Exploring the Interface between Inflammatory and Therapeutic Glucocorticoid Induced Bone and Muscle Loss


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Glucocorticoids and Therapeutic Glucocorticoid Excess
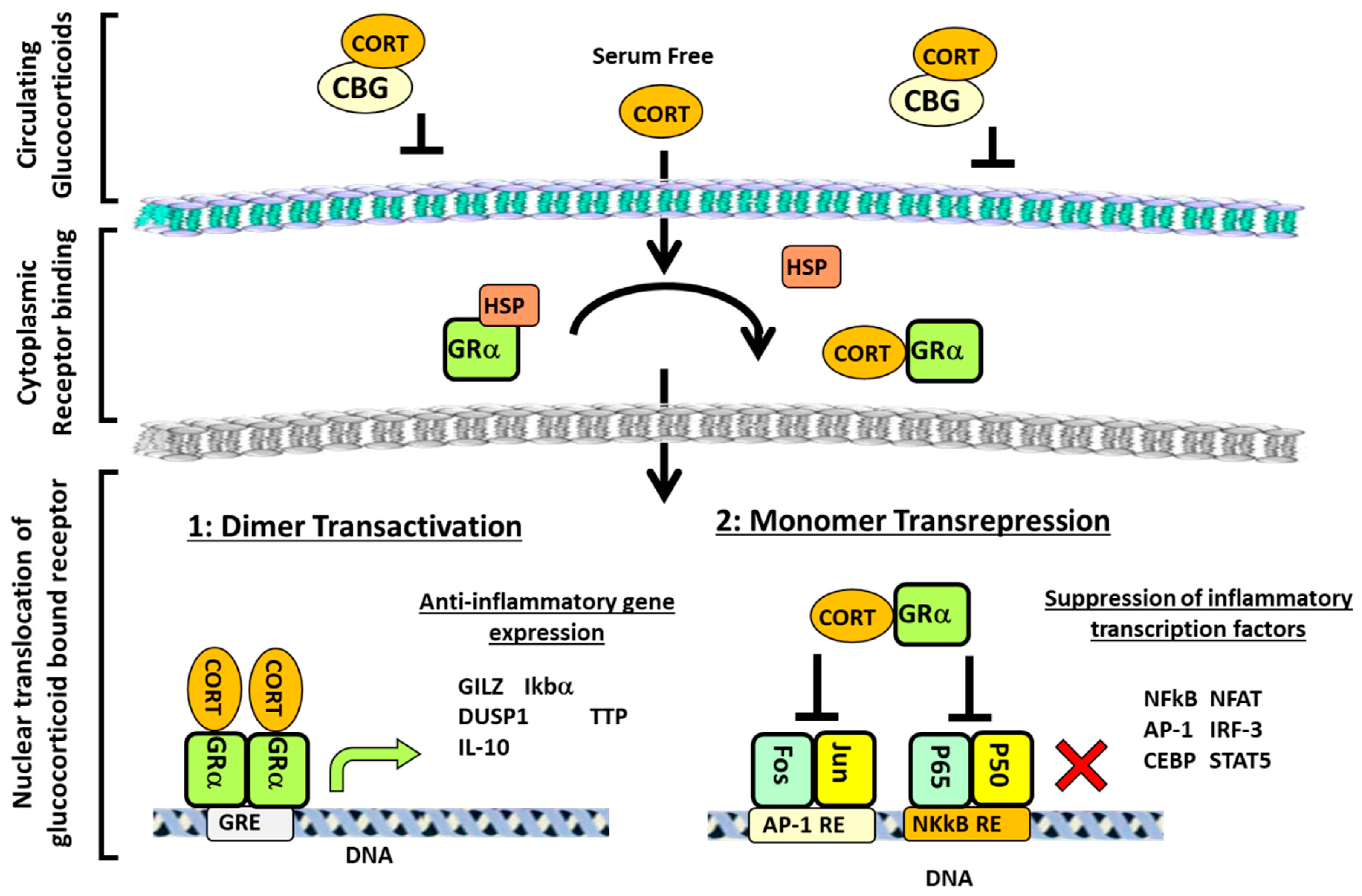
2. Glucocorticoid Signalling and Regulation of Inflammation
3. Bone Metabolism
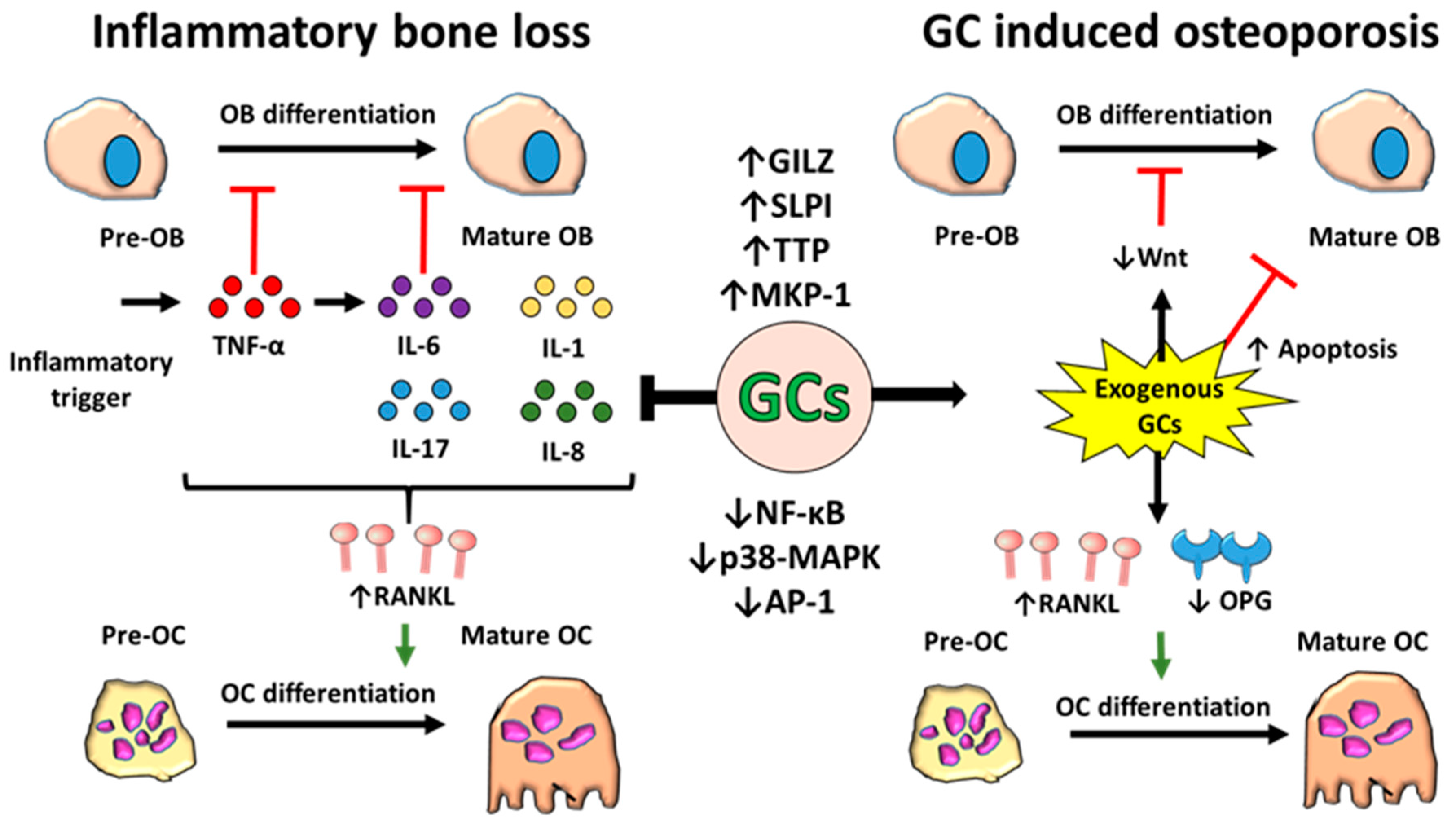
4. Regulation of Bone Metabolism by Inflammation
5. Effects of Glucocorticoids on Bone Metabolism
6. Glucocorticoids, Inflammation and Bone Homeostasis
7. Muscle Mass Related Metabolism
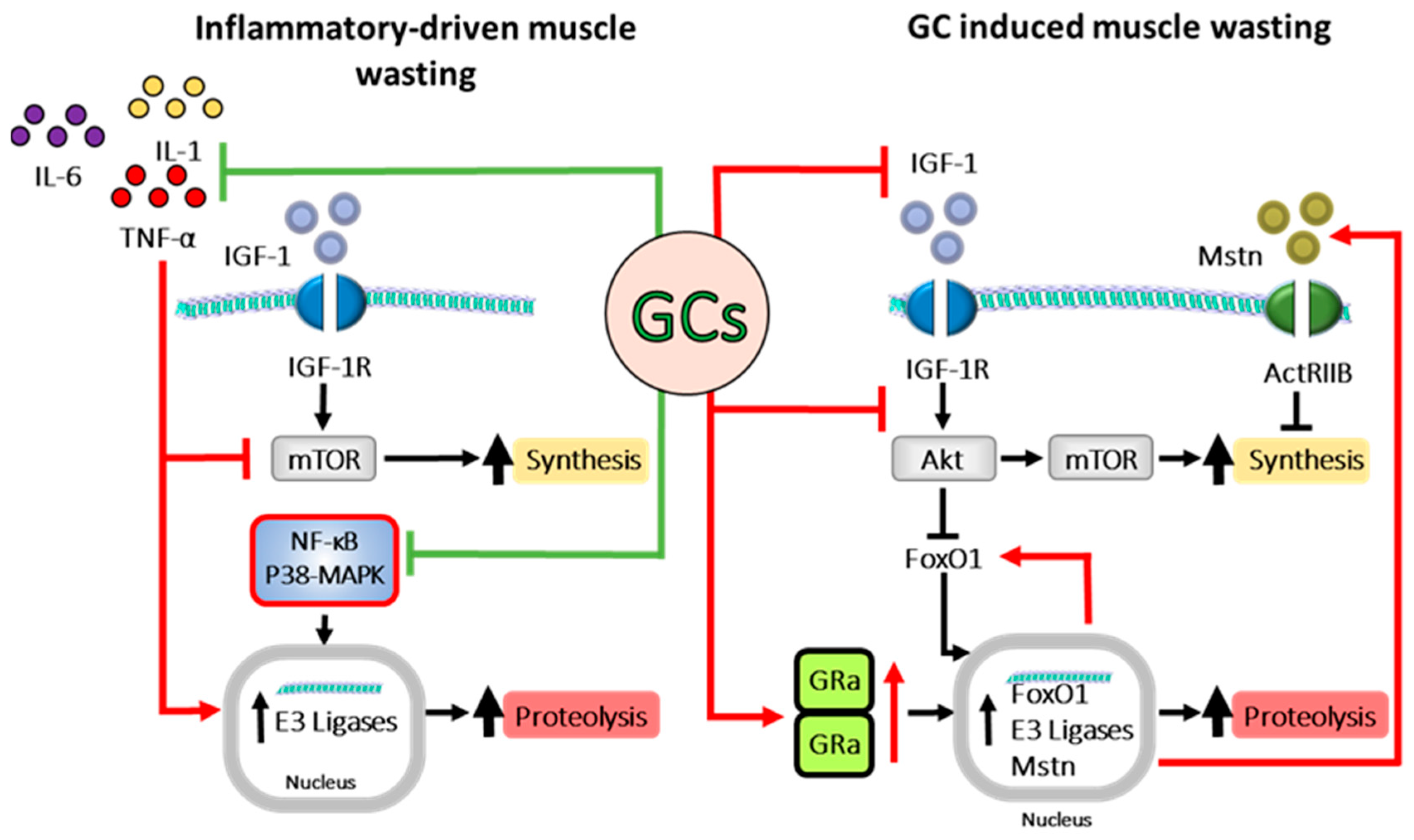
8. Effects of Inflammation of Muscle Metabolism
9. Effects of Glucocorticoids on Muscle Metabolism
10. Interaction between Inflammation and Glucocorticoids in Muscle
11. Insights from Murine Models of Chronic Inflammation Receiving Therapeutic Glucocorticoids
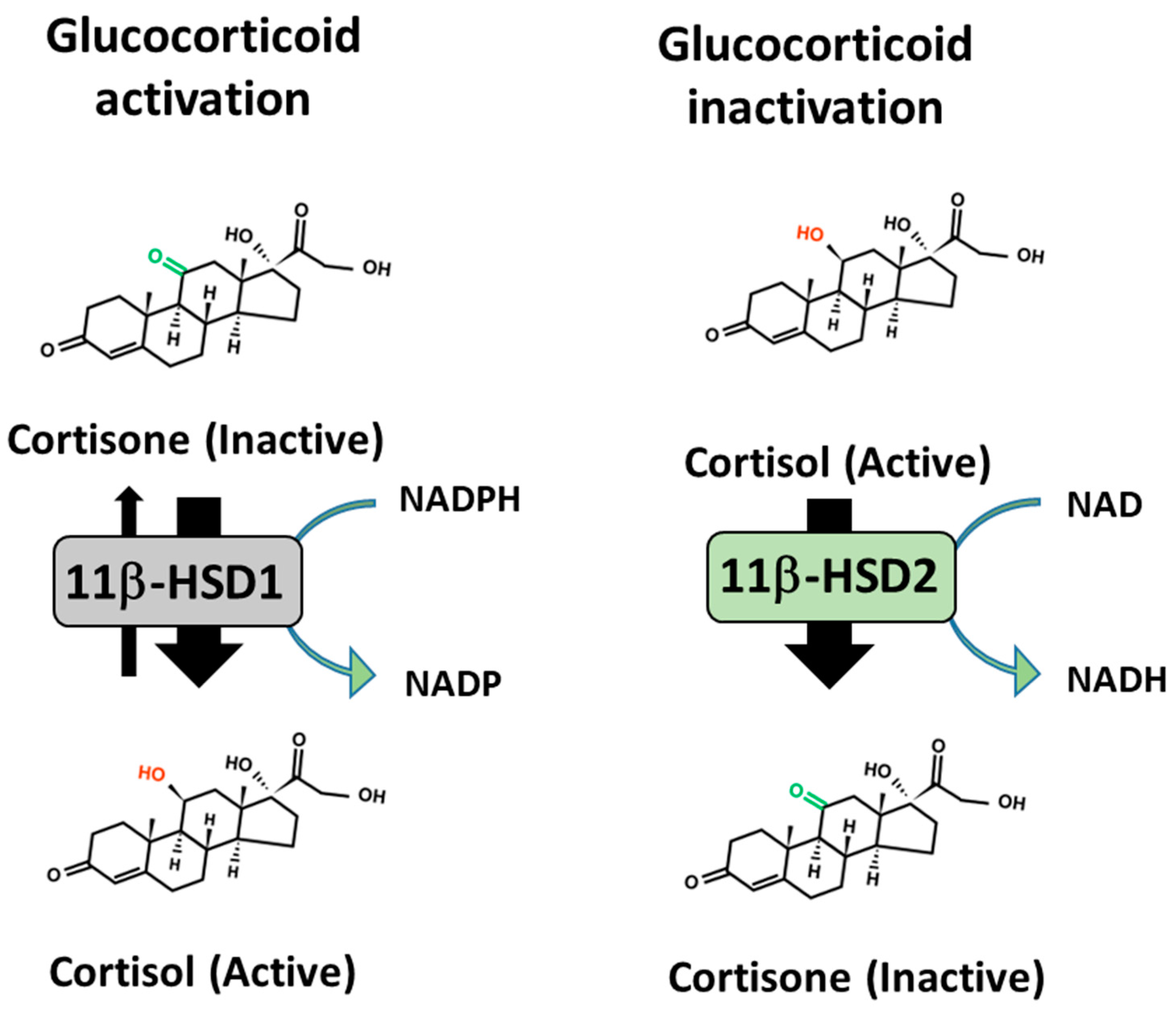
12. Pre-Receptor Regulation of Therapeutic GC Action to Protect against Side Effects
13. Conclusions
Acknowledgments
Conflicts of Interest
References
- Clayton, S.A.; Jones, S.W.; Kurowska-Stolarska, M.; Clark, A.R. The role of microRNAs in glucocorticoid action. J. Biol. Chem. 2018, 293, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.; Saklatvala, J.; Clark, A.R. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Marco, B.D.; Massetti, M.; Bruscoli, S.; Macchiarulo, A.; Virgilio, R.D.; Velardi, E.; Donato, V.; Migliorati, G.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: Role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res. 2007, 35, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Klint, E.A.; Grundtman, C.; Engström, M.; Catrina, A.I.; Makrygiannakis, D.; Klareskog, L.; Andersson, U.; Ulfgren, A.K. Intraarticular glucocorticoid treatment reduces inflammation in synovial cell infiltrations more efficiently than in synovial blood vessels. Arthritis Rheum. 2005, 52, 3880–3889. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, J.S.; Coutinho, A.E.; Cailhier, J.F.; Man, T.Y.; Clay, M.; Thomas, G.; Harris, H.J.; Mullins, J.J.; Seckl, J.R.; Savill, J.S.; et al. Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J. Immunol. 2006, 176, 7605–7611. [Google Scholar] [CrossRef]
- Fardet, L.; Flahault, A.; Kettaneh, A.; Tiev, K.P.; Généreau, T.; Tolédano, C.; Lebbé, C.; Cabane, J. Corticosteroid-induced clinical adverse events: Frequency, risk factors and patient’s opinion. Br. J. Dermatol. 2007, 157, 142–148. [Google Scholar] [CrossRef]
- Feldstein, A.C.; Elmer, P.J.; Nichols, G.A.; Herson, M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos. Int. 2005, 16, 2168–2174. [Google Scholar] [CrossRef]
- Strehl, C.; Bijlsma, J.W.; de Wit, M.; Boers, M.; Caeyers, N.; Cutolo, M.; Dasgupta, B.; Dixon, W.G.; Geenen, R.; Huizinga, T.W.; et al. Defining conditions where long-term glucocorticoid treatment has an acceptably low level of harm to facilitate implementation of existing recommendations: Viewpoints from an EULAR task force. Ann. Rheum. Dis. 2016, 75, 952–957. [Google Scholar] [CrossRef]
- Löfberg, E.; Gutierrez, A.; Wernerman, J.; Anderstam, B.; Mitch, W.E.; Price, S.R.; Bergström, J.; Alvestrand, A. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur. J. Clin. Investig. 2002, 32, 345–353. [Google Scholar] [CrossRef]
- Qi, D.; Pulinilkunnil, T.; An, D.; Ghosh, S.; Abrahani, A.; Pospisilik, J.A.; Brownsey, R.; Wambolt, R.; Allard, M.; Rodrigues, B. Single-dose dexamethasone induces whole-body insulin resistance and alters both cardiac fatty acid and carbohydrate metabolism. Diabetes 2004, 53, 1790–1797. [Google Scholar] [CrossRef]
- Wu, B.; Li, P.; Liu, Y.; Lou, Z.; Ding, Y.; Shu, C.; Ye, S.; Bartlam, M.; Shen, B.; Rao, Z. 3D structure of human FK506-binding protein 52: Implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc. Natl. Acad. Sci. USA 2004, 101, 8348–8353. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br. J. Pharmacol. 2006, 148, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, H.M.; Tuckermann, J.P.; Göttlicher, M.; Vujic, M.; Weih, F.; Angel, P.; Herrlich, P.; Schütz, G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001, 20, 7168–7173. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Schottelius, A.; Döcke, W.D.; Strehlke, P.; Jaroch, S.; Schmees, N.; Rehwinkel, H.; Hennekes, H.; Asadullah, K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc. Natl. Acad. Sci. USA 2004, 101, 227–232. [Google Scholar] [CrossRef]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA Binding of the Glucocorticoid Receptor Is Not Essential for Survival. Cell 1998, 93, 531–541. [Google Scholar] [CrossRef]
- Koenen, M.; Culemann, S.; Vettorazzi, S.; Caratti, G.; Frappart, L.; Baum, W.; Krönke, G.; Baschant, U.; Tuckermann, J.P. Glucocorticoid receptor in stromal cells is essential for glucocorticoid-mediated suppression of inflammation in arthritis. Ann. Rheum. Dis. 2018, 77, 1610–1618. [Google Scholar] [CrossRef]
- Scheschowitsch, K.; Leite, J.A.; Assreuy, J. New Insights in Glucocorticoid Receptor Signaling-More Than Just a Ligand-Binding Receptor. Front. Endocrinol. 2017, 8, 16. [Google Scholar] [CrossRef]
- Abbinante-Nissen, J.M.; Simpson, L.G.; Leikauf, G.D. Corticosteroids increase secretory leukocyte protease inhibitor transcript levels in airway epithelial cells. Am. J. Physiol. 1995, 268 Pt 1, L601–L606. [Google Scholar] [CrossRef]
- Manetsch, M.; Ramsay, E.E.; King, E.M.; Seidel, P.; Che, W.; Ge, Q.; Hibbs, D.E.; Newton, R.; Ammit, A.J. Corticosteroids and β₂-agonists upregulate mitogen-activated protein kinase phosphatase 1: In vitro mechanisms. Br. J. Pharmacol. 2012, 166, 2049–2059. [Google Scholar] [CrossRef]
- Wang, J.C.; Derynck, M.K.; Nonaka, D.F.; Khodabakhsh, D.B.; Haqq, C.; Yamamoto, K.R. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 15603–15608. [Google Scholar] [CrossRef]
- Smoak, K.; Cidlowski, J.A. Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol. Cell. Biol. 2006, 26, 9126–9135. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Frost, H.M. Skeletal structural adaptations to mechanical usage (SATMU): 2. Redefining Wolff’s law: The remodeling problem. Anat. Rec. 1990, 226, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Burra, S.; Nicolella, D.P.; Francis, W.L.; Freitas, C.J.; Mueschke, N.J.; Poole, K.; Jiang, J.X. Dendritic processes of osteocytes are mechanotransducers that induce the opening of hemichannels. Proc. Natl. Acad. Sci. USA 2010, 107, 13648–13653. [Google Scholar] [CrossRef] [PubMed]
- Verborgt, O.; Tatton, N.A.; Majeska, R.J.; Schaffler, M.B. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: Complementary roles in bone remodeling regulation? J. Bone Miner. Res. 2002, 17, 907–914. [Google Scholar] [CrossRef]
- Aguirre, J.I.; Plotkin, L.I.; Stewart, S.A.; Weinstein, R.S.; Parfitt, A.M.; Manolagas, S.C.; Bellido, T. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 2006, 21, 605–615. [Google Scholar] [CrossRef]
- Al-Dujaili, S.A.; Lau, E.; Al-Dujaili, H.; Tsang, K.; Guenther, A.; You, L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J. Cell. Biochem. 2011, 112, 2412–2423. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.I.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell. 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Kukita, T.; Wada, N.; Kukita, A.; Kakimoto, T.; Sandra, F.; Toh, K.; Nagata, K.; Iijima, T.; Horiuchi, M.; Matsusaki, H.; et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J. Exp. Med. 2004, 200, 941–946. [Google Scholar] [CrossRef]
- Wu, H.; Xu, G.; Li, Y.P. Atp6v0d2 is an essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption. J. Bone Miner. Res. 2009, 24, 871–885. [Google Scholar] [CrossRef]
- Väänänen, H.K.; Karhukorpi, E.K.; Sundquist, K.; Wallmark, B.; Roininen, I.; Hentunen, T.; Tuukkanen, J.; Lakkakorpi, P. Evidence for the presence of a proton pump of the vacuolar H(+)-ATPase type in the ruffled borders of osteoclasts. J. Cell Biol. 1990, 111, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C.; Schlesinger, P.H. Purification of a stilbene sensitive chloride channel and reconstitution of chloride conductivity into phospholipid vesicles. Biochem. Biophys. Res. Commun. 1990, 171, 920–925. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Poole, K.E.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005, 19, 1842–1844. [Google Scholar] [CrossRef] [Green Version]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Roca, H.; Phimphilai, M.; Gopalakrishnan, R.; Xiao, G.; Franceschi, R.T. Cooperative interactions between RUNX2 and homeodomain protein-binding sites are critical for the osteoblast-specific expression of the bone sialoprotein gene. J. Biol. Chem. 2005, 280, 30845–30855. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Venton, L.; Sakata, T.; Halloran, B.P. Expression of RANKL and OPG correlates with age-related bone loss in male C57BL/6 mice. J. Bone Miner. Res. 2003, 18, 270–277. [Google Scholar] [CrossRef]
- Andersen, T.L.; Abdelgawad, M.E.; Kristensen, H.B.; Hauge, E.M.; Rolighed, L.; Bollerslev, J.; Kjærsgaard-Andersen, P.; Delaisse, J.M. Understanding coupling between bone resorption and formation: Are reversal cells the missing link? Am. J. Pathol. 2013, 183, 235–246. [Google Scholar] [CrossRef]
- Cowles, E.A.; DeRome, M.E.; Pastizzo, G.; Brailey, L.L.; Gronowicz, G.A. Mineralization and the expression of matrix proteins during in vivo bone development. Calcif. Tissue Int. 1998, 62, 74–82. [Google Scholar] [CrossRef]
- Weiner, S. Organization of extracellularly mineralized tissues: A comparative study of biological crystal growth. CRC Crit. Rev. Biochem. 1986, 20, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Orimo, H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nippon Med. Sch. 2010, 77, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugeberg, G.; Orstavik, R.E.; Uhlig, T.; Falch, J.A.; Halse, J.I.; Kvien, T.K. Bone loss in patients with rheumatoid arthritis: Results from a population-based cohort of 366 patients followed up for two years. Arthritis Rheum. 2002, 46, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Walker-Bone, K. Recognizing and treating secondary osteoporosis. Nat. Rev. Rheumatol. 2012, 8, 480–492. [Google Scholar] [CrossRef]
- Tack, G.J.; Verbeek, W.H.; Schreurs, M.W.; Mulder, C.J. The spectrum of celiac disease: Epidemiology, clinical aspects and treatment. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 204–213. [Google Scholar] [CrossRef]
- Graat-Verboom, L.; Spruit, M.A.; van den Borne, B.E.; Smeenk, F.W.; Martens, E.J.; Lunde, R.; Wouters, E.F. Correlates of osteoporosis in chronic obstructive pulmonary disease: An underestimated systemic component. Respir. Med. 2009, 103, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Parameswaran, V.; Udayan, R.; Burgess, J.; Jones, G. Circulating levels of inflammatory markers predict change in bone mineral density and resorption in older adults: A longitudinal study. J. Clin. Endocrinol. Metab. 2008, 93, 1952–1958. [Google Scholar] [CrossRef] [Green Version]
- Barbour, K.E.; Lui, L.Y.; Ensrud, K.E.; Hillier, T.A.; LeBlanc, E.S.; Ing, S.W.; Hochberg, M.C.; Cauley, J.A. Study of Osteoporotic Fractures (SOF) Research Group. Inflammatory markers and risk of hip fracture in older white women: The study of osteoporotic fractures. J. Bone Miner. Res. 2014, 29, 2057–2064. [Google Scholar] [CrossRef] [Green Version]
- Barbour, K.E.; Boudreau, R.; Danielson, M.E.; Youk, A.O.; Wactawski-Wende, J.; Greep, N.C.; LaCroix, A.Z.; Jackson, R.D.; Wallace, R.B.; Bauer, D.C.; et al. Inflammatory markers and the risk of hip fracture: The Women’s Health Initiative. J. Bone Miner. Res. 2012, 27, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- Pasco, J.A.; Kotowicz, M.A.; Henry, M.J.; Nicholson, G.C.; Spilsbury, H.J.; Box, J.D.; Schneider, H.G. High-sensitivity C-reactive protein and fracture risk in elderly women. JAMA 2006, 296, 1353–1355. [Google Scholar] [CrossRef]
- Hardy, R.; Cooper, M.S. Bone loss in inflammatory disorders. J. Endocrinol. 2009, 201, 309. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmqvist, P.; Persson, E.; Conaway, H.H.; Lerner, U.H. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshitake, F.; Itoh, S.; Narita, H.; Ishihara, K.; Ebisu, S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J. Biol. Chem. 2008, 283, 11535–11540. [Google Scholar] [CrossRef] [Green Version]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef]
- Ragab, A.A.; Nalepka, J.L.; Bi, Y.; Greenfield, E.M. Cytokines synergistically induce osteoclast differentiation: Support by immortalized or normal calvarial cells. Am. J. Physiol. Cell Physiol. 2002, 283, C679–C687. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Matsuyama, T.; Hosokawa, Y.; Makihira, S.; Seki, M.; Karimbux, N.Y.; Goncalves, R.B.; Valverde, P.; Dibart, S.; Li, Y.P.; et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am. J. Pathol. 2006, 169, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef]
- Rifas, L.; Weitzmann, M.N. A novel T cell cytokine, secreted osteoclastogenic factor of activated T cells, induces osteoclast formation in a RANKL-independent manner. Arthritis Rheum. 2009, 60, 3324–3335. [Google Scholar] [CrossRef] [Green Version]
- Abbas, S.; Zhang, Y.H.; Clohisy, J.C.; Abu-Amer, Y. Tumor necrosis factor-alpha inhibits pre-osteoblast differentiation through its type-1 receptor. Cytokine 2003, 22, 33–41. [Google Scholar] [CrossRef]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Parfitt, A.M.; Manolagas, S.C. Osteoblast Programmed Cell Death (Apoptosis): Modulation by Growth Factors and Cytokines. J. Bone Miner. Res. 1998, 13, 793–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneshiro, S.; Ebina, K.; Shi, K.; Higuchi, C.; Hirao, M.; Okamoto, M.; Koizumi, K.; Morimoto, T.; Yoshikawa, H.; Hashimoto, J. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J. Bone Miner. Metab. 2014, 32, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Redlich, K.; Görtz, B.; Hayer, S.; Zwerina, J.; Doerr, N.; Kostenuik, P.; Bergmeister, H.; Kollias, G.; Steiner, G.; Smolen, J.S.; et al. Repair of local bone erosions and reversal of systemic bone loss upon therapy with anti-tumor necrosis factor in combination with osteoprotegerin or parathyroid hormone in tumor necrosis factor-mediated arthritis. Am. J. Pathol. 2004, 164, 543–555. [Google Scholar] [CrossRef] [Green Version]
- van Staa, T.P.; Leufkens, H.G.; Cooper, C. The epidemiology of corticosteroid-induced osteoporosis: A meta-analysis. Osteoporos. Int. 2002, 13, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Mak, W.; Zheng, Y.; Dunstan, C.R.; Seibel, M.J. Osteoblasts directly control lineage commitment of mesenchymal progenitor cells through Wnt signaling. J. Biol. Chem. 2008, 283, 1936–1945. [Google Scholar] [CrossRef] [Green Version]
- Mak, W.; Shao, X.; Dunstan, C.R.; Seibel, M.J.; Zhou, H. Biphasic glucocorticoid-dependent regulation of Wnt expression and its inhibitors in mature osteoblastic cells. Calcif. Tissue Int. 2009, 85, 538–545. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Jia, D.; Plotkin, L.I.; Bellido, T.; Powers, C.C.; Stewart, S.A.; Manolagas, S.C.; Weinstein, R.S. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 2004, 145, 1835–1841. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, G.; Faienza, M.F.; Piacente, L.; Ventura, A.; Oranger, A.; Carbone, C.; Benedetto, A.D.; Colaianni, G.; Gigante, M.; Mori, G.; et al. High dickkopf-1 levels in sera and leukocytes from children with 21-hydroxylase deficiency on chronic glucocorticoid treatment. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E546–E554. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Seitz, S.; Baschant, U.; Schilling, A.F.; Illing, A.; Stride, B.; Kirilov, M.; Takacz, A.; Schmidt-Ullrich, R.; Ostermay, S.; et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010, 11, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; O’Brien, C.A.; Stewart, S.A.; Manolagas, S.C.; Weinstein, R.S. Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology 2006, 147, 5592–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, J.; Biskobing, D.M.; Jadhav, L.; Fan, D.; Nanes, M.S.; Perkins, S.; Fan, X. Dexamethasone promotes expression of membrane-bound macrophage colony-stimulating factor in murine osteoblast-like cells. Endocrinology 1998, 139, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.; Lorentzon, M.; Conaway, H.H.; Lerner, U.H. Glucocorticoid Regulation of Osteoclast Differentiation and Expression of Receptor Activator of Nuclear Factor-κB (NF-κB) Ligand, Osteoprotegerin, and Receptor Activator of NF-κB in Mouse Calvarial Bones. Endocrinology 2006, 147, 3613–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Gori, F.; Riggs, B.L.; Lacey, D.L.; Dunstan, C.R.; Spelsberg, T.C.; Khosla, S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: Potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 1999, 140, 4382–4389. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, E.L.; Williams, J.H.; Davie, M.W.; Marshall, M.J. Effects of dissociated glucocorticoids on OPG and RANKL in osteoblastic cells. Bone 2006, 38, 652–661. [Google Scholar] [CrossRef]
- Faienza, M.F.; Brunetti, G.; Colucci, S.; Piacente, L.; Ciccarelli, M.; Giordani, L.; Del Vecchio, G.C.; D’Amore, M.; Albanese, L.; Cavallo, L.; et al. Osteoclastogenesis in children with 21-hydroxylase deficiency on long-term glucocorticoid therapy: The role of receptor activator of nuclear factor-kappaB ligand/osteoprotegerin imbalance. J. Clin. Endocrinol. Metab. 2009, 94, 2269–2276. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yoon, S.H.; Grynpas, M.D.; Mitchell, J. Pre-treatment with Pamidronate Improves Bone Mechanical Properties in Mdx Mice Treated with Glucocorticoids. Calcif. Tissue Int. 2019, 104, 182–192. [Google Scholar] [CrossRef]
- Kim, H.J.; Zhao, H.; Kitaura, H.; Bhattacharyya, S.; Brewer, J.A.; Muglia, L.J.; Ross, F.P.; Teitelbaum, S.L. Glucocorticoids suppress bone formation via the osteoclast. J. Clin. Investig. 2006, 116, 2152–2160. [Google Scholar] [CrossRef] [Green Version]
- Fenton, C.G.; Webster, J.M.; Martin, C.S.; Fareed, S.; Wehmeyer, C.; Mackie, H.; Jones, R.; Seabright, A.P.; Lewis, J.W.; Lai, Y.C.; et al. Therapeutic glucocorticoids prevent bone loss but drive muscle wasting when administered in chronic polyarthritis. Arthritis Res. Ther. 2019, 21, 182. [Google Scholar] [CrossRef] [Green Version]
- Güler-Yüksel, M.; Bijsterbosch, J.; Goekoop-Ruiterman, Y.P.M.; de Vries-Bouwstra, J.K.; Hulsmans, H.M.J.; de Beus, W.M.; Han, K.H.; Breedveld, F.C.; Dijkmans, B.A.C.; Allaart, C.F.; et al. Changes in bone mineral density in patients with recent onset, active rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, P.N.; Cohen, M.L.; Eisman, J.A.; Pocock, N.A.; Champion, G.D.; Yeates, M.G. Effects of low dose corticosteroids on bone mass in rheumatoid arthritis: A longitudinal study. Ann. Rheum. Dis. 1989, 48, 535–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, P.N.; Eisman, J.A.; Yeates, M.G.; Pocock, N.A.; Eberl, S.; Champion, G.D. Osteoporosis in rheumatoid arthritis: Safety of low dose corticosteroids. Ann. Rheum. Dis. 1986, 45, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijbrandts, C.A.; Klaasen, R.; Dijkgraaf, M.G.W.; Gerlag, D.M.; van Eck-Smit, B.L.F.; Tak, P.P. Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: Arrest of bone loss. Ann. Rheum. Dis. 2009, 68, 373–376. [Google Scholar] [CrossRef]
- Haugeberg, G.; Uhlig, T.; Falch, J.A.; Halse, J.I.; Kvien, T.K. Bone mineral density and frequency of osteoporosis in female patients with rheumatoid arthritis: Results from 394 patients in the Oslo County rheumatoid arthritis register. Arthritis Rheum. 2000, 43, 522–530. [Google Scholar] [CrossRef]
- Pereira, R.M.; Corrente, J.E.; Chahade, W.H.; Yoshinari, N.H. Evaluation by dual X-ray absorptiometry (DXA) of bone mineral density in children with juvenile chronic arthritis. Clin. Exp. Rheumatol. 1998, 16, 495–501. [Google Scholar]
- Varonos, S.; Ansell, B.M.; Reeve, J. Vertebral collapse in juvenile chronic arthritis: Its relationship with glucocorticoid therapy. Calcif. Tissue Int. 1987, 41, 75–78. [Google Scholar] [CrossRef]
- Bergström, J.; Hermansen, L.; Hultman, E.; Saltin, B. Diet, Muscle Glycogen and Physical Performance. Acta Physiol. Scand. 1967, 71, 140–150. [Google Scholar] [CrossRef]
- Coleman, M.E.; DeMayo, F.; Yin, K.C.; Lee, H.M.; Geske, R.; Montgomery, C.; Schwartz, R.J. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J. Biol. Chem. 1995, 270, 12109–12116. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.M.V.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopoulos, G.D.; Glass, D.J. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol. Cell. Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef] [Green Version]
- Harwood, A.J. Regulation of GSK-3: A Cellular Multiprocessor. Cell 2001, 105, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Tseng, B.S.; FlÜCk, M.; Carson, J.A. Molecular and cellular adaptation of muscle in response to physical training. Acta Physiol. Scand. 1998, 162, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Pallafacchina, G.; Calabria, E.; Serrano, A.L.; Kalhovde, J.M.; Schiaffino, S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc. Nat. Acad. Sci. USA 2002, 99, 9213–9218. [Google Scholar] [CrossRef] [Green Version]
- Brunn, G.J.; Hudson, C.C.; Sekulić, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Sonenberg, N. eIF4 Initiation Factors: Effectors of mRNA Recruitment to Ribosomes and Regulators of Translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Hardt Stefan, E.; Tomita, H.; Katus Hugo, A.; Sadoshima, J. Phosphorylation of Eukaryotic Translation Initiation Factor 2Bε by Glycogen Synthase Kinase-3β Regulates β-Adrenergic Cardiac Myocyte Hypertrophy. Circ. Res. 2004, 94, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Latres, E.; Amini, A.R.; Amini, A.A.; Griffiths, J.; Martin, F.J.; Wei, Y.; Lin, H.C.; Yancopoulos, G.D.; Glass, D.J. Insulin-like Growth Factor-1 (IGF-1) Inversely Regulates Atrophy-induced Genes via the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin (PI3K/Akt/mTOR) Pathway. J. Biol. Chem. 2005, 280, 2737–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Autophagy in skeletal muscle. FEBS Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Evenson, A.R.; Fareed, M.U.; Menconi, M.J.; Mitchell, J.C.; Hasselgren, P.O. GSK-3beta inhibitors reduce protein degradation in muscles from septic rats and in dexamethasone-treated myotubes. Int. J. Biochem. Cell Biol. 2005, 37, 2226–2238. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Heng, A.E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Béchet, D.; Matondo, M.; Uttenweiler-Joseph, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef] [PubMed]
- Lokireddy, S.; Wijesoma, I.W.; Sze, S.K.; McFarlane, C.; Kambadur, R.; Sharma, M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am. J. Physiol. Cell Physiol. 2012, 303, C512–C529. [Google Scholar] [CrossRef] [Green Version]
- Csibi, A.; Cornille, K.; Leibovitch, M.P.; Poupon, A.; Tintignac, L.A.; Sanchez, A.M.; Leibovitch, S.A. The translation regulatory subunit eIF3f controls the kinase-dependent mTOR signaling required for muscle differentiation and hypertrophy in mouse. PLoS ONE 2010, 5, e8994. [Google Scholar] [CrossRef]
- Lagirand-Cantaloube, J.; Cornille, K.; Csibi, A.; Batonnet-Pichon, S.; Leibovitch, M.P.; Leibovitch, S.A. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE 2009, 4, e4973. [Google Scholar] [CrossRef] [Green Version]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2003, 15, 1101–1111. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Xu, Y.; Yu, D.; Li, D.; Liu, X.; Du, W. Insulin-like growth factor-1 (IGF-1) promotes myoblast proliferation and skeletal muscle growth of embryonic chickens via the PI3K/Akt signalling pathway. Cell Biol. Int. 2015, 39, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Florini, J.R.; Ewton, D.Z.; Magri, K.A. Hormones, growth factors, and myogenic differentiation. Annu. Rev. Physiol. 1991, 53, 201–216. [Google Scholar] [CrossRef]
- Thomas, M.; Langley, B.; Berry, C.; Sharma, M.; Kirk, S.; Bass, J.; Kambadur, R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 2000, 275, 40235–40243. [Google Scholar] [CrossRef] [Green Version]
- Rios, R.; Carneiro, I.; Arce, V.M.; Devesa, J. Myostatin is an inhibitor of myogenic differentiation. Am. J. Physiol. Cell Physiol. 2002, 282, C993–C999. [Google Scholar] [CrossRef] [Green Version]
- Windner, S.E.; Doris, R.A.; Ferguson, C.M.; Nelson, A.C.; Valentin, G.; Tan, H.; Oates, A.C.; Wardle, F.C.; Devoto, S.H. Tbx6, Mesp-b and Ripply1 regulate the onset of skeletal myogenesis in zebrafish. Development 2015, 142, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Biolo, G.; Maggi, S.P.; Williams, B.D.; Tipton, K.D.; Wolfe, R.R. Increased rates of muscle protein turnover and amino acid transport after resistance exercise in humans. Am. J. Physiol. 1995, 268 Pt 1, E514–E520. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.M. The science of muscle hypertrophy: Making dietary protein count. Proc. Nutr. Soc. 2011, 70, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Rudnicki, M.A.; Schnegelsberg, P.N.J.; Stead, R.H.; Braun, T.; Arnold, H.-H.; Jaenisch, R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 1993, 75, 1351–1359. [Google Scholar] [CrossRef]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esuml, E.; Li, S.; Nonaka, I.; Nabeshima, Y.I. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Kassar-Duchossoy, L.; Gayraud-Morel, B.; Gomès, D.; Rocancourt, D.; Buckingham, M.; Shinin, V.; Tajbakhsh, S. Mrf4 determines skeletal muscle identity in Myf5:Myod double-mutant mice. Nature 2004, 431, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, S.; Rocancourt, D.; Cossu, G.; Buckingham, M. Redefining the Genetic Hierarchies Controlling Skeletal Myogenesis: Pax-3 and Myf-5 Act Upstream of MyoD. Cell 1997, 89, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Roubenoff, R.; Roubenoff, R.A.; Ward, L.M.; Holland, S.M.; Hellmann, D.B. Rheumatoid cachexia: Depletion of lean body mass in rheumatoid arthritis. Possible association with tumor necrosis factor. J. Rheumatol. 1992, 19, 1505–1510. [Google Scholar] [PubMed]
- Londhe, P.; Guttridge, D.C. Inflammation induced loss of skeletal muscle. Bone 2015, 80, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.; Bruunsgaard, H.; Weis, N.; Hendel, H.W.; Andreassen, B.U.; Eldrup, E.; Dela, F.; Pedersen, B.K. Circulating levels of TNF-alpha and IL-6-relation to truncal fat mass and muscle mass in healthy elderly individuals and in patients with type-2 diabetes. Mech. Ageing Dev. 2003, 124, 495–502. [Google Scholar] [CrossRef]
- Jonsdottir, I.H.; Schjerling, P.; Ostrowski, K.; Asp, S.; Richter, E.A.; Pedersen, B.K. Muscle contractions induce interleukin-6 mRNA production in rat skeletal muscles. J. Physiol. 2000, 528, 157–163. [Google Scholar] [CrossRef]
- Goodman, M.N. Interleukin-6 induces skeletal muscle protein breakdown in rats. Proc. Soc. Exp. Biol. Med. 1994, 205, 182–185. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E., Jr.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKβ/NF-κB Activation Causes Severe Muscle Wasting in Mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layne, M.D.; Farmer, S.R. Tumor necrosis factor-alpha and basic fibroblast growth factor differentially inhibit the insulin-like growth factor-I induced expression of myogenin in C2C12 myoblasts. Exp. Cell Res. 1999, 249, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Reid, M.B. NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Celemin, L.; Pasko, N.; Blomart, V.; Thissen, J.P. Inhibition of muscle insulin-like growth factor I expression by tumor necrosis factor-alpha. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E1279–E1290. [Google Scholar] [CrossRef]
- Frost, R.A.; Lang, C.H.; Gelato, M.C. Transient exposure of human myoblasts to tumor necrosis factor-alpha inhibits serum and insulin-like growth factor-I stimulated protein synthesis. Endocrinology 1997, 138, 4153–4159. [Google Scholar] [CrossRef]
- Zhang, L.; Rajan, V.; Lin, E.; Hu, Z.; Han, H.Q.; Zhou, X.; Song, Y.; Min, H.; Wang, X.; Du, J.; et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011, 25, 1653–1663. [Google Scholar] [CrossRef] [Green Version]
- Brandt, C.; Nielsen, A.R.; Fischer, C.P.; Hansen, J.; Pedersen, B.K.; Plomgaard, P. Plasma and muscle myostatin in relation to type 2 diabetes. PLoS ONE 2012, 7, e37236. [Google Scholar] [CrossRef] [Green Version]
- Lecker, S.H.; Solomon, V.; Mitch, W.E.; Goldberg, A.L. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J. Nutr. 1999, 129 (Suppl. S1), 227S–237S. [Google Scholar] [CrossRef] [Green Version]
- Hall-Angeras, M.; Angeras, U.; Zamir, O.; Hasselgren, P.O.; Fischer, J.E. Effect of the glucocorticoid receptor antagonist RU 38486 on muscle protein breakdown in sepsis. Surgery 1991, 109, 468–473. [Google Scholar]
- Schakman, O.; Dehoux, M.; Bouchuari, S.; Delaere, S.; Lause, P.; Decroly, N.; Shoelson, S.E.; Thissen, J.P. Role of IGF-I and the TNFalpha/NF-kappaB pathway in the induction of muscle atrogenes by acute inflammation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E729–E739. [Google Scholar] [CrossRef] [Green Version]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- de Theije, C.C.; Schols, A.M.; Lamers, W.H.; Ceelen, J.J.; van Gorp, R.H.; Hermans, J.R.; Köhler, S.E.; Langen, R.C. Glucocorticoid Receptor Signaling Impairs Protein Turnover Regulation in Hypoxia-Induced Muscle Atrophy in Male Mice. Endocrinology 2018, 159, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.J.; Alamdari, N.; O’Neal, P.; Gonnella, P.; Aversa, Z.; Hasselgren, P.O. Sepsis increases the expression and activity of the transcription factor Forkhead Box O 1 (FOXO1) in skeletal muscle by a glucocorticoid-dependent mechanism. Int. J. Biochem. Cell Biol. 2010, 42, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schakman, O.; Gilson, H.; de Coninck, V.; Lause, P.; Verniers, J.; Havaux, X.; Ketelslegers, J.M.; Thissen, J.P. Insulin-like growth factor-I gene transfer by electroporation prevents skeletal muscle atrophy in glucocorticoid-treated rats. Endocrinology 2005, 146, 1789–1797. [Google Scholar] [CrossRef] [Green Version]
- Tomas, F.M.; Munro, H.N.; Young, V.R. Effect of glucocorticoid administration on the rate of muscle protein breakdown in vivo in rats, as measured by urinary excretion of N tau-methylhistidine. Biochem. J. 1979, 178, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Hilton-Jones, D. Diagnosis and treatment of inflammatory muscle diseases. J. Neurol. Neurosurg. Psychiatry 2003, 74 (Suppl. 2), ii25–ii31. [Google Scholar] [CrossRef] [Green Version]
- Gayan-Ramirez, G.; Vanderhoydonc, F.; Verhoeven, G.; Decramer, M. Acute treatment with corticosteroids decreases IGF-1 and IGF-2 expression in the rat diaphragm and gastrocnemius. Am. J. Respir. Crit. Care Med. 1999, 159, 283–289. [Google Scholar] [CrossRef]
- Fournier, M.; Huang, Z.S.; Li, H.; Da, X.; Cercek, B.; Lewis, M.I. Insulin-like growth factor I prevents corticosteroid-induced diaphragm muscle atrophy in emphysematous hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R34–R43. [Google Scholar] [CrossRef]
- Wang, H.; Kubica, N.; Ellisen, L.W.; Jefferson, L.S.; Kimball, S.R. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J. Biol. Chem. 2006, 281, 39128–39134. [Google Scholar] [CrossRef] [Green Version]
- Imae, M.; Fu, Z.; Yoshida, A.; Noguchi, T.; Kato, H. Nutritional and hormonal factors control the gene expression of FoxOs, the mammalian homologues of DAF-16. J. Mol. Endocrinol. 2003, 30, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Artaza, J.N.; Bhasin, S.; Mallidis, C.; Taylor, W.; Ma, K.; Gonzalez-Cadavid, N.F. Endogenous expression and localization of myostatin and its relation to myosin heavy chain distribution in C2C12 skeletal muscle cells. J. Cell. Physiol. 2002, 190, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Mallidis, C.; Artaza, J.; Taylor, W.; Gonzalez-Cadavid, N.; Bhasin, S. Characterization of 5′-regulatory region of human myostatin gene: Regulation by dexamethasone in vitro. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1128–E1136. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Mallidis, C.; Bhasin, S.; Mahabadi, V.; Artaza, J.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E363–E371. [Google Scholar] [CrossRef] [PubMed]
- Ponyi, A.; Borgulya, G.; Constantin, T.; Vancsa, A.; Gergely, L.; Danko, K. Functional outcome and quality of life in adult patients with idiopathic inflammatory myositis. Rheumatology 2005, 44, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, C.; Pegoraro, E.; Turella, E.; Intino, M.T.; Pini, A.; Costa, C. Deflazacort in Duchenne dystrophy: Study of long-term effect. Muscle Nerve 1994, 17, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Brooke, M.H.; Fenichel, G.M.; Griggs, R.C.; Mendell, J.R.; Moxley, R.T., 3rd; Miller, J.P.; Kaiser, K.K.; Florence, J.M.; Pandya, S.; Signore, L.; et al. Clinical investigation of Duchenne muscular dystrophy. Interesting results in a trial of prednisone. Arch. Neurol. 1987, 44, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Fenichel, G.M.; Mendell, J.R.; Moxley, R.T., 3rd; Griggs, R.C.; Brooke, M.H.; Miller, J.P.; Pestronk, A.; Robison, J.; King, W.; Signore, L.; et al. A comparison of daily and alternate-day prednisone therapy in the treatment of Duchenne muscular dystrophy. Arch. Neurol. 1991, 48, 575–579. [Google Scholar] [CrossRef]
- Fenichel, G.M.; Florence, J.M.; Pestronk, A.; Mendell, J.R.; Moxley, R.T., 3rd; Griggs, R.C.; Brooke, M.H.; Miller, J.P.; Robison, J.; King, W.; et al. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology 1991, 41, 1874–1877. [Google Scholar] [CrossRef]
- Griggs, R.C.; Moxley, R.T., 3rd; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch. Neurol. 1991, 48, 383–388. [Google Scholar] [CrossRef]
- Lemmey, A.B.; Wilkinson, T.J.; Perkins, C.M.; Nixon, L.A.; Sheikh, F.; Jones, J.G.; Ahmad, Y.A.; O’Brien, T.D. Muscle loss following a single high-dose intramuscular injection of corticosteroids to treat disease flare in patients with rheumatoid arthritis. Eur. J. Rheumatol. 2018, 5, 160–164. [Google Scholar] [CrossRef]
- Gibson, J.N.; Poyser, N.L.; Morrison, W.L.; Scrimgeour, C.M.; Rennie, M.J. Muscle protein synthesis in patients with rheumatoid arthritis: Effect of chronic corticosteroid therapy on prostaglandin F2 alpha availability. Eur. J. Clin. Investig. 1991, 21, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Tiao, G.; Fagan, J.; Roegner, V.; Lieberman, M.; Wang, J.J.; Fischer, J.E.; Hasselgren, P.O. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Investig. 1996, 97, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gathercole, L.L.; Lavery, G.G.; Morgan, S.A.; Cooper, M.S.; Sinclair, A.J.; Tomlinson, J.W.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase 1: Translational and therapeutic aspects. Endocr. Rev. 2013, 34, 525–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, R.S.; Doig, C.L.; Hussain, Z.; O’leary, M.; Morgan, S.A.; Pearson, M.J.; Naylor, A.; Jones, S.W.; Filer, A.; Stewart, P.M.; et al. 11beta-hydroxysteroid dehydrogenase type 1 within muscle protects against the adverse effects of local inflammation. J. Pathol. 2016, 240, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Bujalska, I.; Shimojo, M.; Howie, A.; Stewart, P.M. Human 11 beta-hydroxysteroid dehydrogenase: Studies on the stably transfected isoforms and localization of the type 2 isozyme within renal tissue. Steroids 1997, 62, 77–82. [Google Scholar] [CrossRef]
- Fenton, C.G.; Doig, C.L.; Fareed, S.; Naylor, A.; Morrell, A.P.; Addison, O.; Wehmeyer, C.; Buckley, C.D.; Cooper, M.S.; Lavery, G.G.; et al. 11beta-HSD1 plays a critical role in trabecular bone loss associated with systemic glucocorticoid therapy. Arthritis Res. Ther. 2019, 21, 188. [Google Scholar] [CrossRef] [Green Version]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Nat. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef] [Green Version]
- Feig, P.U.; Shah, S.; Hermanowski-Vosatka, A.; Plotkin, D.; Springer, M.S.; Donahue, S.; Thach, C.; Klein, E.J.; Lai, E.; Kaufman, K.D. Effects of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes. Metab. 2011, 13, 498–504. [Google Scholar] [CrossRef]
- Rosenstock, J.; Banarer, S.; Fonseca, V.A.; Inzucchi, S.E.; Sun, W.; Yao, W.; Hollis, G.; Flores, R.; Levy, R.; Williams, W.V.; et al. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 2010, 33, 1516–1522. [Google Scholar] [CrossRef] [Green Version]
- Ahasan, M.M.; Hardy, R.; Jones, C.; Kaur, K.; Nanus, D.; Juarez, M.; Morgan, S.A.; Hassan-Smith, Z.; Bénézech, C.; Caamano, J.H.; et al. Inflammatory regulation of glucocorticoid metabolism in mesenchymal stromal cells. Arthritis Rheum. 2012, 64, 2404–2413. [Google Scholar] [CrossRef]
- Kaur, K.; Hardy, R.; Ahasan, M.M.; Eijken, M.; Van Leeuwen, J.P.; Filer, A.; Thomas, A.M.; Raza, K.; Buckley, C.D.; Stewart, P.M.; et al. Synergistic induction of local glucocorticoid generation by inflammatory cytokines and glucocorticoids: Implications for inflammation associated bone loss. Ann. Rheum. Dis. 2010, 69, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.E.; Gray, M.; Brownstein, D.G.; Salter, D.M.; Sawatzky, D.A.; Clay, S.; Gilmour, J.S.; Seckl, J.R.; Savill, J.S.; Chapman, K.E. 11beta-Hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology 2012, 153, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, R.S.; Fenton, C.; Croft, A.P.; Naylor, A.J.; Begum, R.; Desanti, G.; Buckley, C.D.; Lavery, G.; Cooper, M.S.; Raza, K. 11 Beta-hydroxysteroid dehydrogenase type 1 regulates synovitis, joint destruction, and systemic bone loss in chronic polyarthritis. J. Autoimmun. 2018, 92, 104–113. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webster, J.M.; Fenton, C.G.; Langen, R.; Hardy, R.S. Exploring the Interface between Inflammatory and Therapeutic Glucocorticoid Induced Bone and Muscle Loss. Int. J. Mol. Sci. 2019, 20, 5768. https://doi.org/10.3390/ijms20225768
Webster JM, Fenton CG, Langen R, Hardy RS. Exploring the Interface between Inflammatory and Therapeutic Glucocorticoid Induced Bone and Muscle Loss. International Journal of Molecular Sciences. 2019; 20(22):5768. https://doi.org/10.3390/ijms20225768
Chicago/Turabian StyleWebster, Justine M., Chloe G. Fenton, Ramon Langen, and Rowan S. Hardy. 2019. "Exploring the Interface between Inflammatory and Therapeutic Glucocorticoid Induced Bone and Muscle Loss" International Journal of Molecular Sciences 20, no. 22: 5768. https://doi.org/10.3390/ijms20225768