How Oxygen Availability Affects the Antimicrobial Efficacy of Host Defense Peptides: Lessons Learned from Studying the Copper-Binding Peptides Piscidins 1 and 3

 ,
,
Abstract
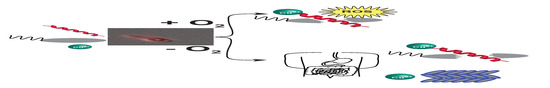
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
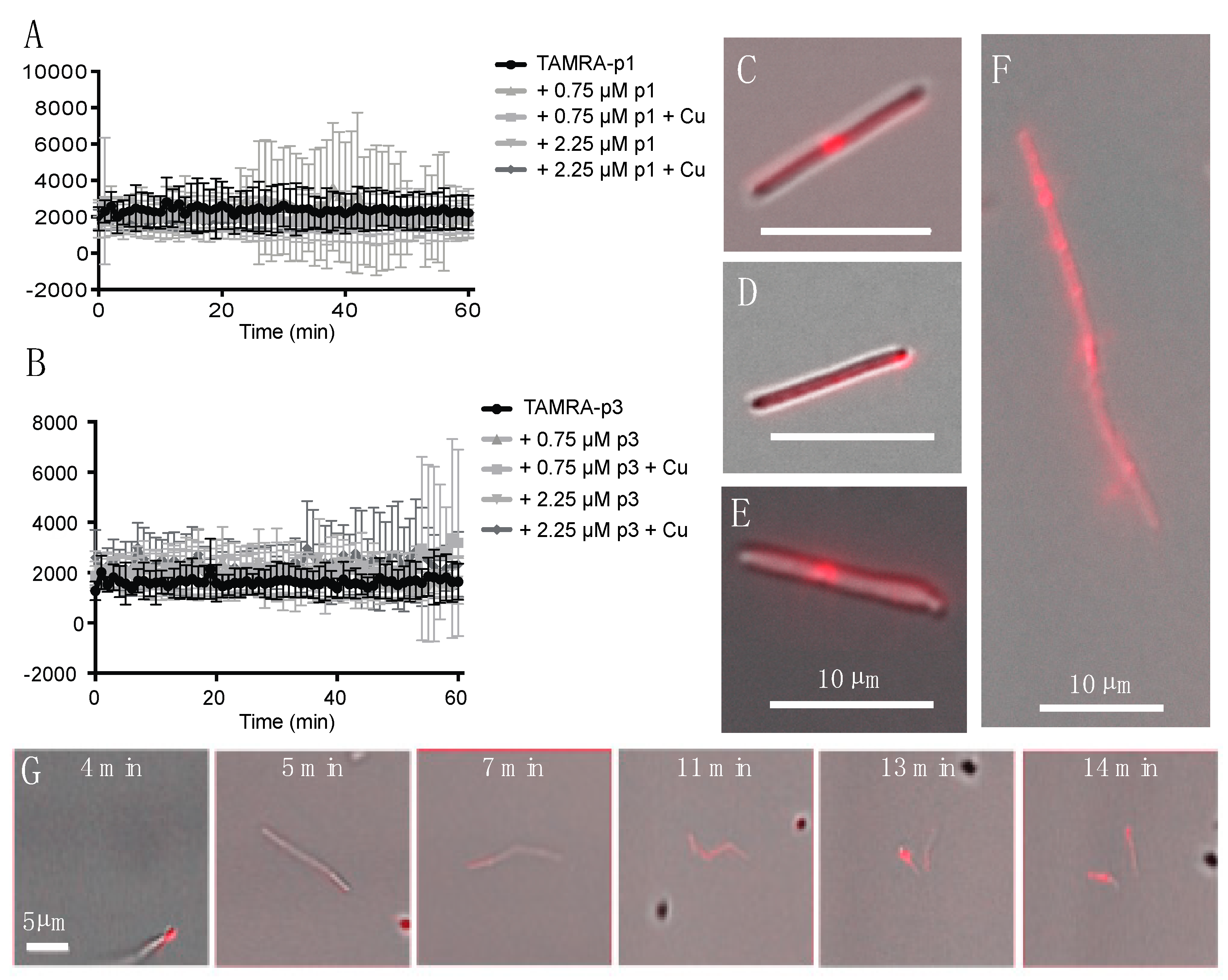
2.1. Piscidins Are Incorporated into C. difficile and Appear to Localize to Sites of Membrane Curvature
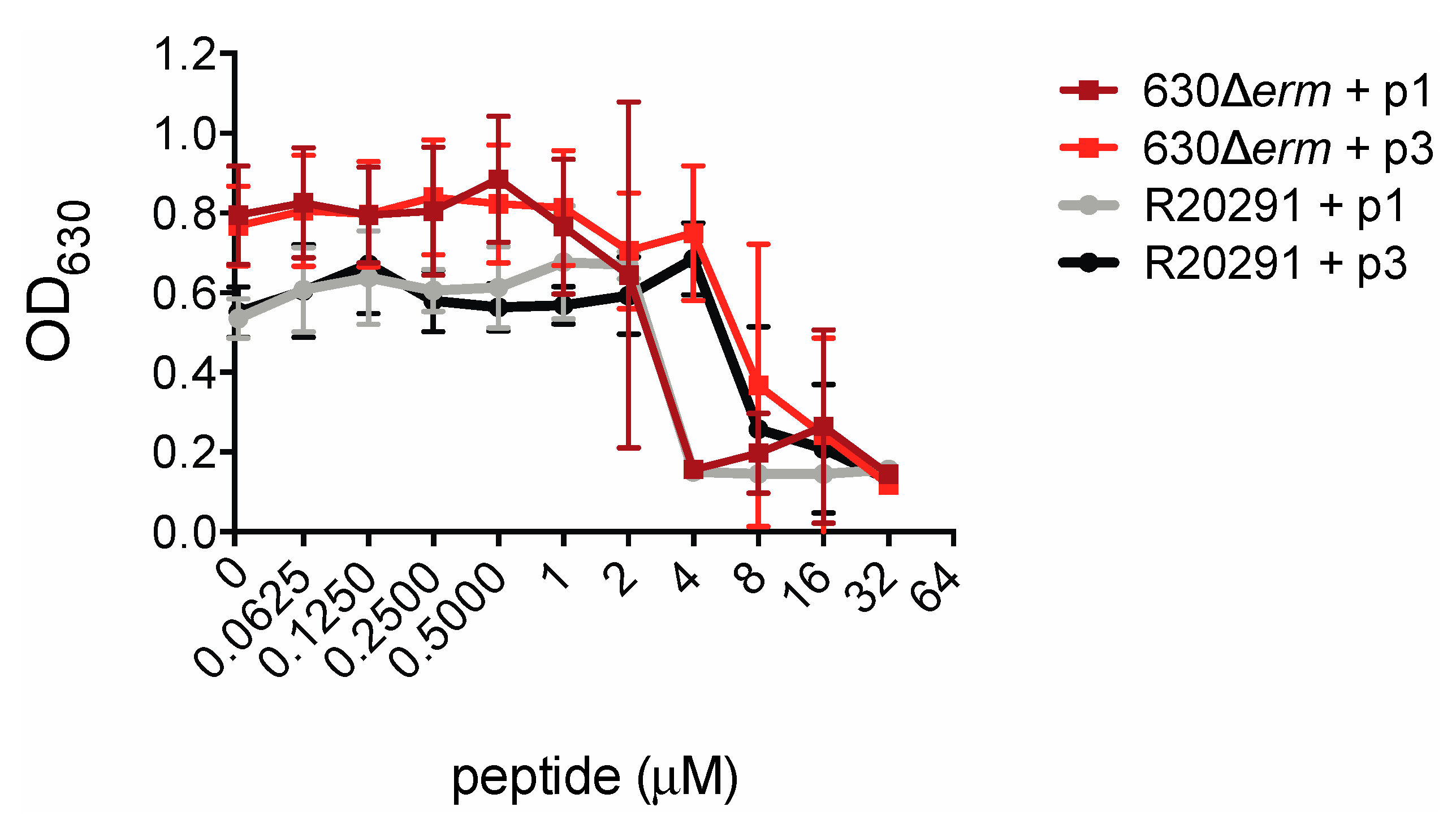
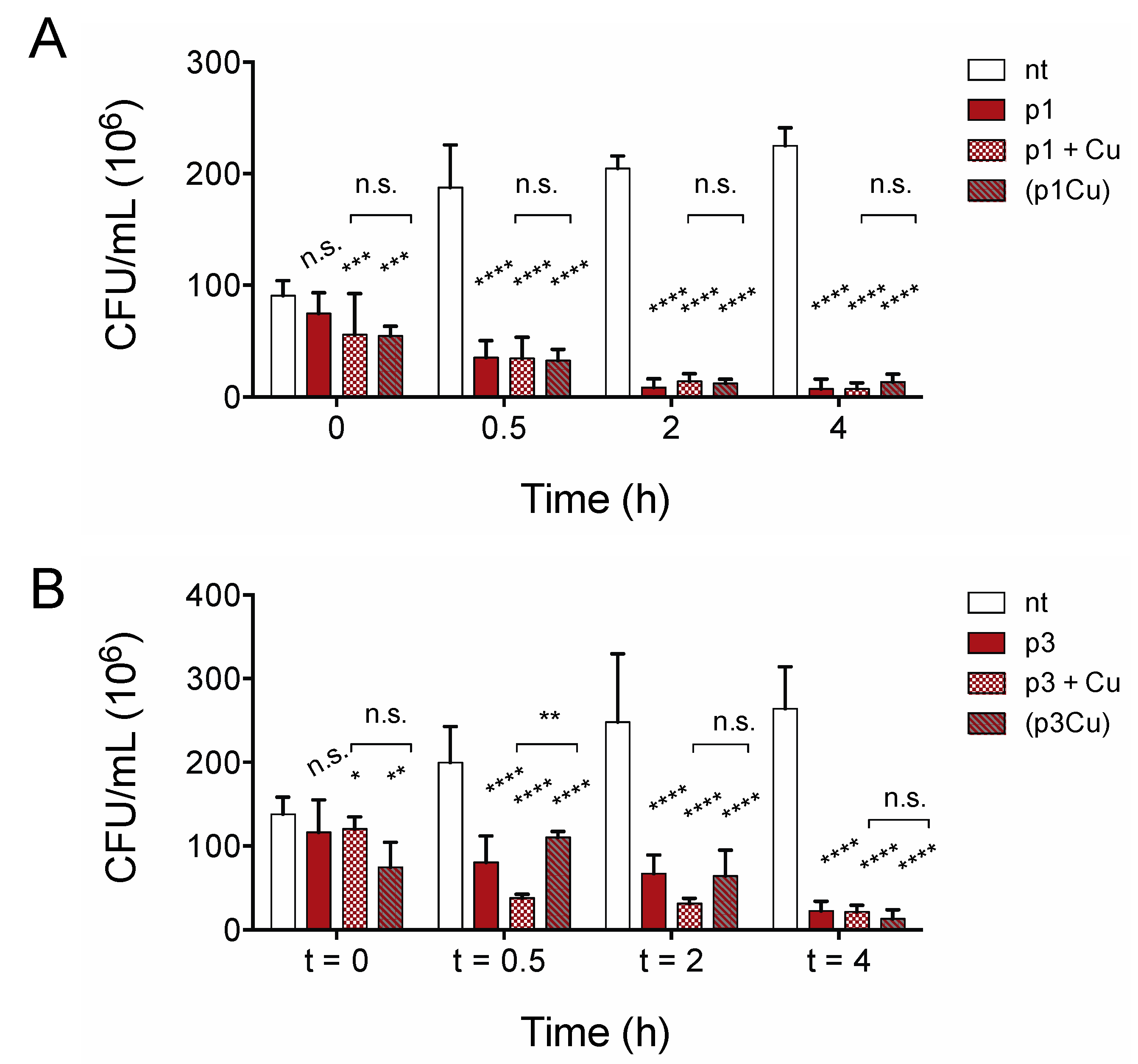
2.2. Piscidins Prevent C. difficile Proliferation
2.3. Piscidins Reduce Established C. difficile Populations
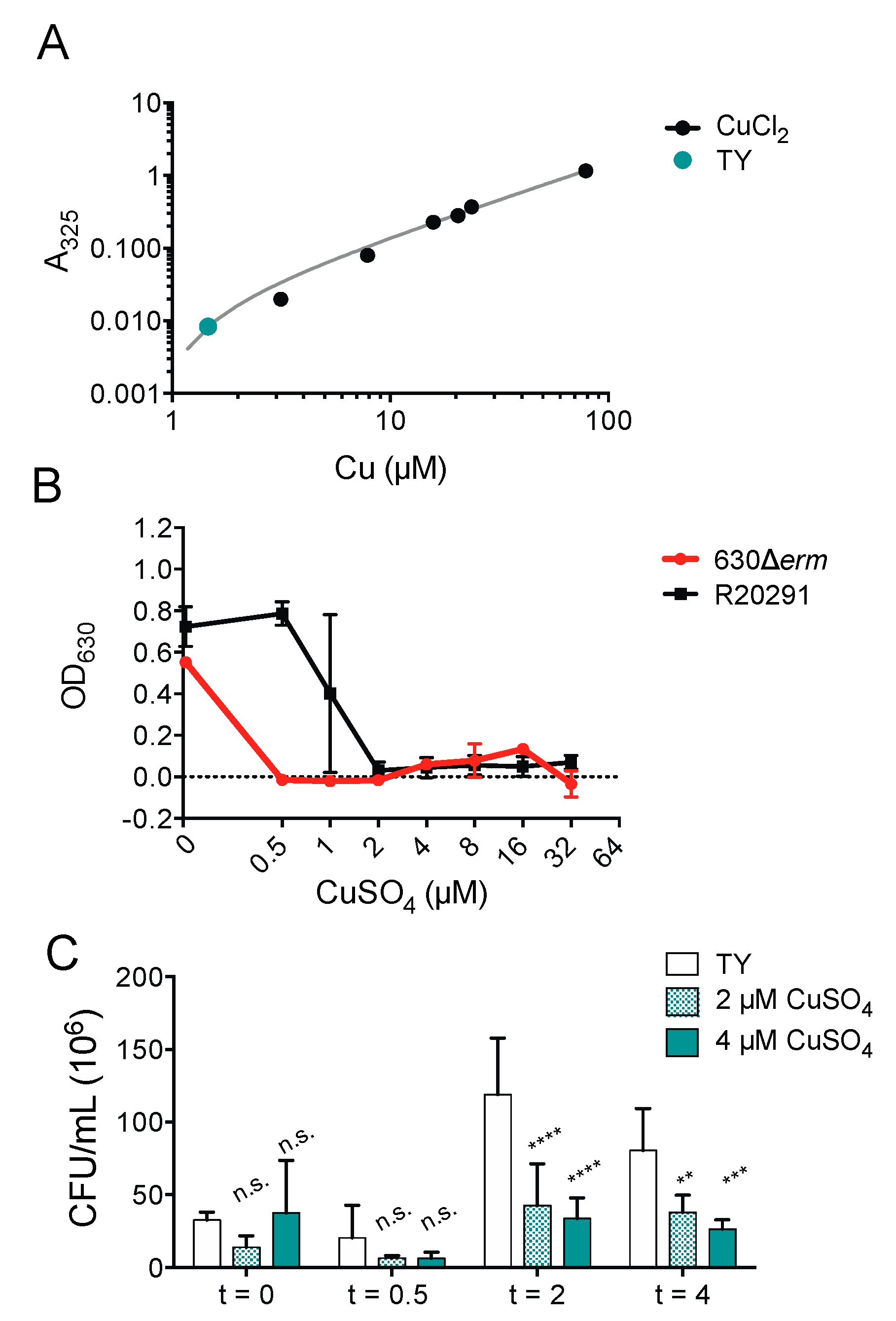
2.4. Copper Is Toxic to Anaerobically Growing C. difficile
3. Discussion
4. Materials and Methods
4.1. Materials, Chemicals, Bacterial Strains and Growth Conditions
4.2. Peptide Synthesis
4.3. Microscopy
4.4. Growth Inhibition Assays
4.5. Time-Kill Assays
4.6. Atomic Absorption Spectroscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, M. Fidaxomicin (OPT-80) for the treatment of Clostridium difficile infection. Expert Opin. Pharm. 2010, 11, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Nearly Half a Million Americans Suffered from Clostridium difficile Infections in a Single Year. Available online: https://www.cdc.gov/media/releases/2015/p0225-clostridium-difficile.html (accessed on 10 October 2019).
- Denève, C.; Janoir, C.; Poilane, I.; Fantinato, C.; Collignon, A. New trends in Clostridium difficile virulence and pathogenesis. Int. J. Antimicrob. Agents 2009, 33, S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Khanna, S. Community-acquired Clostridium difficile infection: An increasing public health threat. Infect Drug Resist. 2014, 7, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Loo, V.G.; Poirier, L.; Miller, M.A.; Oughton, M.; Libman, M.D.; Michaud, S.; Bourgault, A.M.; Nguyen, T.; Frenette, C.; Kelly, M.; et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N. Engl. J. Med. 2005, 353, 2442–2449. [Google Scholar] [CrossRef]
- Bartlett, J.G. Clostridium difficile: Progress and challenges. Ann. N. Y. Acad. Sci. 2010, 1213, 62–69. [Google Scholar] [CrossRef]
- Smits, W.K. Hype or hypervirulence: A reflection on problematic C. difficile strains. Virulence 2013, 4, 592–596. [Google Scholar] [CrossRef]
- Louie, T.J.; Miller, M.A.; Mullane, K.M.; Weiss, K.; Lentnek, A.; Golan, Y.; Gorbach, S.; Sears, P.; Shue, Y.K. Fidaxomicin versus vancomycin for Clostridium difficile infection. N. Engl. J. Med. 2011, 364, 422–431. [Google Scholar] [CrossRef]
- Drekonja, D.M.; Butler, M.; MacDonald, R.; Bliss, D.; Filice, G.A.; Rector, T.S.; Wilt, T.J. Comparative effectiveness of Clostridium difficile treatments: A systematic review. Ann. Intern. Med. 2011, 155, 839–847. [Google Scholar] [CrossRef]
- Boyle, M.L.; Ruth-Sahd, L.A.; Zhou, Z. Fecal microbiota transplant to treat recurrent Clostridium difficile infections. Crit. Care Nurse 2015, 35, 51–64. [Google Scholar] [CrossRef]
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 2018, 66, 987–994. [Google Scholar] [CrossRef]
- Van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, G.; Ianiro, G.; Gasbarrini, A. Fecal microbiota transplantation for the treatment of Clostridium difficile infection: A systematic review. J. Clin. Gastroenterol. 2014, 48, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Palacios, A.; Lejeune, J.T. Moist-heat resistance, spore aging, and superdormancy in Clostridium difficile. Appl. Env. Microbiol. 2011, 77, 3085–3091. [Google Scholar] [CrossRef]
- Edwards, A.N.; Karim, S.T.; Pascual, R.A.; Jowhar, L.M.; Anderson, S.E.; McBride, S.M. Chemical and stress resistances of Clostridium difficile spores and vegetative cells. Front. Microbiol. 2016, 7, 1698. [Google Scholar] [CrossRef] [PubMed]
- Sorg, J.A.; Sonenshein, A.L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008, 190, 2505–2512. [Google Scholar] [CrossRef]
- Howerton, A.; Ramirez, N.; Abel-Santos, E. Mapping interactions between germinants and Clostridium difficile spores. J. Bacteriol. 2011, 193, 274–282. [Google Scholar] [CrossRef]
- Sarker, M.R.; Paredes-Sabja, D. Molecular basis of early stages of Clostridium difficile infection: Germination and colonization. Future Microbiol. 2012, 7, 933–943. [Google Scholar] [CrossRef]
- Cochetière, M.-F.; Montassier, E.; Hardouin, J.-B.; Carton, T.; Vacon, F.; Durand, T.; Lalande, V.; Petit, J.; Potel, G.; Beaugerie, L. Human Intestinal Microbiota Gene Risk Factors for Antibiotic-Associated Diarrhea: Perspectives for Prevention. Microb. Ecol. 2010, 59, 830–837. [Google Scholar] [CrossRef] [Green Version]
- Manges, A.R.; Labbe, A.; Loo, V.G.; Atherton, J.K.; Behr, M.A.; Masson, L.; Tellis, P.A.; Brousseau, R. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridum difficile-associated disease. J. Infect. Dis. 2010, 202, 1877–1884. [Google Scholar] [CrossRef]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.B.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013, 502, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef] [PubMed]
- Frädrich, C.; Beer, L.-A.; Gerhard, R. Reactive oxygen species as additional determinants for cytotoxicity of Clostridium difficile Toxins A and B. Toxins 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, G.H.; Agerberth, B. Neutrophil antibacterial peptides, multifunctional effector molecules in the mammalian immune system. J. Immunol. Methods 1999, 232, 45–54. [Google Scholar] [CrossRef]
- McBride, S.M.; Sonenshein, A.L. The dlt operon confers resistance to cationic antimicrobial peptides in Clostridium difficile. Microbiology 2011, 157, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Nicolas, P.; Rosenstein, Y. Multifunctional host defense peptides. Febs. J. 2009, 276, 6464. [Google Scholar] [CrossRef]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals (Basel) 2013, 6, 1543–1575. [Google Scholar] [CrossRef]
- Yount, N.Y.; Yeaman, M.R. Immunocontinuum: Perspectives in antimicrobial peptide mechanisms of action and resistance. Protein Pept. Lett. 2005, 12, 49–67. [Google Scholar] [CrossRef]
- Wong, C.C.; Zhang, L.; Ren, S.X.; Shen, J.; Chan, R.L.; Cho, C.H. Antibacterial peptides and gastrointestinal diseases. Curr. Pharm. Des. 2011, 17, 1583–1586. [Google Scholar] [CrossRef]
- Nuding, S.; Frasch, T.; Schaller, M.; Stange, E.F.; Zabel, L.T. Synergistic effects of antimicrobial peptides and antibiotics against Clostridium difficile. Antimicrob. Agents Chemother. 2014, 58, 5719–5725. [Google Scholar] [CrossRef]
- Lauth, X.; Shike, H.; Burns, J.C.; Westerman, M.E.; Ostland, V.E.; Carlberg, J.M.; Van Olst, J.C.; Nizet, V.; Taylor, S.W.; Shimizu, C.; et al. Discovery and characterization of two isoforms of moronecidin, a novel antimicrobial peptide from hybrid striped bass. J. Biol. Chem. 2002, 277, 5030–5039. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, Y.K.; Lim, S.S.; Zhu, W.L.; Ko, H.; Shin, S.Y.; Hahm, K.S.; Kim, Y. Solution structure and cell selectivity of piscidin 1 and its analogues. Biochemistry 2007, 46, 3653–3663. [Google Scholar] [CrossRef]
- Wang, G.; Watson, K.M.; Peterkofsky, A.; Buckheit, R.W. Identification of novel human immunodeficiency virus type 1-inhibitory peptides based on the antimicrobial peptide database. Antimicrob. Agents Chemother. 2010, 54, 1343–1346. [Google Scholar] [CrossRef]
- Chen, W.; Cotten, M.L. Expression, purification, and micelle reconstitution of antimicrobial piscidin 1 and piscidin 3 for NMR studies. Protein Expr. Purif. 2014, 102, 63–68. [Google Scholar] [CrossRef]
- Hayden, R.M.; Goldberg, G.K.; Ferguson, B.M.; Schoeneck, M.W.; Libardo, M.D.; Mayeux, S.E.; Shrestha, A.; Bogardus, K.A.; Hammer, J.; Pryshchep, S.; et al. Complementary effects of host defense peptides Piscidin 1 and Piscidin 3 on DNA and lipid membranes: Biophysical insights into contrasting biological activities. J. Phys. Chem. B 2015, 119, 15235–15246. [Google Scholar] [CrossRef]
- Libardo, M.D.J.; Bahar, A.A.; Ma, B.; Fu, R.; McCormick, L.E.; Zhao, J.; McCallum, S.A.; Nussinov, R.; Ren, D.; Angeles-Boza, A.M.; et al. Nuclease activity gives an edge to host-defense peptide piscidin 3 over piscidin 1, rendering it more effective against persisters and biofilms. Febs. J. 2017, 284, 3662–3683. [Google Scholar] [CrossRef]
- Comert, F.; Greenwood, A.; Maramba, J.; Acevedo, R.; Lucas, L.; Kulasinghe, T.; Cairns, L.S.; Wen, Y.; Fu, R.; Hammer, J.; et al. The host-defense peptide piscidin P1 reorganizes lipid domains in membranes and alters activation energies in mechanosensitive ion channels. J. Biol. Chem. 2019, in press. [Google Scholar] [CrossRef]
- Kim, S.Y.; Zhang, F.; Gong, W.; Chen, K.; Xia, K.; Liu, F.; Gross, R.; Wang, J.M.; Linhardt, R.J.; Cotten, M.L. Copper regulates the interactions of antimicrobial piscidin peptides from fish mast cells with formyl peptide receptors and heparin. J. Biol. Chem. 2018, 293, 15381–15396. [Google Scholar] [CrossRef] [Green Version]
- Joyner, J.C.; Reichfield, J.; Cowan, J.A. Factors influencing the DNA nuclease activity of iron, cobalt, nickel, and copper chelates. J. Am. Chem. Soc. 2011, 133, 15613–15626. [Google Scholar] [CrossRef]
- Joyner, J.C.; Hodnick, W.F.; Cowan, A.S.; Tamuly, D.; Boyd, R.; Cowan, J.A. Antimicrobial metallopeptides with broad nuclease and ribonuclease Activity. Chem. Commun. 2013, 49, 2118–2120. [Google Scholar] [CrossRef]
- Libardo, M.D.; Nagella, S.; Lugo, A.; Pierce, S.; Angeles-Boza, A.M. Copper-binding tripeptide motif increases potency of the antimicrobial peptide Anoplin via Reactive Oxygen Species generation. Biochem. Biophys. Res. Commun. 2015, 456, 446–451. [Google Scholar] [CrossRef]
- Hillmann, F.; Fischer, R.J.; Saint-Prix, F.; Girbal, L.; Bahl, H. PerR acts as a switch for oxygen tolerance in the strict anaerobe Clostridium acetobutylicum. Mol. Microbiol. 2008, 68, 848–860. [Google Scholar] [CrossRef]
- Riebe, O.; Fischer, R.J.; Wampler, D.A.; Kurtz, D.M.; Bahl, H. Pathway for H2O2 and O2 detoxification in Clostridium acetobutylicum. Microbiology 2009, 155, 16–24. [Google Scholar] [CrossRef]
- Zhang, L.; Nie, X.; Ravcheev, D.A.; Rodionov, D.A.; Sheng, J.; Gu, Y.; Yang, S.; Jiang, W.; Yang, C. Redox-responsive repressor Rex modulates alcohol production and oxidative stress tolerance in Clostridium acetobutylicum. J. Bacteriol. 2014, 196, 3949–3963. [Google Scholar] [CrossRef]
- McQuade, R.; Roxas, B.; Viswanathan, V.K.; Vedantam, G. Clostridium difficile clinical isolates exhibit variable susceptibility and proteome alterations upon exposure to mammalian cationic antimicrobial peptides. Anaerobe 2012, 18, 614–620. [Google Scholar] [CrossRef]
- Corminboeuf, O.; Leroy, X. FPR2/ALXR agonists and the resolution of inflammation. J. Med. Chem. 2015, 58, 537–559. [Google Scholar] [CrossRef]
- Le, Y.; Murphy, P.M.; Wang, J.M. Formyl-peptide receptors revisited. Trends Immunol. 2002, 23, 541–548. [Google Scholar] [CrossRef]
- Migeotte, I.; Communi, D.; Parmentier, M. Formyl peptide receptors: A promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006, 17, 501–519. [Google Scholar] [CrossRef]
- Pundir, P.; Catalli, A.; Leggiadro, C.; Douglas, S.E.; Kulka, M. Pleurocidin, a novel antimicrobial peptide, induces human mast cell activation through the FPRL1 receptor. Mucosal. Immunol. 2014, 7, 177–187. [Google Scholar] [CrossRef]
- Park, Y.J.; Lee, S.K.; Jung, Y.S.; Lee, M.; Lee, H.Y.; Kim, S.D.; Park, J.S.; Koo, J.; Hwang, J.S.; Bae, Y.S. Promotion of formyl peptide receptor 1-mediated neutrophil chemotactic migration by antimicrobial peptides isolated from the centipede Scolopendra subspinipes mutilans. BMB Rep. 2016, 49, 520–525. [Google Scholar] [CrossRef]
- Chen, W.F.; Huang, S.Y.; Liao, C.Y.; Sung, C.S.; Chen, J.Y.; Wen, Z.H. The use of the antimicrobial peptide piscidin (PCD)-1 as a novel anti-nociceptive agent. Biomaterials 2015, 53, 1–11. [Google Scholar] [CrossRef]
- Lee, E.; Shin, A.; Jeong, K.W.; Jin, B.; Jnawali, H.N.; Shin, S.; Shin, S.Y.; Kim, Y. Role of phenylalanine and valine10 residues in the antimicrobial activity and cytotoxicity of piscidin-1. PLoS ONE 2014, 9, e114453. [Google Scholar] [CrossRef]
- Mansour, S.C.; de la Fuente-Nunez, C.; Hancock, R.E. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 2013, 9, 761–768. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Unifying themes in host defence effector polypeptides. Nat. Rev. Microbiol. 2007, 5, 727–740. [Google Scholar] [CrossRef]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef]
- Hale, J.D.; Hancock, R.E. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti. Infect 2007, 5, 951–959. [Google Scholar] [CrossRef]
- Hing, T.C.; Ho, S.; Shih, D.Q.; Ichikawa, R.; Cheng, M.; Chen, J.; Chen, X.; Law, I.; Najarian, R.; Kelly, C.P.; et al. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut 2013, 62, 1295–1305. [Google Scholar] [CrossRef]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Buckley, A.M.; Jukes, C.; Candlish, D.; Irvine, J.J.; Spencer, J.; Fagan, R.P.; Roe, A.J.; Christie, J.M.; Fairweather, N.F.; Douce, G.R. Lighting up Clostridium difficile: Reporting gene expression using fluorescent LOV domains. Sci. Rep. 2016, 6, 23463. [Google Scholar] [CrossRef]
- Courson, D.S.; Pokhrel, A.; Scott, C.; Madrill, M.; Rinehold, A.J.; Tamayo, R.; Cheney, R.E.; Purcell, E.B. Single cell analysis of nutrient regulation of Clostridioides (Clostridium) difficile motility. Anaerobe 2019, 59, 205–211. [Google Scholar] [CrossRef]
- Hussain, H.A.; Roberts, A.P.; Mullany, P. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J. Med. Microbiol. 2005, 54, 137–141. [Google Scholar] [CrossRef]
- Stabler, R.A.; He, M.; Dawson, L.; Martin, M.; Valiente, E.; Corton, C.; Lawley, T.D.; Sebaihia, M.; Quail, M.A.; Rose, G.; et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009, 10, R102. [Google Scholar] [CrossRef]
- Lachowicz, D.; Pituch, H.; Obuch-Woszczatyński, P. Antimicrobial susceptibility patterns of Clostridium difficile strains belonging to different polymerase chain reaction ribotypes isolated in Poland in 2012. Anaerobe 2015, 31, 37–41. [Google Scholar] [CrossRef]
- Kuijper, E.J.; Coignard, B.; Tull, P. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin. Microbiol. Infect. 2006, 12, 2–18. [Google Scholar] [CrossRef]
- Drudy, D.; Kyne, L.; O’Mahony, R.; Fanning, S. gyrA Mutations in Fluoroquinolone-resistant Clostridium difficile PCR-027. Emerg. Infect. Dis. 2007, 13, 504–505. [Google Scholar] [CrossRef]
- Banawas, S.S. Clostridium difficile infections: A global overview of drug sensitivity and resistance mechanisms. Biomed. Res. Int. 2018, 2018, 9. [Google Scholar] [CrossRef]
- Libardo, M.D.; Cervantes, J.L.; Salazar, J.C.; Angeles-Boza, A.M. Improved bioactivity of antimicrobial peptides by addition of amino-terminal copper and nickel (ATCUN) binding motifs. Chem. Med. Chem. 2014, 9, 1892–1901. [Google Scholar] [CrossRef]
- Jin, Y.; Lewis, M.A.; Gokhale, N.H.; Long, E.C.; Cowan, J.A. Influence of stereochemistry and redox potentials on the single- and double-strand DNA cleavage efficiency of Cu(II) and Ni(II) Lys-Gly-His-derived ATCUN metallopeptides. J. Am. Chem. Soc. 2007, 129, 8353–8361. [Google Scholar] [CrossRef]
- Fernandez-Mazarrasa, C.; Mazarrasa, O.; Calvo, J.; del Arco, A.; Martinez-Martinez, L. High concentrations of manganese in Mueller-Hinton agar increase MICs of tigecycline determined by Etest. J. Clin. Microbiol. 2009, 47, 827–829. [Google Scholar] [CrossRef]
- Manteca, A.; Alvarez, R.; Salazar, N.; Yague, P.; Sanchez, J. Mycelium differentiation and antibiotic production in submerged cultures of Streptomyces coelicolor. Appl. Env. Microbiol. 2008, 74, 3877–3886. [Google Scholar] [CrossRef]
- Poole, K. Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 2012, 67, 2069–2089. [Google Scholar] [CrossRef] [Green Version]
- Sorg, J.A.; Dineen, S.S. Laboratory maintenance of Clostridium difficile. Curr. Protoc. Microbiol. 2009, 12, 1–10. [Google Scholar] [CrossRef]
- Purcell, E.B.; McKee, R.W.; McBride, S.M.; Waters, C.M.; Tamayo, R. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J. Bact. 2012, 194, 3307–3316. [Google Scholar] [CrossRef]
- Chekmenev, E.Y.; Vollmar, B.S.; Forseth, K.T.; Manion, M.N.; Jones, S.M.; Wagner, T.J.; Endicott, R.M.; Kyriss, B.P.; Homem, L.M.; Pate, M.; et al. Investigating molecular recognition and biological function at interfaces using piscidins, antimicrobial peptides from fish. Biochim. Biophys. Acta 2006, 1758, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Perrin, B.S.; Tian, Y.; Fu, R.; Grant, C.V.; Chekmenev, E.Y.; Wieczorek, W.E.; Dao, A.E.; Hayden, R.M.; Burzynski, C.M.; Venable, R.M.; et al. High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion. J. Am. Chem. Soc. 2014, 136, 3491–3504. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Ghaedi, M.; Ahmadi, F.; Shokrollahi, A. Simultaneous preconcentration and determination of copper, nickel, cobalt and lead ions content by flame atomic absorption spectrometry. J. Hazard. Mater. 2007, 142, 272–278. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oludiran, A.; Courson, D.S.; Stuart, M.D.; Radwan, A.R.; Poutsma, J.C.; Cotten, M.L.; Purcell, E.B. How Oxygen Availability Affects the Antimicrobial Efficacy of Host Defense Peptides: Lessons Learned from Studying the Copper-Binding Peptides Piscidins 1 and 3. Int. J. Mol. Sci. 2019, 20, 5289. https://doi.org/10.3390/ijms20215289
Oludiran A, Courson DS, Stuart MD, Radwan AR, Poutsma JC, Cotten ML, Purcell EB. How Oxygen Availability Affects the Antimicrobial Efficacy of Host Defense Peptides: Lessons Learned from Studying the Copper-Binding Peptides Piscidins 1 and 3. International Journal of Molecular Sciences. 2019; 20(21):5289. https://doi.org/10.3390/ijms20215289
Chicago/Turabian StyleOludiran, Adenrele, David S. Courson, Malia D. Stuart, Anwar R. Radwan, John C. Poutsma, Myriam L. Cotten, and Erin B. Purcell. 2019. "How Oxygen Availability Affects the Antimicrobial Efficacy of Host Defense Peptides: Lessons Learned from Studying the Copper-Binding Peptides Piscidins 1 and 3" International Journal of Molecular Sciences 20, no. 21: 5289. https://doi.org/10.3390/ijms20215289