Altered Genome-Wide DNA Methylation in Peripheral Blood of South African Women with Gestational Diabetes Mellitus


Abstract
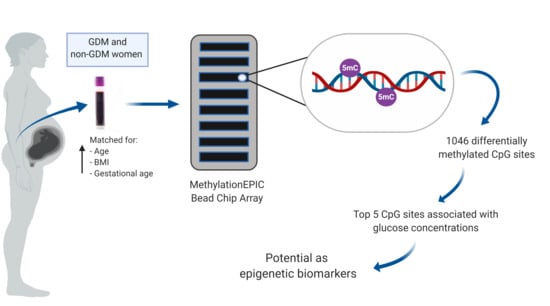
:
1. Introduction
2. Results
2.1. Study Participants
2.2. Genome-Wide DNA Methylation Profiling
2.3. Functional Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Study Participants
4.2. DNA Extraction
4.3. Genome-Wide DNA Methylation Profiling
4.4. Processing and Analysis of the Human Methylation EPIC Bead Chip Array
4.5. Functional Enrichment Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GDM | Gestational diabetes mellitus |
| CAMTA1 | Calmodulin binding transcription activator 1 |
| MAPK | Mitogen activated protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| T2D | Type 2 diabetes |
| CpG | Cytosine-phosphate-guanine |
| OGTT | Oral glucose tolerance test |
| HIV | Human immunodeficiency virus |
| BMI | Body mass index |
| HOMA | Homeostatic model of assessment |
| CRP | c-Reactive protein |
| HbA1c | Glycated hemoglobin |
| PCA | Principal component analysis |
| FDR | False discovery rate |
| UTR | Untranslated regions |
| CDS | Coding domain sequences |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| NRG1 | Neuregulin 1 |
| SNIP1 | smad Nuclear Interacting Protein 1 |
| PPFIBP2 | Protein-tyrosine phosphatase, receptor-type, f polypeptide-interacting protein-binding protein 2 |
| SWAP70 | Switching b cell complex subunit swap70 |
| SEMA6D | Semiphorin 6d |
| CDH8 | Cadherin 8 |
| WNT6 | Wnt family member 6 |
| RFTN1 | Raftlin, lipid raft linker 1 |
| UNC5C | Unc-5 netrin receptor c |
| NUDT6 | Nucleoside diphosphate-linked moiety x motif 6 |
| STOX2 | Storkhead box |
| MSH5 | Muts protein homolog 5 |
| KHDRBS2 | KH RNA binding domain containing, signal transduction associated 2 |
| NRG1 | Neuregulin 1 |
| SLC9A3 | Solute carrier family 9 member a3 |
| MEA1 | Male-enhanced antigen 1 |
| KLHDC3 | Kelch domain-containing protein 3 |
| RASA3 | RAS p21 protein activator 3 |
| CYP26B1 | Cytochrome p450 family 26 subfamily b member 1 |
| IADPSG | International association of diabetes in pregnancy study group |
| WHO | World Health Organisation |
| HAPO | Hyperglycemia and adverse pregnancy outcomes |
| T1D | Type 1 diabetes |
| EDTA | Ethylenediaminetetraacetic acid |
| NOOB | Normal-exponential out-of-band |
| SEM | Standard error of the mean |
| ANOVA | One-way analysis of variance |
References
- International Diabetes Federation IDF diabetes atlas—Across the globe. Available online: http://diabetesatlas.org/across-the-globe.html (accessed on 6 July 2018).
- Jiwani, A.; Marseille, E.; Lohse, N.; Damm, P.; Hod, M.; Kahn, J.G. Gestational diabetes mellitus: Results from a survey of country prevalence and practices. J. Matern. Fetal. Neonatal. Med. 2012, 25, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Hadar, E.; Hod, M. Maternal complications of GDM; Diapedia: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Moore, L.E. Fetal and neonatal consequences of maternal diabetes. In Diabetes in Pregnancy: The Complete Guide to Management; Moore, L.E., Ed.; Springer International Publishing: New York, NY, USA, 2018; pp. 7–16. [Google Scholar]
- Hod, M.; Merlob, P.; Friedman, S.; Schoenfeld, A.; Ovadia, J. Gestational Diabetes Mellitus: A Survey of Perinatal Complications in the 1980s. Diabetes 1991, 40, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Newton, K.M.; Knopp, R.H. Gestational diabetes and the incidence of type 2 diabetes: A systematic review. Diabetes Care 2002, 25, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.D.; Mathiesen, E.R.; Hansen, T.; Pedersen, O.; Jensen, D.M.; Lauenborg, J.; Damm, P. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes: The role of intrauterine hyperglycemia. Diabetes Care 2008, 31, 340–346. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, E.; Qiao, Y.; Katzmarzyk, P.T.; Chaput, J.-P.; Fogelholm, M.; Johnson, W.D.; Kuriyan, R.; Kurpad, A.; Lambert, E.V.; et al. Maternal gestational diabetes and childhood obesity at age 9–11: Results of a multinational study. Diabetologia 2016, 59, 2339–2348. [Google Scholar] [CrossRef]
- Damm, P.; Houshmand-Oeregaard, A.; Kelstrup, L.; Lauenborg, J.; Mathiesen, E.R.; Clausen, T.D. Gestational diabetes mellitus and long-term consequences for mother and offspring: A view from Denmark. Diabetologia 2016, 59, 1396–1399. [Google Scholar] [CrossRef]
- Smith, C.J.; Ryckman, K.K. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab. Syndr. Obes. 2015, 8, 295–302. [Google Scholar]
- Lim, D.H.K.; Maher, E.R. DNA methylation: A form of epigenetic control of gene expression. Obstet. Gynaecol. 2010, 12, 37–42. [Google Scholar] [CrossRef]
- Reichetzeder, C.; Dwi Putra, S.E.; Pfab, T.; Slowinski, T.; Neuber, C.; Kleuser, B.; Hocher, B. Increased global placental DNA methylation levels are associated with gestational diabetes. Clin. Epigenetics 2016, 8, 82. [Google Scholar] [CrossRef]
- El Hajj, N.; Pliushch, G.; Schneider, E.; Dittrich, M.; Müller, T.; Korenkov, M.; Aretz, M.; Zechner, U.; Lehnen, H.; Haaf, T. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes 2013, 62, 1320–1328. [Google Scholar] [CrossRef]
- Willmer, T.; Johnson, R.; Louw, J.; Pheiffer, C. Blood-based DNA methylation biomarkers for type 2 diabetes: Potential for clinical applications. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lee, C.-N.; Li, H.-Y.; Hsu, K.-H.; Lin, S.-Y. Genome-wide DNA methylation variation in maternal and cord blood of gestational diabetes population. Diabetes Res. Clin. Pract. 2017, 132, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Enquobahrie, D.A.; Moore, A.; Muhie, S.; Tadesse, M.G.; Lin, S.; Williams, M.A. Early Pregnancy Maternal Blood DNA Methylation in Repeat Pregnancies and Change in Gestational Diabetes Mellitus Status—A Pilot Study. Reprod. Sci. 2015, 22, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Farrell, W.E.; Haworth, K.E.; Emes, R.D.; Kitchen, M.O.; Glossop, J.R.; Hanna, F.W.; Fryer, A.A. Maternal genome-wide DNA methylation profiling in gestational diabetes shows distinctive disease-associated changes relative to matched healthy pregnancies. Epigenetics 2018, 13, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Haertle, L.; El Hajj, N.; Dittrich, M.; Müller, T.; Nanda, I.; Lehnen, H.; Haaf, T. Epigenetic signatures of gestational diabetes mellitus on cord blood methylation. Clin. Epigenetics 2017, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.; Adam, S.; Wyk, N.V.; Rheeder, P.; Louw, J.; Pheiffer, C. Global DNA methylation profiling in peripheral blood cells of South African women with gestational diabetes mellitus. Biomarkers 2019, 24, 225–231. [Google Scholar] [CrossRef]
- Moen, G.-H.; Sommer, C.; Prasad, R.B.; Sletner, L.; Groop, L.; Qvigstad, E.; Birkeland, K.I. Mechanisms in endocrinology: Epigenetic modifications and gestational diabetes: A systematic review of published literature. Eur. J. Endocrinol. 2017, 176, 247–267. [Google Scholar] [CrossRef]
- Nakabayashi, K. Illumina HumanMethylation BeadChip for Genome-Wide DNA Methylation Profiling: Advantages and Limitations. In Handbook of Nutrition, Diet, and Epigenetics; Patel, V., Preedy, V., Eds.; Springer International Publishing: New York, NY, USA, 2017; pp. 1–15. [Google Scholar]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef]
- McCartney, D.L.; Walker, R.M.; Morris, S.W.; McIntosh, A.M.; Porteous, D.J.; Evans, K.L. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data 2016, 9, 22–24. [Google Scholar] [CrossRef]
- Ranchod, H.A.; Vaughan, J.E.; Jarvis, P. Incidence of gestational diabetes at Northdale Hospital, Pietermaritzburg. S. Afr. Med. J. 1991, 80, 14–16. [Google Scholar]
- Adam, S.; Rheeder, P. Screening for gestational diabetes mellitus in a South African population: Prevalence, comparison of diagnostic criteria and the role of risk factors. South Afr. Med. J. 2017, 107, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, L.; Ling, C. The potential use of DNA methylation biomarkers to identify risk and progression of type 2 diabetes. Front. Endocrinol. 2015, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Mollet, I.G.; Malm, H.A.; Wendt, A.; Orho-Melander, M.; Eliasson, L. Integrator of stress responses Calmodulin Binding Transcription Activator 1 (Camta1) regulates miR-212/miR-132 expression and insulin secretion. J. Biol. Chem. 2016, 291, 18440–18452. [Google Scholar] [CrossRef]
- Soriano-Tárraga, C.; Jiménez-Conde, J.; Giralt-Steinhauer, E.; Ois, Á.; Rodríguez-Campello, A.; Cuadrado-Godia, E.; Fernández-Cadenas, I.; Montaner, J.; Lucas, G.; Elosua, R.; et al. DNA isolation method is a source of global DNA methylation variability measured with LUMA. Experimental analysis and a systematic review. PLoS ONE 2013, 8, e60750. [Google Scholar] [CrossRef]
- Hjort, L.; Martino, D.; Grunnet, L.G.; Naeem, H.; Maksimovic, J.; Olsson, A.H.; Zhang, C.; Ling, C.; Olsen, S.F.; Saffery, R.; et al. Gestational diabetes and maternal obesity are associated with epigenome-wide methylation changes in children. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Huang, R.C.; Garratt, E.S.; Pan, H.; Wu, Y.; Davis, E.A.; Barton, S.J.; Burdge, G.C.; Godfrey, K.M.; Holbrook, J.D.; Lillycrop, K.A. Genome-wide methylation analysis identifies differentially methylated CpG loci associated with severe obesity in childhood. Epigenetics 2015, 10, 995–1005. [Google Scholar] [CrossRef]
- Wang, X.; Yang, T.; Miao, J.; Liu, H.; Wu, K.; Guo, J.; Chen, J.; Li, T. Correlation between maternal and fetal insulin resistance in pregnant women with gestational diabetes mellitus. Clin. Lab. 2018, 64, 945–953. [Google Scholar] [CrossRef]
- Kang, J.; Lee, C.-N.; Li, H.-Y.; Hsu, K.-H.; Wang, S.-H.; Lin, S.-Y. Association of interleukin-10 methylation levels with gestational diabetes in a Taiwanese population. Front. Genet. 2018, 9, 222. [Google Scholar] [CrossRef]
- Matsha, T.E.; Pheiffer, C.; Mutize, T.; Erasmus, R.T.; Kengne, A.P. Glucose Tolerance, MTHFR C677T and NOS3 G894T Polymorphisms, and Global DNA Methylation in Mixed Ancestry African Individuals. J. Diabetes Res. 2016, 2016, 8738072. [Google Scholar] [CrossRef]
- Simar, D.; Versteyhe, S.; Donkin, I.; Liu, J.; Hesson, L.; Nylander, V.; Fossum, A.; Barrès, R. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metab. Clin. Exp. 2014, 63, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- WHO. Diagnostic Criteria and Classification of Hyperglycaemia First Detected in Pregnancy; WHO Guidelines Approved by the Guidelines Review Committee; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- HAPO Study Cooperative Research Group; Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [PubMed]
- Alwan, N.; Tuffnell, D.J.; West, J. Treatments for gestational diabetes. Cochrane Database Syst. Rev. 2009, 8, CD003395. [Google Scholar] [CrossRef] [PubMed]
- Świderska, E.; Strycharz, J.; Wróblewski, A.; Szemraj, J.; Drzewoski, J.; Śliwińska, A. Role of PI3K/AKT Pathway in Insulin-Mediated Glucose Uptake. In Glucose Transport; IntechOpen: London, UK, 2018. [Google Scholar]
- Peng, Y.-S.; Lin, J.-R.; Cheng, B.-H.; Ho, C.; Lin, Y.-H.; Shen, C.-H.; Tsai, M.-H. Incidence and relative risk for developing cancers in women with gestational diabetes mellitus: A nationwide cohort study in Taiwan. BMJ Open 2019, 9, e024583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.-M.M.; O’Brien, K.M.; Zhao, S.; Weinberg, C.R.; Baird, D.D.; Sandler, D.P. Gestational diabetes mellitus may be associated with increased risk of breast cancer. Br. J. Cancer 2017, 116, 960–963. [Google Scholar] [CrossRef] [Green Version]
- Sella, T.; Chodick, G.; Barchana, M.; Heymann, A.D.; Porath, A.; Kokia, E.; Shalev, V. Gestational diabetes and risk of incident primary cancer: A large historical cohort study in Israel. Cancer Causes Control 2011, 22, 1513. [Google Scholar] [CrossRef]
- Monteiro, L.J.; Norman, J.E.; Rice, G.E.; Illanes, S.E. Fetal programming and gestational diabetes mellitus. Placenta 2016, 48, S54–S60. [Google Scholar] [CrossRef] [Green Version]
- Reinius, L.E.; Acevedo, N.; Joerink, M.; Pershagen, G.; Dahlén, S.-E.; Greco, D.; Söderhäll, C.; Scheynius, A.; Kere, J. Differential DNA Methylation in Purified Human Blood Cells: Implications for Cell Lineage and Studies on Disease Susceptibility. PLoS ONE 2012, 7, e41361. [Google Scholar] [CrossRef]
- Metzger, B.E.; Persson, B.; Buchanan, T.A.; Catalano, P.A.; Damm, P.; Dyer, A.R.; de Leiva, A.; Hod, M.; Kitzmiler, J.L.; Lowe, L.P.; et al. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 2010, 33, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Illumina, Inc Ininium® HD Assay Methylation Protocol Guide. Available online: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/infinium_assays/infinium_hd_methylation/infinium-hd-methylation-guide-15019519-01.pdf (accessed on 3 June 2019).
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450 k methylation array data improves replication in large cancer studies. Genome Biol 2014, 15, 503. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew E Jaffe FlowSorted.Blood.450k. Available online: http://bioconductor.org/packages/FlowSorted.Blood.450k/ (accessed on 9 May 2019).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Non-GDM (n = 12) | GDM (n = 12) | p-Value | |
|---|---|---|---|---|
| Age (years) a | 27.3 (0.3) | 27.3 (0.3) | 1.00 | |
| Gestational age (weeks) a | 19.3 (1.5) | 19.3 (2.0) | 1.00 | |
| BMI (kg/m2) a | 27.1 (1.3) | 27.6 (1.1) | 0.77 | |
| Fasting glucose (mmol/L) a | 4.3 (0.1) | 5.5 (0.1) | <0.001 | |
| 1hr OGTT (mmol/L) a | 5.2 (0.3) | 6.6 (0.4) | 0.01 | |
| 2hr OGTT (mmol/L) a | 5.2 (0.3) | 5.8 (0.3) | 0.07 | |
| HbA1c (%) a | 5.1 (0.1) | 5.1 (0.1) | 0.85 | |
| Fasting insulin (mIU/L) b | 8 (7.5-9.0) | 10.2 (6.3-12.7) | 0.65 | |
| HOMA b | 1.6 (1.6-1.8) | 2.6 (1.5-2.9) | 0.31 | |
| Adiponectin (µg/mL) b | 10.4 (7.3-23.8) | 9.7 (4.7-12.0) | 0.28 | |
| C-reactive protein (mg/L) a | 7.1 (1.2) | 7.7 (1.1) | 0.75 | |
| Risk factors: n (%) c | None | 10 (83.3) | 7 (58.3) | 0.37 |
| ≥1 risk factor | 2 (16.7) | 5 (41.8) | ||
| * Education: n (%) c | <grade 12 | 7 (63.6) | 5 (41.7) | 0.29 |
| ≥grade 12 | 4 (36.4) | 7 (58.3) | ||
| Employment: n (%) c | None | 8 (66.7) | 7 (58.3) | 1.00 |
| Formal/informal employment | 4 (33.3) | 5 (41.7) | ||
| Probe ID | Location | Gene Symbol | Gene Name | Region | p-Value | Methylation |
|---|---|---|---|---|---|---|
| cg22985016 | Chr5:492187–524227 | SLC9A3 | Solute Carrier Family 9 Member A3 | Intron | 1.84 × 10−7 | ↑ |
| cg21910650 | Chr6:42976841–42986722 | MEA1; KLHDC3 | Male-Enhanced Antigen 1; Kelch domain-containing protein 3 | Promoter/5’UTR | 3.23 × 10−6 | ↓ |
| g23643951 | Chr1:7151432–7309551 | CAMTA1 | Calmodulin Binding Transcription Activator 1 | Intron | 4.46 × 10−6 | ↓ |
| cg16306629 | Chr8:119121060–119129059 | COLECT10 * | Collectin Subfamily member 10* | Enhancer * | 9.22 × 10−6 | ↑ |
| 07966372 | Chr13:114782770–114898099 | RASA3 | RAS P21 Protein Activator 3 | 5’UTR/Intron | 9.75 × 10−6 | ↓ |
| CpG Site | a Univariate | b Multivariate | ||||
|---|---|---|---|---|---|---|
| Coefficient | 95% CI | p-Value | Coefficient | 95% CI | p-Value | |
| cg22985016 (SLC93A) | 0.028 | 0.019; 0.037 | <0.001 | 0.028 | 0.019; 0.037 | <0.001 |
| cg21910650 (MEA1;KLHDC3) | −0.088 | −0.117; −0.058 | <0.001 | −0.087 | −0.118; −0.056 | <0.001 |
| cg23643951 (CAMTA1) | −0.056 | −0.070; −0.042 | <0.001 | −0.056 | −0.071; −0.042 | <0.001 |
| cg16306629 (Unknown) | 0.274 | 0.183; 0.366 | <0.001 | 0.275 | 0.192; 0.359 | <0.001 |
| cg07966372 (RASA3) | −0.015 | −0.025; −0.004 | 0.006 | −0.015 | −0.026; −0.004 | 0.008 |
| Variable | cg22985016 (SLC93A) | cg21910650 (MEA1; KLHDC3) | cg23643951 (CAMTA1) | cg16306629 (Unknown) | cg07966372 (RASA3) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rho | p-Value | Rho | p-Value | Rho | p-Value | Rho | p-Value | Rho | p-Value | |
| Fasting glucose (mmol/L) | 0.728 | <0.001 | −0.694 | <0.001 | −0.735 | <0.001 | 0.724 | <0.001 | −0.452 | 0.026 |
| 1 h OGTT (mmol/L) | 0.502 | 0.012 | −0.377 | 0.069 | −0.399 | 0.053 | 0.559 | 0.004 | 0.016 | 0.939 |
| 2 h OGTT (mmol/L) | 0.297 | 0.168 | −0.249 | 0.250 | −0.338 | 0.115 | 0.266 | 0.219 | 0.098 | 0.658 |
| Fasting insulin (mIU/L) | −0.037 | 0.888 | −0.103 | 0.691 | −0.204 | 0.433 | 0.109 | 0.674 | −0.495 | 0.043 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, S.; Adam, S.; Rheeder, P.; Louw, J.; Pheiffer, C. Altered Genome-Wide DNA Methylation in Peripheral Blood of South African Women with Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 5828. https://doi.org/10.3390/ijms20235828
Dias S, Adam S, Rheeder P, Louw J, Pheiffer C. Altered Genome-Wide DNA Methylation in Peripheral Blood of South African Women with Gestational Diabetes Mellitus. International Journal of Molecular Sciences. 2019; 20(23):5828. https://doi.org/10.3390/ijms20235828
Chicago/Turabian StyleDias, Stephanie, Sumaiya Adam, Paul Rheeder, Johan Louw, and Carmen Pheiffer. 2019. "Altered Genome-Wide DNA Methylation in Peripheral Blood of South African Women with Gestational Diabetes Mellitus" International Journal of Molecular Sciences 20, no. 23: 5828. https://doi.org/10.3390/ijms20235828