HBx and c-MYC Cooperate to Induce URI1 Expression in HBV-Related Hepatocellular Carcinoma

 ,
,
Abstract
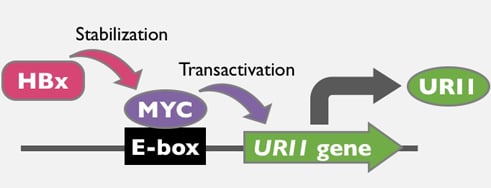
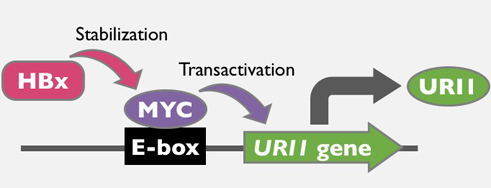
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
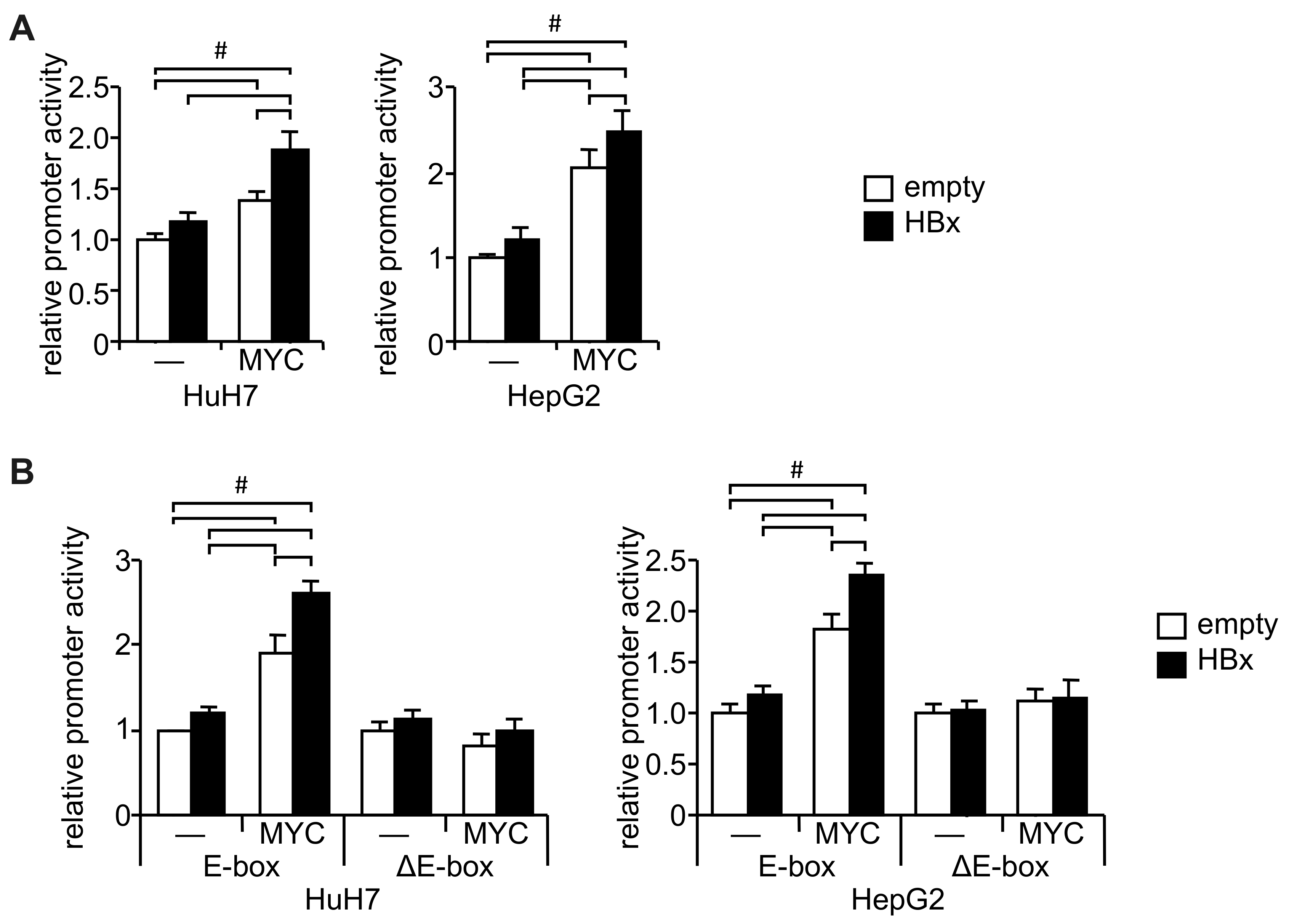
2.1. URI1 Promoter Activation by HBx and c-MYC
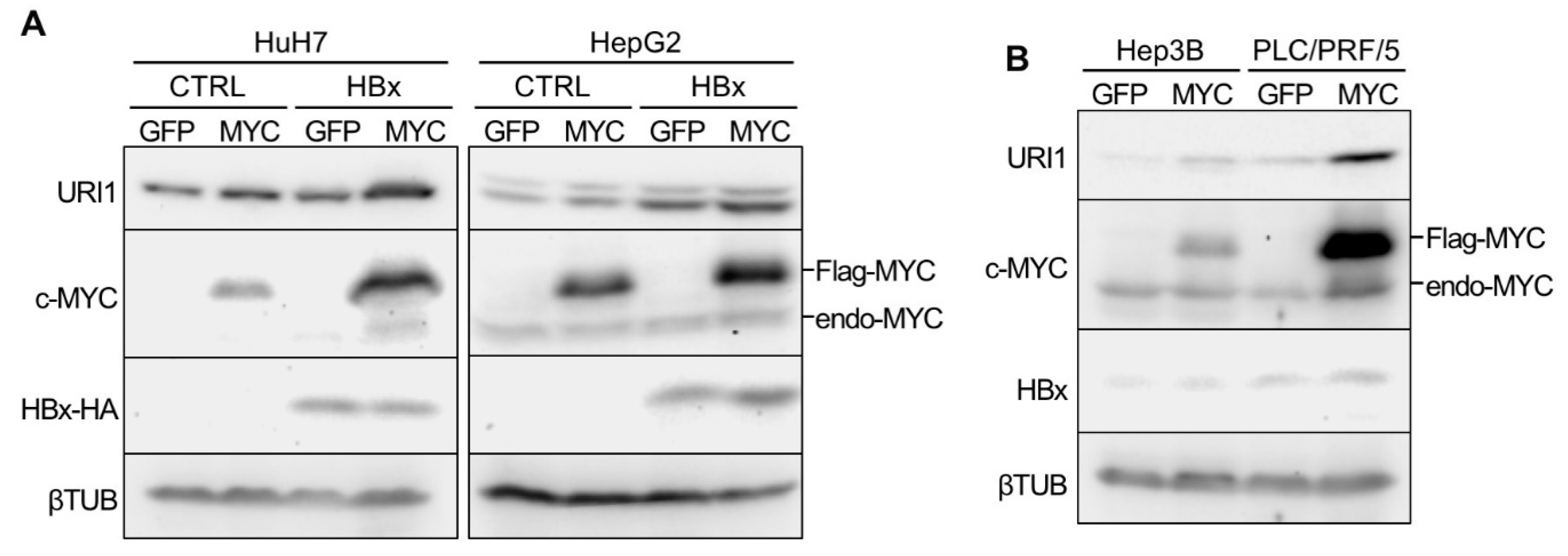
2.2. Induction of URI1 Expression by HBx and c-MYC
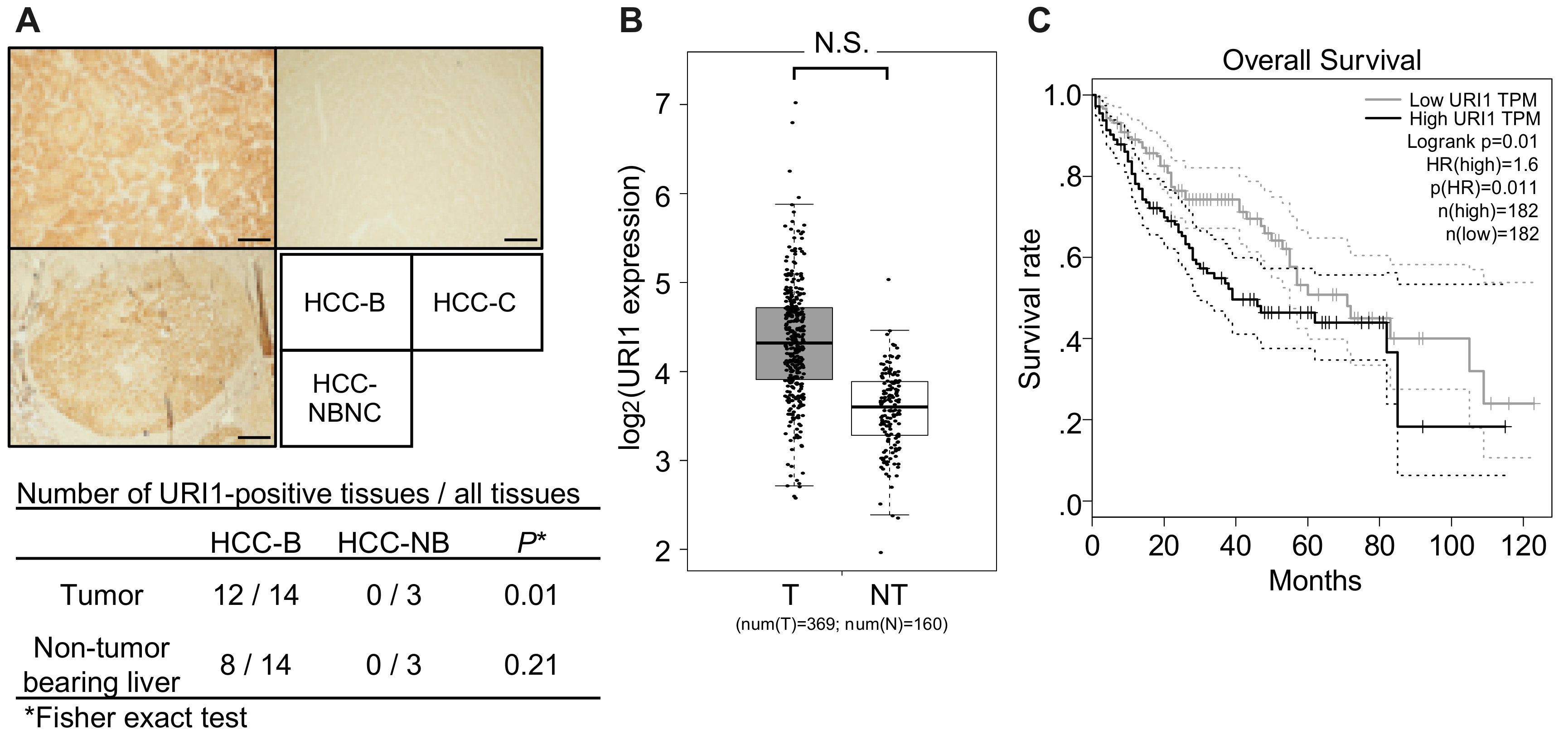
2.3. URI1 Expression in HCC-B Tissues
2.4. Involvement of c-MYC in URI1 Expression in HCC-B
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Immunohistochemistry and ChIP Assay
4.4. Plasmid Construction
4.5. Reporter Assay, Western Blotting, qPCR
4.6. Analysis of TCGA Data Set
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ChIP | chromatin immunoprecipitation |
| GTEx | genotype-tissue expression |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HCC-B | hepatitis B virus-related hepatocellular carcinoma |
| HCC-NB | non-hepatitis B virus-related hepatocellular carcinoma |
| qPCR | quantitative polymerase chain reaction |
| TCGA | The Cancer Genome Atlas |
| URI | unconventional prefoldin RPB5 interactor |
References
- Liu, Z.; Jiang, Y.; Yuan, H.; Fang, Q.; Cai, N.; Suo, C.; Jin, L.; Zhang, T. The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J. Hepatol. 2019, 70, 674–683. [Google Scholar] [CrossRef]
- Tateishi, R.; Uchino, K.; Fujiwara, N.; Takehara, T.; Okanoue, T.; Seike, M.; Yoshiji, H.; Yatsuhashi, H.; Shimizu, M.; Torimura, T.; et al. A nationwide survey on non-B, non-C hepatocellular carcinoma in Japan: 2011–2015 update. J. Gastroenterol. 2019, 54, 367–376. [Google Scholar] [CrossRef]
- Papatheodoridis, G.V.; Idilman, R.; Dalekos, G.N.; Buti, M.; Chi, H.; van Boemmel, F.; Calleja, J.L.; Sypsa, V.; Goulis, J.; Manolakopoulos, S.; et al. The risk of hepatocellular carcinoma decreases after the first 5 years of entecavir or tenofovir in Caucasians with chronic hepatitis B. Hepatology 2017, 66, 1444–1453. [Google Scholar] [CrossRef]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Kalra, N.; Kumar, V. The X protein of hepatitis B virus binds to the F box protein Skp2 and inhibits the ubiquitination and proteasomal degradation of c-Myc. FEBS Lett. 2006, 580, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, W.; Ko, C.; Ryu, W.S. Hepatitis B virus X protein enhances Myc stability by inhibiting SCF(Skp2) ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis. Oncogene 2016, 35, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Terradillos, O.; Billet, O.; Renard, C.A.; Levy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yang, Y.; Zhang, L.; Tang, G.; Wang, Y.; Xue, G.; Zhou, W.; Sun, S. Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015, 61, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Tummala, K.S.; Gomes, A.L.; Yilmaz, M.; Graña, O.; Bakiri, L.; Ruppen, I.; Ximénez-Embún, P.; Sheshappanavar, V.; Rodriguez-Justo, M.; Pisano, D.G.; et al. Inhibition of de novo NAD(+) synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell 2014, 26, 826–839. [Google Scholar] [CrossRef]
- Gomes, A.L.; Teijeiro, A.; Burén, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pan, Y.F.; Ding, Z.W.; Yang, G.Z.; Tan, Y.X.; Yang, C.; Jiang, T.Y.; Liu, L.J.; Zhang, B.; Han, T.; et al. RMP promotes venous metastases of hepatocellular carcinoma through promoting IL-6 transcription. Oncogene 2015, 34, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, Y.; Zhou, W.; Zhong, L.; Wen, Z.; Yu, H.; Chen, S.; Shen, J.; Chen, H.; She, Q.; et al. The viral oncoprotein HBx of Hepatitis B virus promotes the growth of hepatocellular carcinoma through cooperating with the cellular oncoprotein RMP. Int. J. Biol. Sci. 2014, 10, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.H.; Iyer, V.R. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS ONE 2008, 3, e1798. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Chèneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2016, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed]
- Natri, H.M.; Wilson, M.A.; Buetow, K.H. Distinct molecular etiologies of male and female hepatocellular carcinoma. BMC Cancer 2019, 19, 951. [Google Scholar] [CrossRef]
- Guerrieri, F.; Belloni, L.; D’Andrea, D.; Pediconi, N.; Le Pera, L.; Testoni, B.; Scisciani, C.; Floriot, O.; Zoulim, F.; Tramontano, A.; et al. Genome-wide identification of direct HBx genomic targets. BMC Genom. 2017, 18, 184. [Google Scholar] [CrossRef]
- Feld, J.J.; Krassenburg, L.A.P. What Comes First: Treatment of Viral Hepatitis or Liver Cancer? Dig. Dis. Sci. 2019, 64, 1041–1049. [Google Scholar] [CrossRef]
- Jones, S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004, 5, 226. [Google Scholar] [CrossRef]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Lüscher, B.; Larsson, L.G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bell, J.L.; Carter, D.; Gherardi, S.; Poulos, R.C.; Milazzo, G.; Wong, J.W.; Al-Awar, R.; Tee, A.E.; Liu, P.Y.; et al. WDR5 Supports an N-Myc Transcriptional Complex That Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res. 2015, 75, 5143–5154. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Suzuki, H.; Honda, M.; Okada, H.; Kaneko, S.; Inoue, I.; Ebisui, E.; Hashimoto, K.; Carninci, P.; Kanki, K.; et al. Prevention of hepatocellular carcinoma by targeting MYCN-positive liver cancer stem cells with acyclic retinoid. Proc. Natl. Acad. Sci. USA 2018, 115, 4969–4974. [Google Scholar] [CrossRef] [PubMed]
- Dorjsuren, D.; Lin, Y.; Wei, W.; Yamashita, T.; Nomura, T.; Hayashi, N.; Murakami, S. RMP, a novel RNA polymerase II subunit 5-interacting protein, counteracts transactivation by hepatitis B virus X protein. Mol. Cell Biol. 1998, 18, 7546–7555. [Google Scholar] [CrossRef]
- Lin, Y.; Nomura, T.; Cheong, J.H.; Dorjsuren, D.; Iida, K.; Murakami, S. Hepatitis B virus X protein is a transcriptional modulator that com-municates with transcriptional factor IIB and RNA polymerase II subunit 5. J. Biol. Chem. 1997, 272, 7132–7139. [Google Scholar] [CrossRef]
- Burén, S.; Gomes, A.L.; Teijeiro, A.; Fawal, M.A.; Yilmaz, M.; Tummala, K.S.; Perez, M.; Rodriguez-Justo, M.; Campos-Olivas, R.; Megías, D.; et al. Regulation of OGT by URI in Response to Glucose Confers c-MYC-Dependent Survival Mechanisms. Cancer Cell 2016, 30, 290–307. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Minner, S.; Guldvik, I.J.; Sandmann, M.J.; Tsourlakis, M.C.; Berge, V.; Svindland, A.; Schlomm, T.; Mills, I.G. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 2013, 73, 5277–5287. [Google Scholar] [CrossRef]
- Chou, T.Y.; Hart, G.W.; Dang, C.V. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J. Biol. Chem. 1995, 270, 18961–18965. [Google Scholar] [CrossRef]
- Shim, H.; Chun, Y.S.; Lewis, B.C.; Dang, C.V. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl. Acad. Sci. USA 1998, 95, 1511–1516. [Google Scholar] [CrossRef]
- Wang, M.D.; Wu, H.; Huang, S.; Zhang, H.L.; Qin, C.J.; Zhao, L.H.; Fu, G.B.; Zhou, X.; Wang, X.M.; Tang, L.; et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget 2016, 7, 6711–6726. [Google Scholar] [CrossRef]
- Labeur, T.A.; Ten Cate, D.W.G.; Takkenberg, R.B.; Azahaf, H.; van Oijen, M.G.H.; van Delden, O.M.; de Man, R.A.; van Vugt, J.L.; IJzermans, J.N.M.; Eskens, F.A.L.M.; et al. Are we SHARP enough? The importance of adequate patient selection in sorafenib treatment for hepatocellular carcinoma. Acta Oncol. 2018, 57, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutkowska, J.; Strzadala, L.; Rapak, A. Sorafenib in Combination with Betulinic Acid Synergistically Induces Cell Cycle Arrest and Inhibits Clonogenic Activity in Pancreatic Ductal Adenocarcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3234. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, L.; Liu, P.; Jairam, K.; Yin, Y.; Chen, K.; Sprengers, D.; Peppelenbosch, M.P.; Pan, Q.; Smits, R. Blocking Wnt Secretion Reduces Growth of Hepatocellular Carcinoma Cell Lines Mostly Independent of β-Catenin Signaling. Neoplasia 2016, 18, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/β-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, M.Y.; Kim, C.M.; Park, Y.M.; Ryu, W.S. Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology 2004, 39, 1683–1993. [Google Scholar] [CrossRef]
- Pan, S.C.; Cui, H.H.; Qiu, C.G. HOTAIR promotes myocardial fibrosis through regulating URI1 expression via Wnt pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6983–6990. [Google Scholar] [CrossRef]
- Liu, P.; Xie, S.H.; Hu, S.; Cheng, X.; Gao, T.; Zhang, C.; Song, Z. Age-specific sex difference in the incidence of hepatocellular carcinoma in the United States. Oncotarget 2017, 8, 68131–68137. [Google Scholar] [CrossRef] [Green Version]
- Buschow, S.I.; Biesta, P.J.; Groothuismink, Z.M.A.; Erler, N.S.; Vanwolleghem, T.; Ho, E.; Najera, I.; Ait-Goughoulte, M.; de Knegt, R.J.; Boonstra, A.; et al. TLR7 polymorphism, sex and chronic HBV infection influence plasmacytoid DC maturation by TLR7 ligands. Antivir. Res. 2018, 157, 27–37. [Google Scholar] [CrossRef]
- Cheung, P.F.; Yip, C.W.; Ng, L.W.; Lo, K.W.; Wong, N.; Choy, K.W.; Chow, C.; Chan, K.F.; Cheung, T.T.; Poon, R.T.; et al. Establishment and characterization of a novel primary hepatocellular carcinoma cell line with metastatic ability in vivo. Cancer Cell Int. 2014, 14, 103. [Google Scholar] [CrossRef] [Green Version]
- Forgues, M.; Marrogi, A.J.; Spillare, E.A.; Wu, C.G.; Yang, Q.; Yoshida, M.; Wang, X.W. Interaction of the hepatitis B virus X protein with the Crm1-dependent nuclear export pathway. J. Biol. Chem. 2001, 276, 22797–22803. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, H.; da Costa, K.A.; Lee, S.; Renga, B.; Jaeschke, H.; Yang, Z.; Orena, S.J.; Goedken, M.J.; Zhang, Y.; Kong, B.; et al. Interactions Between Nuclear Receptor SHP and FOXA1 Maintain Oscillatory Homocysteine Homeostasis in Mice. Gastroenterology 2015, 148, 1012–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, H.; Oura, S. Involvement of MAFB and MAFF in Retinoid-Mediated Suppression of Hepatocellular Carcinoma Invasion. Int. J. Mol. Sci. 2018, 19, 1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Boxi, K.; Chenwei, L.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuchiya, H.; Amisaki, M.; Takenaga, A.; Honjo, S.; Fujiwara, Y.; Shiota, G. HBx and c-MYC Cooperate to Induce URI1 Expression in HBV-Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5714. https://doi.org/10.3390/ijms20225714
Tsuchiya H, Amisaki M, Takenaga A, Honjo S, Fujiwara Y, Shiota G. HBx and c-MYC Cooperate to Induce URI1 Expression in HBV-Related Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2019; 20(22):5714. https://doi.org/10.3390/ijms20225714
Chicago/Turabian StyleTsuchiya, Hiroyuki, Masataka Amisaki, Ai Takenaga, Soichiro Honjo, Yoshiyuki Fujiwara, and Goshi Shiota. 2019. "HBx and c-MYC Cooperate to Induce URI1 Expression in HBV-Related Hepatocellular Carcinoma" International Journal of Molecular Sciences 20, no. 22: 5714. https://doi.org/10.3390/ijms20225714