinflammatory cytokine tumor necrosis factor-α. J. Neurosci. Res. 2012, 90, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.-H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress–dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A–associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, S.A. Betaine in human nutrition. Am. J. Clin. Nutr. 2004, 80, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, T.V.; Dong, X.; Sawyer, G.J.; Caldwell, A.; Halket, J.; Sherwood, R.; Quaglia, A.; Dew, T.; Anilkumar, N.; Burr, S. NADPH oxidase 4 regulates homocysteine metabolism and protects against acetaminophen-induced liver damage in mice. Free Radic. Biol. Med. 2015, 89, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Mudd, S.H.; Cantoni, G. Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers’ yeast. J. Biol. Chem. 1958, 231, 481–492. [Google Scholar]
- Mato, J.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C.; Gukovsky, I.; Lugea, A.; Reyes, C.N.; Huang, Z.-Z.; Chen, L.; Mato, J.M.; Bottiglieri, T.; Pandol, S.J. Role of S-adenosylmethionine in two experimental models of pancreatitis. FASEB J. 2003, 17, 56–58. [Google Scholar] [CrossRef] [Green Version]
- Shimizu-Saito, K.; Horikawa, S.; Kojima, N.; Shiga, J.; Senoo, H.; Tsukada, K. Differential expression of S-adenosylmethionine synthetase isozymes in different cell types of rat liver. Hepatology 1997, 26, 424–431. [Google Scholar] [CrossRef]
- Ubagai, T.; Lei, K.-J.; Huang, S.; Mudd, S.H.; Levy, H.L.; Chou, J.Y. Molecular mechanisms of an inborn error of methionine pathway. Methionine adenosyltransferase deficiency. J. Clin. Investig. 1995, 96, 1943–1947. [Google Scholar] [CrossRef] [Green Version]
- Schnackenberg, L.K.; Chen, M.; Sun, J.; Holland, R.D.; Dragan, Y.; Tong, W.; Welsh, W.; Beger, R.D. Evaluations of the trans-sulfuration pathway in multiple liver toxicity studies. Toxicol. Appl. Pharmacol. 2009, 235, 25–32. [Google Scholar] [CrossRef]
- Shimizu, K.; Abe, M.; Yokoyama, S.; Takahashi, H.; Sawada, N.; Mori, M.; Tsukada, K. Decreased activities of S-adenosylmethionine synthetase isozymes in hereditary hepatitis in Long-Evans rats. Life Sci. 1990, 46, 1837–1842. [Google Scholar] [CrossRef]
- Okada, G.; Teraoka, H.; Tsukada, K. Multiple species of mammalian S-adenosylmethionine synthetase. Partial purification and characterization. Biochemistry 1981, 20, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.M.; Hoffman, J.L. Fractionation and kinetic properties of rat liver and kidney methionine adenosyltransferase isozymes. Biochemistry 1983, 22, 1636–1641. [Google Scholar] [CrossRef]
- Gromadzka, G.; Rudnicka, M.; Chabik, G.; Przybyłkowski, A.; Członkowska, A. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson’s disease. J. Hepatol. 2011, 55, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Chen, Z.; Rozen, R.; Matthews, R.G. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc. Natl. Acad. Sci. USA 2001, 98, 14853–14858. [Google Scholar] [CrossRef] [Green Version]
- Böttiger, A.K.; Hurtig-Wennlöf, A.; Sjöström, M.; Yngve, A.; Nilsson, T.K. Association of total plasma homocysteine with methylenetetrahydrofolate reductase genotypes 677C> T, 1298A> C, and 1793G> A and the corresponding haplotypes in Swedish children and adolescents. Int. J. Mol. Med. 2007, 19, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Mato, J.M.; Martínez-Chantar, M.L.; Lu, S.C. S-adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2015, 12, 183–189. [Google Scholar] [CrossRef]
- Robert, K.; Nehmé, J.; Bourdon, E.; Pivert, G.; Friguet, B.; Delcayre, C.; Delabar, J.M.; Janel, N. Cystathionine β synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterology 2005, 128, 1405–1415. [Google Scholar] [CrossRef] [Green Version]
- Ereño-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martínez-Cruz, L.A. Structural insight into the molecular mechanism of allosteric activation of human cystathionine β-synthase by S-adenosylmethionine. Proc. Natl. Acad. Sci. USA 2014, 111, E3845–E3852. [Google Scholar] [CrossRef] [Green Version]
- Bethin, K.E.; Cimato, T.R.; Ettinger, M.J. Copper binding to mouse liver S-adenosylhomocysteine hydrolase and the effects of copper on its levels. J. Biol. Chem. 1995, 270, 20703–20711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethin, K.E.; Petrovic, N.; Ettinger, M.J. Identification of a major hepatic copper binding protein as S-adenosylhomocysteine hydrolase. J. Biol. Chem. 1995, 270, 20698–20702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, Y.; Chen, J.; Wei, W.; Pan, X.; Liu, J.; Liu, Q.; Leu, W.; Zhang, L.; Yang, X. Copper ions inhibit S-adenosylhomocysteine hydrolase by causing dissociation of NAD+ cofactor. Biochemistry 2007, 46, 11451–11458. [Google Scholar] [CrossRef] [PubMed]
- Fuenzalida, M.; Arias, H.R. Role of nicotinic and muscarinic receptors on synaptic plasticity and neurological diseases. Curr. Pharm. Des. 2016, 22, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.L.; Sigala, B.; Soeda, J.; Cordero, P.; Nguyen, V.; McKee, C.; Mouraliderane, A.; Vinciguerra, M.; Oben, J.A. Acetylcholine induces fibrogenic effects via M2/M3 acetylcholine receptors in non-alcoholic steatohepatitis and in primary human hepatic stellate cells. J. Gastroenterol. Hepatol. 2016, 31, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, H.; Terada, T. Mast cell, myofibroblast and nerve terminal complexes in carbon tetrachloride-induced cirrhotic rat livers. J. Hepatol. 1998, 29, 112–119. [Google Scholar] [CrossRef]
- Nishio, T.; Taura, K.; Iwaisako, K.; Koyama, Y.; Tanabe, K.; Yamamoto, G.; Okuda, Y.; Ikeno, Y.; Yoshino, K.; Kasai, Y. Hepatic vagus nerve regulates Kupffer cell activation via α7 nicotinic acetylcholine receptor in nonalcoholic steatohepatitis. J. Gastroenterol. 2017, 52, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Fernstrom, J.D.; Fernstrom, M.H. Tyrosine, phenylalanine, and catecholamine synthesis and function in the brain. J. Nutr. 2007, 137, 1539S–1547S. [Google Scholar] [CrossRef]
- Ceberio, L.; Hermida, Á.; Venegas, E.; Arrieta, F.; Morales, M.; Forga, M.; Gonzalo, M. Phenylketonuria in the adult patient. Expert Opin. Orphan Drugs 2019, in press. [Google Scholar] [CrossRef]
- Bilder, D.A.; Kobori, J.A.; Cohen-Pfeffer, J.L.; Johnson, E.M.; Jurecki, E.R.; Grant, M.L. Neuropsychiatric comorbidities in adults with phenylketonuria: A retrospective cohort study. Mol. Genet. Metab. 2017, 121, 1–8. [Google Scholar] [CrossRef]
- Russo, P.A.; Mitchell, G.A.; Tanguay, R.M. Tyrosinemia: A review. Pediatric Dev. Pathol. 2001, 4, 212–221. [Google Scholar] [CrossRef] [PubMed]
- van Ginkel, W.G.; Jahja, R.; Huijbregts, S.C.; van Spronsen, F.J. Neurological and neuropsychological problems in tyrosinemia type I patients. In Hereditary Tyrosinemia; Springer: Berlin/Heidelberg, Germany, 2017; pp. 111–122. [Google Scholar]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the nervous system. Physiol. Rev. 2008, 88, 1183–1241. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, M.E.; MacMillan, L.; Stevens, J.R.; Brosnan, J.T. Division of labour: How does folate metabolism partition between one-carbon metabolism and amino acid oxidation? Biochem. J. 2015, 472, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyberg, P.; Gottfries, C.G.; Holmgren, G.; Persson, S.; Roos, B.E.; Winblad, B. Advanced catecholaminergic disturbances in the brain in a case of Wilson’s disease. Acta Neurol. Scand. 1982, 65, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Iso, H.; Kitanaka, N.; Kitanaka, J.; Eguchi, H.; Ookawara, T.; Ozawa, K.; Shimoda, S.; Yoshihara, D.; Takemura, M. Effects of copper metabolism on neurological functions in Wistar and Wilson’s disease model rats. Biochem. Biophys. Res. Commun. 2006, 349, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Brůha, R.; Dušek, P. Pathogenesis of Wilson disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 142, pp. 43–55. [Google Scholar]
- Barthel, H.; Hermann, W.; Kluge, R.; Hesse, S.; Collingridge, D.R.; Wagner, A.; Sabri, O. Concordant pre-and postsynaptic deficits of dopaminergic neurotransmission in neurologic Wilson disease. Am. J. Neuroradiol. 2003, 24, 234–238. [Google Scholar]
- Snow, B.J.; Bhatt, M.; Martin, W.; Li, D.; Calne, D.B. The nigrostriatal dopaminergic pathway in Wilson’s disease studied with positron emission tomography. J. Neurol. Neurosurg. Psychiatry 1991, 54, 12–17. [Google Scholar] [CrossRef]
- Westermark, K.; Tedroff, J.; Thuomas, K.Å.; Hartvig, P.; Långström, B.; Andersson, Y.; Hörnfeldt, K.; Aquilonius, S.M. Neurological Wilson’s disease studied with magnetic resonance imaging and with positron emission tomography using dopaminergic markers. Mov. Disord. Off. J. Mov. Disord. Soc. 1995, 10, 596–603. [Google Scholar] [CrossRef]
- Schlaug, G.; Hefter, H.; Engelbrecht, V.; Kuwert, T.; Arnold, S.; Stöcklin, G.; Seitz, R.J. Neurological impairment and recovery in Wilson’s disease: Evidence from PET and MRI. J. Neurol. Sci. 1996, 136, 129–139. [Google Scholar] [CrossRef]
- Oder, W.; Brücke, T.; Kollegger, H.; Spatt, J.; Asenbaum, S.; Deecke, L. Dopamine D2 receptor binding is reduced in Wilson’s disease: Correlation of neurological deficits with striatal 123 I-Iodobenzamide binding. J. Neural Transm. 1996, 103, 1093–1103. [Google Scholar] [CrossRef]
- Zlomuzica, A.; Dere, D.; Binder, S.; Silva, M.A.D.S.; Huston, J.P.; Dere, E. Neuronal histamine and cognitive symptoms in Alzheimer’s disease. Neuropharmacology 2016, 106, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, K.; Ito, C.; Tashiro, M.; Kato, M.; Kano, M.; Itoh, M.; Iwata, R.; Matsuoka, H.; Sato, M.; Yanai, K. Histamine H1 receptors in schizophrenic patients measured by positron emission tomography. Eur. Neuropsychopharmacol. 2005, 15, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Fukudo, S.; Tashiro, A.; Utsumi, A.; Tamura, D.; Itoh, M.; Iwata, R.; Tashiro, M.; Mochizuki, H.; Funaki, Y. Decreased histamine H1 receptor binding in the brain of depressed patients. Eur. J. Neurosci. 2004, 20, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Rimoin, D.L.; Pyeritz, R.E.; Korf, B. Emery and Rimoin’s Essential Medical Genetics; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Baumgartner, M.R.; Valle, D.; Dionisi-Vici, C. Disorders of Ornithine and Proline Metabolism. In Inborn Metabolic Diseases; Springer: Berlin/Heidelberg, Germany, 2016; pp. 321–331. [Google Scholar]
- Seessle, J.; Gohdes, A.; Gotthardt, D.N.; Pfeiffenberger, J.; Eckert, N.; Stremmel, W.; Reuner, U.; Weiss, K.H. Alterations of lipid metabolism in Wilson disease. Lipids Health Dis. 2011, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Farina, A.; Dumonceau, J.-M.; Lescuyer, P. Proteomic analysis of human bile and potential applications for cancer diagnosis. Expert Rev. Proteom. 2009, 6, 285–301. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332. [Google Scholar] [CrossRef] [Green Version]
- Kerner, J.; Hoppel, C. Fatty acid import into mitochondria. Biochim. Et Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 2000, 1486, 1–17. [Google Scholar] [CrossRef]
- Violante, S.; IJlst, L.; te Brinke, H.; Koster, J.; de Almeida, I.T.; Wanders, R.J.; Ventura, F.V.; Houten, S.M. Peroxisomes contribute to the acylcarnitine production when the carnitine shuttle is deficient. Biochim. Et Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 2013, 1831, 1467–1474. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, C.; Chen, L.; Han, X.; Ji, L. Human serum acylcarnitine profiles in different glucose tolerance states. Diabetes Res. Clin. Pract. 2014, 104, 376–382. [Google Scholar] [CrossRef]
- Adams, S.H.; Hoppel, C.L.; Lok, K.H.; Zhao, L.; Wong, S.W.; Minkler, P.E.; Hwang, D.H.; Newman, J.W.; Garvey, W.T. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid β-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 2009, 139, 1073–1081. [Google Scholar] [CrossRef]
- Ramos-Roman, M.A.; Sweetman, L.; Valdez, M.J.; Parks, E.J. Postprandial changes in plasma acylcarnitine concentrations as markers of fatty acid flux in overweight and obesity. Metabolism 2012, 61, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Rinaldo, P.; Cowan, T.M.; Matern, D. Acylcarnitine profile analysis. Genet. Med. 2008, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Schooneman, M.G.; Achterkamp, N.; Argmann, C.A.; Soeters, M.R.; Houten, S.M. Plasma acylcarnitines inadequately reflect tissue acylcarnitine metabolism. Biochim. Et Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 2014, 1841, 987–994. [Google Scholar] [CrossRef]
- Soeters, M.R.; Sauerwein, H.P.; Duran, M.; Wanders, R.J.; Ackermans, M.T.; Fliers, E.; Houten, S.M.; Serlie, M.J. Muscle acylcarnitines during short-term fasting in lean healthy men. Clin. Sci. 2009, 116, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Stuehr, D.J. Enzymes of the L-arginine to nitric oxide pathway. J. Nutr. 2004, 134, 2748S–2751S. [Google Scholar] [CrossRef]
- Racké, K.; Warnken, M. L-arginine metabolic pathways. Open Nitric Oxide J. 2010, 2, 9–19. [Google Scholar] [CrossRef]
- Ferenci, P.; Caca, K.; Loudianos, G.; Mieli-Vergani, G.; Tanner, S.; Sternlieb, I.; Schilsky, M.; Cox, D.; Berr, F. Diagnosis and phenotypic classification of Wilson disease 1. Liver Int. 2003, 23, 139–142. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523. [Google Scholar] [CrossRef]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743. [Google Scholar] [CrossRef]
- Aittokallio, T.; Schwikowski, B. Graph-based methods for analysing networks in cell biology. Brief. Bioinform. 2006, 7, 243–255. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazi, T.A.; Sarode, G.V.; Czlonkowska, A.; Litwin, T.; Kim, K.; Shibata, N.M.; Medici, V. Dysregulated Choline, Methionine, and Aromatic Amino Acid Metabolism in Patients with Wilson Disease: Exploratory Metabolomic Profiling and Implications for Hepatic and Neurologic Phenotypes. Int. J. Mol. Sci. 2019, 20, 5937. https://doi.org/10.3390/ijms20235937
Mazi TA, Sarode GV, Czlonkowska A, Litwin T, Kim K, Shibata NM, Medici V. Dysregulated Choline, Methionine, and Aromatic Amino Acid Metabolism in Patients with Wilson Disease: Exploratory Metabolomic Profiling and Implications for Hepatic and Neurologic Phenotypes. International Journal of Molecular Sciences. 2019; 20(23):5937. https://doi.org/10.3390/ijms20235937
Chicago/Turabian StyleMazi, Tagreed A., Gaurav V. Sarode, Anna Czlonkowska, Tomasz Litwin, Kyoungmi Kim, Noreene M. Shibata, and Valentina Medici. 2019. "Dysregulated Choline, Methionine, and Aromatic Amino Acid Metabolism in Patients with Wilson Disease: Exploratory Metabolomic Profiling and Implications for Hepatic and Neurologic Phenotypes" International Journal of Molecular Sciences 20, no. 23: 5937. https://doi.org/10.3390/ijms20235937