Development and Progression of Non-Alcoholic Fatty Liver Disease: The Role of Advanced Glycation End Products


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
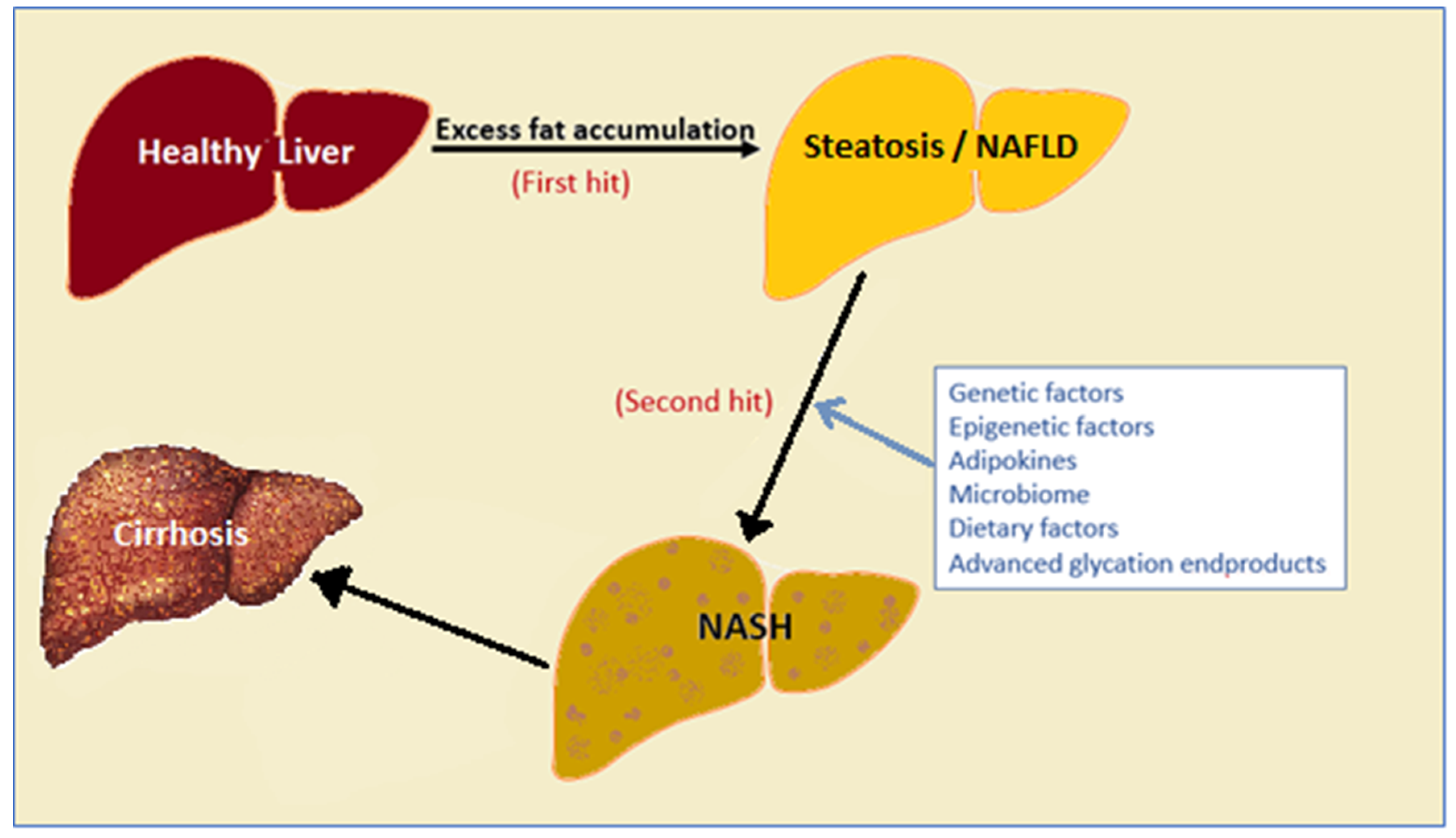
:1. Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Prevalence
2. Factors That Drive NAFLD Progression
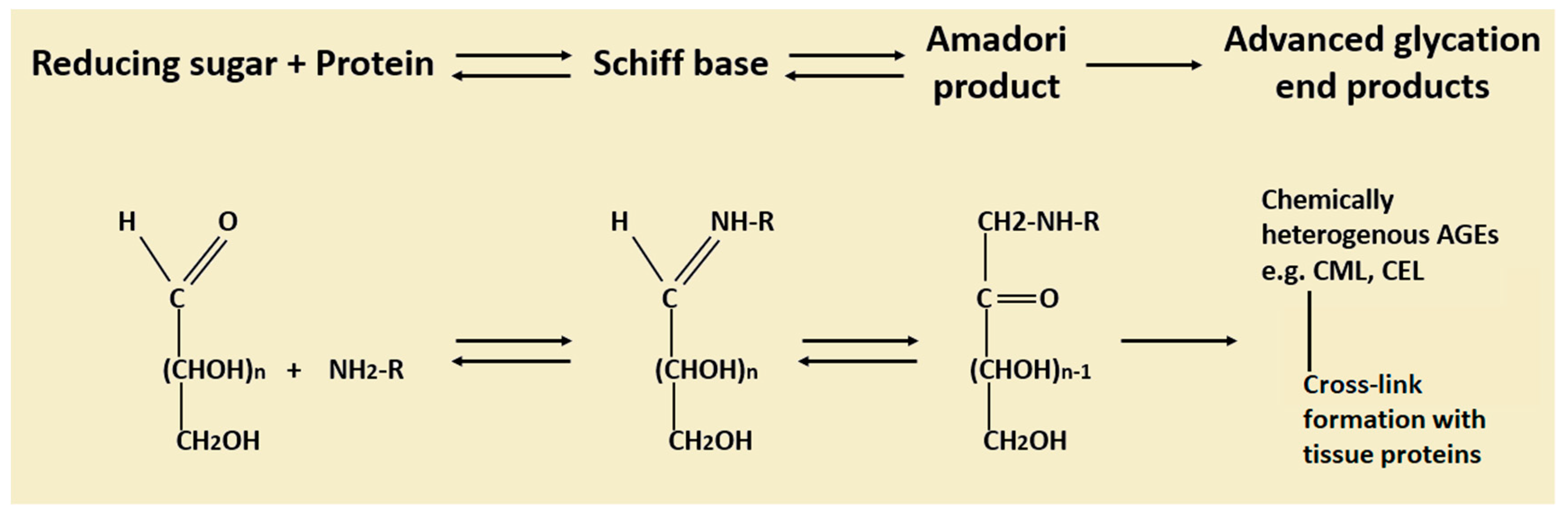
3. Advanced Glycation End Products (AGEs): Formation, Metabolism and Their Role in Diseases
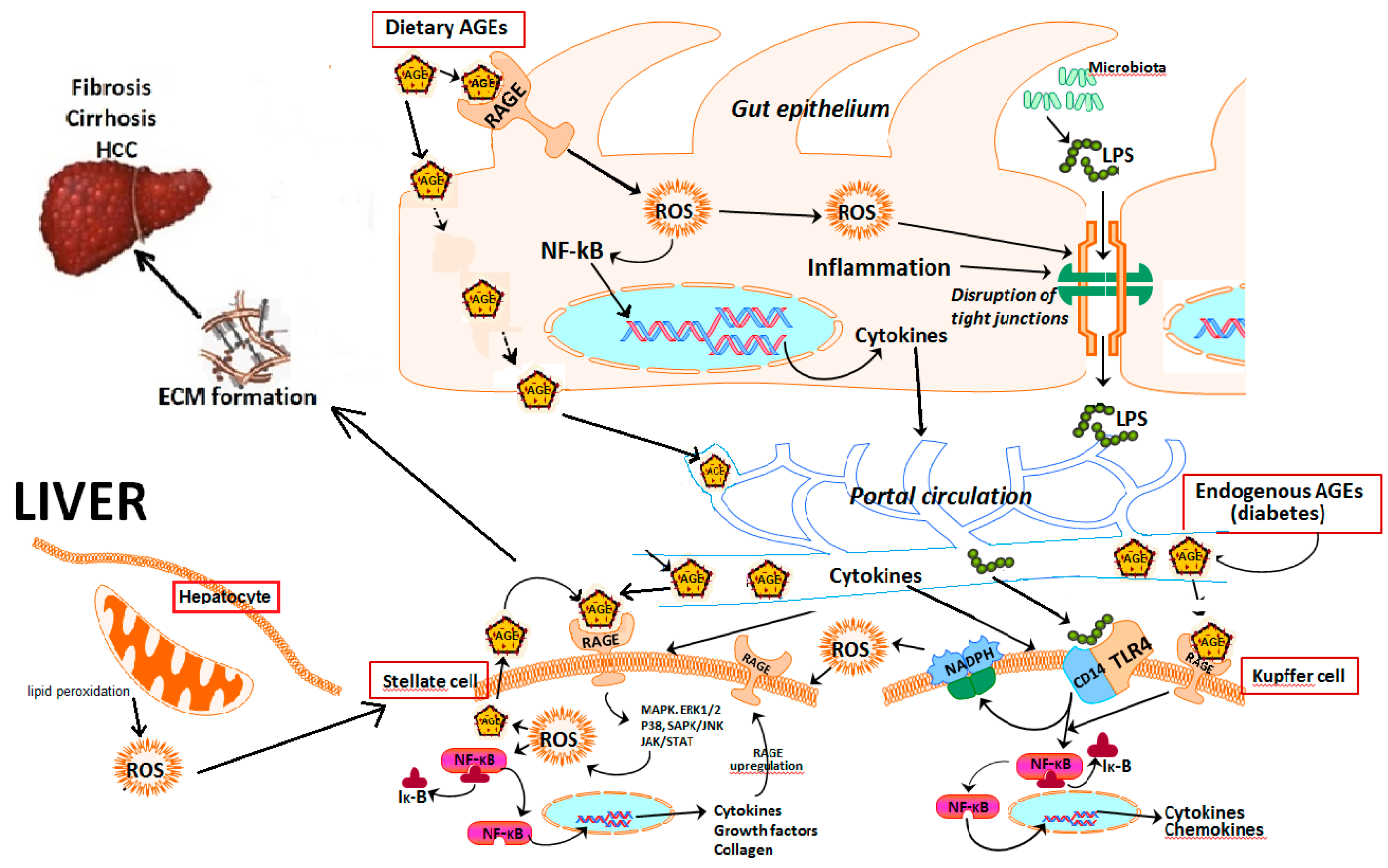
4. Advanced Glycation End Products Receptors (RAGEs) and Cell Signaling
5. Role of Hepatic Cells in AGE–RAGE Axis-Induced NAFLD Progression
6. Primary Sources of Advanced Glycation End Products
6.1. Endogenous AGEs
6.2. Exogenous AGEs
7. Role of Advanced Glycation End Products in NAFLD Progression
8. Targeting the AGE/RAGE Axis in NAFLD Progression to Liver Fibrosis
8.1. Impact of Dietary Modifications on NAFLD Progression
8.2. AGE Inhibitors
8.3. Inhibitors of RAGE or AGE/RAGE Binding
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.G.; Kim, S.U.; Wong, V.W.S. New trends on obesity and NAFLD in Asia. J. Hepatol. 2017, 67, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Yeh, M.M.; Brunt, E.M. Pathology of Nonalcoholic Fatty Liver Disease. Am. J. Clin. Pathol. 2007, 128, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolisml 2016, 65, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Wilson, L.; Kleiner, D.E.; Cummings, O.W.; Brunt, E.M.; Ünalp, A. Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Heurtier, A.; Gombert, S.; Giral, P.; Bruckert, E.; Grimaldi, A.; Capron, F.; Poynard, T. Sampling Variability of Liver Biopsy in Nonalcoholic Fatty Liver Disease. Gastroenterology 2005, 128, 1898–1906. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two hits? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Dajani, A.; AbuHammour, A. Treatment of nonalcoholic fatty liver disease: Where do we stand? an overview. Saudi J. Gastroenterol. 2016, 22, 91–105. [Google Scholar] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L.J.D. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Franke, S.; Rüster, C.; Wolf, G. Advanced glycation end-products and the kidney. Eur. J. Clin. Investig. 2010, 40, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilbaud, A.; Niquet-Leridon, C.; Boulanger, E.; Tessier, F.J. How Can Diet Affect the Accumulation of Advanced Glycation End-Products in the Human Body? Foods 2016, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Araki, N.; Higashi, T.; Mori, T.; Shibayama, R.; Kawabe, Y.; Kodama, T.; Takahashi, K.; Shichiri, M.; Horiuchi, S. Macrophage Scavenger Receptor Mediates the Endocytic Uptake and Degradation of Advanced Glycation End Products of the Maillard Reaction. Eur. J. Biochem. 1995, 230, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Ueda, Y.; Horie, K.; Nangaku, M.; Tanaka, S.; de Strihou, C.V.Y.; Kurokawa, K. Renal catabolism of advanced glycation end products: The fate of pentosidine. Kidney Int. 1998, 53, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Smedsrød, B.; Melkko, J.; Araki, N.; Sano, H.; Horiuchi, S. Advanced glycation end products are eliminated by scavenger-receptor-mediated endocytosis in hepatic sinusoidal Kupffer and endothelial cells. Biochem. J. 1997, 322, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Svistounov, D.; Oteiza, A.; Zykova, S.N.; Sørensen, K.K.; McCourt, P.; McLachlan, A.J.; McCuskey, R.S.; Smedsrød, B. Hepatic disposal of advanced glycation end products during maturation and aging. Exp. Gerontol. 2013, 48, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Hofmann, M.A.; Nawroth, P.P.; Ziegler, R. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc. Res. 1998, 37, 586–600. [Google Scholar] [CrossRef]
- Sims, T.J.; Rasmussen, L.M.; Oxlund, H.; Bailey, A.J.J.D. The role of glycation cross-links in diabetic vascular stiffening. Diabetologia 1996, 39, 946. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Glomb, M.; Elgawish, A.; Sell, D.R. The mechanism of collagen cross-linking in diabetes: A puzzle nearing resolution. Diabetes 1996, 45, S67–S72. [Google Scholar] [CrossRef] [PubMed]
- Gautieri, A.; Passini, F.S.; Silván, U.; Guizar-Sicairos, M.; Carimati, G.; Volpi, P.; Moretti, M.; Schoenhuber, H.; Redaelli, A.; Berli, M.; et al. Advanced glycation end-products: Mechanics of aged collagen from molecule to tissue. Matrix Biol. 2017, 59, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J. Clin. Investig. 1991, 87, 432–438. [Google Scholar] [CrossRef]
- Aso, Y.; Inukai, T.; Tayama, K.; Takemura, Y.J.A.D. Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol. 2000, 37, 87–92. [Google Scholar] [CrossRef]
- Hyogo, H.; Yamagishi, S.I. Advanced Glycation End Products (AGEs) and their Involvement in Liver Disease. Curr. Pharm. Des. 2008, 14, 969–972. [Google Scholar] [CrossRef]
- Stitt, A.W.; He, C.; Vlassara, H. Characterization of the Advanced Glycation End-Product Receptor Complex in Human Vascular Endothelial Cells. Biochem. Biophys. Res. Commun. 1999, 256, 549–556. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar] [PubMed]
- Younessi, P.; Yoonessi, A. Advanced glycation end-products and their receptor-mediated roles: Inflammation and oxidative stress. Iran. J. Med. Sci. 2011, 36, 154–166. [Google Scholar] [PubMed]
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-Associated Sustained Activation of the Transcription Factor Nuclear Factor-κB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed]
- Schwenger, V.; Morath, C.; Salava, A.; Amann, K.; Seregin, Y.; Deppisch, R.; Ritz, E.; Bierhaus, A.; Nawroth, P.P.; Zeier, M. Damage to the Peritoneal Membrane by Glucose Degradation Products Is Mediated by the Receptor for Advanced Glycation End-Products. J. Am. Soc. Nephrol. 2006, 17, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S.; et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 2003, 162, 1123–1137. [Google Scholar] [CrossRef]
- Adams, L.A.; Angulo, P. Treatment of non-alcoholic fatty liver disease. Postgrad. Med. J. 2006, 82, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.I.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef]
- Shen, C.; Ma, Y.; Zeng, Z.; Yin, Q.; Hong, Y.; Hou, X.; Liu, X. RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem. Res. 2017, 42, 2902–2911. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, G.; Cao, X.; Dong, J.; Song, F.; Niu, Y. Blocking AGE-RAGE Signaling Improved Functional Disorders of Macrophages in Diabetic Wound. J. Diabetes Res. 2017, 2017, 1428537. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Herold, K.C.; Schmidt, A.M. Receptor for advanced glycation end products and its ligands: Initiators or amplifiers of joint inflammation—A bit of both? Arthritis Rheum. 2006, 54, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, Y.; Lagache, S.M.M.; Schnack, L.; Godfrey, R.; Kahles, F.; Bruemmer, D.; Waltenberger, J.; Findeisen, H.M. mTOR-Dependent Oxidative Stress Regulates oxLDL-Induced Trained Innate Immunity in Human Monocytes. Front. Immunol. 2019, 9, 3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Kakehashi, A.; Ishii, N.; Fujioka, M.; Gi, M.; Wanibuchi, H. mTOR Activation in Liver Tumors Is Associated with Metabolic Syndrome and Non-Alcoholic Steatohepatitis in Both Mouse Models and Humans. Cancers 2018, 10, 465. [Google Scholar] [CrossRef]
- Zhuang, A.; Forbes, J.M. Diabetic kidney disease: A role for advanced glycation end-product receptor 1 (AGE-R1)? Glycoconj. J. 2016, 33, 645–652. [Google Scholar] [CrossRef]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A.; et al. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004, 23, 4096–4105. [Google Scholar] [CrossRef]
- Forough, R.; Lindner, L.; Partridge, C.; Jones, B.; Guy, G.; Clark, G. Elevated 80K-H protein in breast cancer: A role for FGF-1 stimulation of 80K-H. Int. J. Biol. Mark. 2003, 18, 89–98. [Google Scholar] [CrossRef]
- Strieter, R.M.; Keane, M.P. Innate immunity dictates cytokine polarization relevant to the development of pulmonary fibrosis. J. Clin. Investig. 2004, 114, 165–168. [Google Scholar] [CrossRef]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef] [Green Version]
- Barrera, F.; George, J. The Role of Diet and Nutritional Intervention for the Management of Patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.; Bolender, R.P.; Weibel, E.R. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J. Cell Biol. 1977, 72, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Geerts, A.; Niki, T.; Hellemans, K.; De Craemer, D.; Van Den Berg, K.; Lazou, J.M.; Stange, G.; Van De Winkel, M.; De Bleser, P. Purification of rat hepatic stellate cells by side scatter–activated cell sorting. Hepatology 1998, 27, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J. Submicrosc. Cytol. 1992, 24, 193–203. [Google Scholar]
- Fehrenbach, H.; Weiskirchen, R.; Kasper, M.; Gressner, A.M. Up-regulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology 2001, 34, 943–952. [Google Scholar] [CrossRef]
- Guimarães, E.L.M.; Empsen, C.; Geerts, A.; van Grunsven, L.A. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J. Hepatol. 2010, 52, 389–397. [Google Scholar] [CrossRef]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Leung, C.; Herath, C.B.; Jia, Z.; Andrikopoulos, S.; Brown, B.E.; Davies, M.J.; Rivera, L.R.; Furness, J.B.; Forbes, J.M.; Angus, P.W. Dietary advanced glycation end-products aggravate non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 8026–8040. [Google Scholar] [CrossRef] [Green Version]
- Lanthier, N. Targeting Kupffer cells in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Why and how? World J. Hepatol. 2015, 7, 2184–2188. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [PubMed]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenfeng, Z.; Wu, Y.; Di, M.; Gong, J.; Chuanxin, W.; Chun, H. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef]
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Takeuchi, M. The Relevance of Toxic AGEs (TAGE) Cytotoxicity to NASH Pathogenesis: A Mini-Review. Nutrients 2019, 11, 462. [Google Scholar] [CrossRef] [PubMed]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 2015, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.J.; Niessen, P.M.G.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.M.; Driessen, A.; Wolfs, M.G.M.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of Nε-(carboxymethyl) lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol. 2012, 56, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Gkogkolou, P.; Böhm, M. Advanced glycation end products: Key players in skin aging? Derm. Endocrinol. 2012, 4, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Y.; Reis, E.D.; Dore, A.T.; Lu, M.; Ghodsi, N.; Fallon, J.T.; Fisher, E.A.; Vlassara, H. Lowering of dietary advanced glycation endproducts (AGE) reduces neointimal formation after arterial injury in genetically hypercholesterolemic mice. Atherosclerosis 2002, 163, 303–311. [Google Scholar] [CrossRef]
- Jenkins, D.J.; Wolever, T.M.; Taylor, R.H.; Barker, H.; Fielden, H.; Baldwin, J.M.; Bowling, A.C.; Newman, H.C.; Jenkins, A.L.; Goff, D.V. Glycemic index of foods: A physiological basis for carbohydrate exchange. Am. J. Clin. Nutr. 1981, 34, 362–366. [Google Scholar] [CrossRef]
- Abid, A.; Taha, O.; Nseir, W.; Farah, R.; Grosovski, M.; Assy, N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J. Hepatol. 2009, 51, 918–924. [Google Scholar] [CrossRef]
- Nseir, W.; Nassar, F.; Assy, N. Soft drinks consumption and nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 2579–2588. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Liverini, G.; Iossa, S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur. J. Nutr. 2013, 52, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, E.A.; Chiu, C.-J.; Taylor, A. Dietary glycemia as a determinant of health and longevity. Mol. Asp. Med. 2015, 46, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiki, T.; Weikel, K.A.; Jiao, W.; Shang, F.; Caceres, A.; Pawlak, D.; Handa, J.T.; Brownlee, M.; Nagaraj, R.; Taylor, A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 2012, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kostolanska, J.; Jakuš, V.; Barak, L. HbA1c and serum levels of advanced glycation and oxidation protein products in poorly and well controlled children and adolescents with type 1 diabetes mellitus. J. Pediatr. Endocrinol. Metab. 2009, 22, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Baynes, J.W. Role of Oxidative Stress in Development of Complications in Diabetes. Diabetes 1991, 40, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.M.; Requena, J.R.; Crowley, J.R.; Thorpe, S.R.; Heinecke, J.W. The myeloperoxidase system of human phagocytes generates Nε-(carboxymethyl)lysine on proteins: A mechanism for producing advanced glycation end products at sites of inflammation. J. Clin. Investig. 1999, 104, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. 2007, 62, 427–433. [Google Scholar] [CrossRef]
- Leslie, R.D.G.; Beyan, H.; Sawtell, P.; Boehm, B.O.; Spector, T.D.; Snieder, H. Level of an Advanced Glycated End Product Is Genetically Determined. Study Norm. Twins 2003, 52, 2441–2444. [Google Scholar] [Green Version]
- Lund, M.N.; Ray, C.A. Control of Maillard Reactions in Foods: Strategies and Chemical Mechanisms. J. Agric. Food Chem. 2017, 65, 4537–4552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ames, J.M. Control of the Maillard reaction in food systems. Trends Food Sci. Technol. 1990, 1, 150–154. [Google Scholar] [CrossRef]
- Van Boekel, M.A.J.S. Kinetic aspects of the Maillard reaction: A critical review. Food/Nahr. 2001, 45, 150–159. [Google Scholar] [CrossRef]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Acad. Nutr. Diet. 2004, 104, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Coughlan, M.T. Effect of diet-derived advanced glycation end products on inflammation. Nutr. Rev. 2015, 73, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Šebeková, K.N.; Kupčová, V.; Schinzel, R.; Heidland, A. Markedly elevated levels of plasma advanced glycation end products in patients with liver cirrhosis—Amelioration by liver transplantation. J. Hepatol. 2002, 36, 66–71. [Google Scholar] [CrossRef]
- Yagmur, E.; Tacke, F.; Weiss, C.; Lahme, B.; Manns, M.P.; Kiefer, P.; Trautwein, C.; Gressner, A.M. Elevation of Nε-(carboxymethyl)lysine-modified advanced glycation end products in chronic liver disease is an indicator of liver cirrhosis. Clin. Biochem. 2006, 39, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Yamagishi, S.I.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 1112–1119. [Google Scholar] [CrossRef]
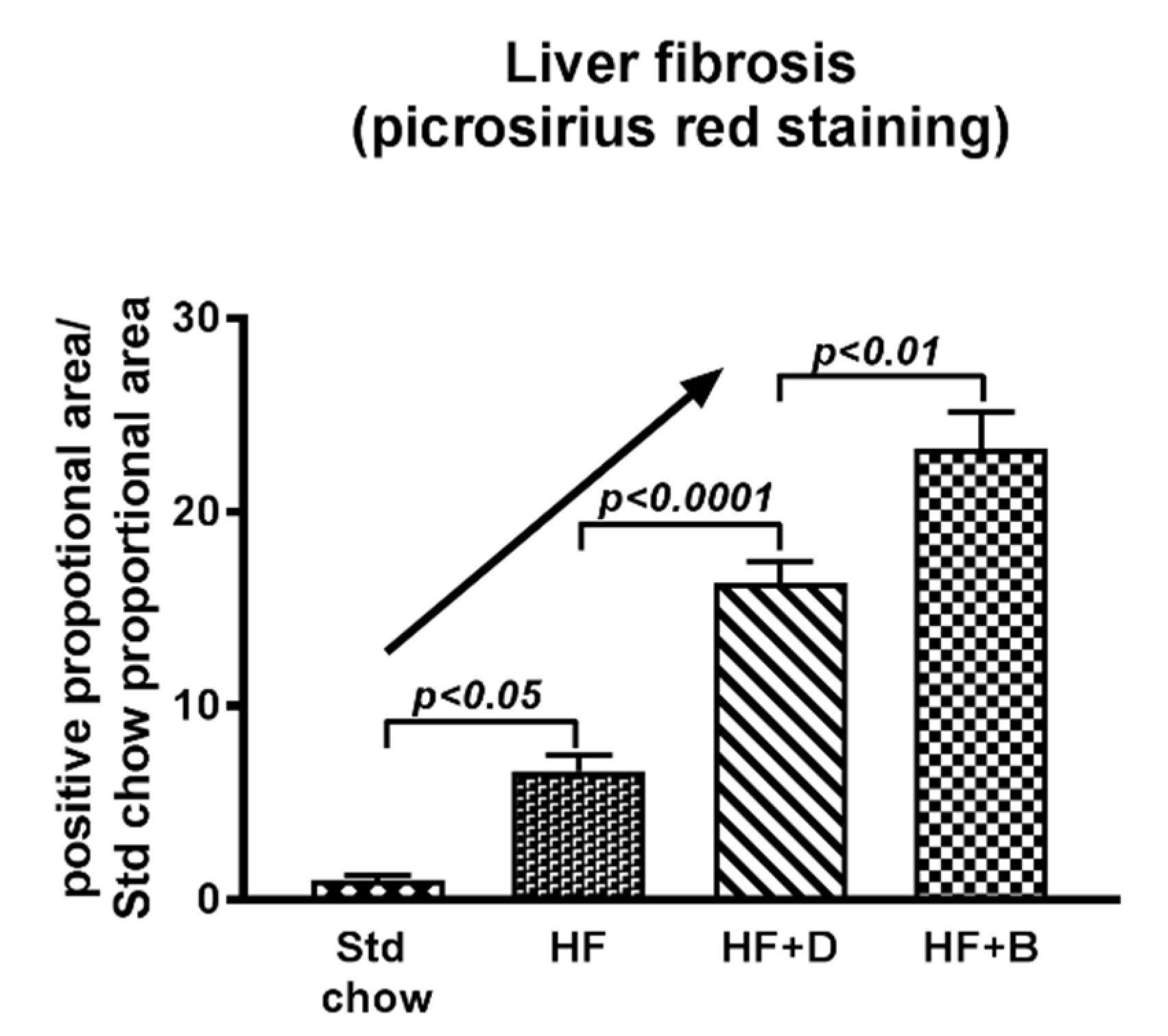
- Goodwin, M.; Herath, C.; Jia, Z.; Leung, C.; Coughlan, M.T.; Forbes, J.; Angus, P. Advanced glycation end products augment experimental hepatic fibrosis. J. Gastroenterol. Hepatol. 2013, 28, 369–376. [Google Scholar] [CrossRef]
- Sayej, W.N.; Knight Iii, P.R.; Guo, W.A.; Mullan, B.; Ohtake, P.J.; Davidson, B.A.; Khan, A.; Baker, R.D.; Baker, S.S. Advanced Glycation End Products Induce Obesity and Hepatosteatosis in CD-1 Wild-Type Mice. BioMed Res. Int. 2016, 2016, 7867852. [Google Scholar] [CrossRef] [PubMed]
- Izushi, Y.; Teshigawara, K.; Liu, K.; Wang, D.; Wake, H.; Takata, K.; Yoshino, T.; Takahashi, H.K.; Mori, S.; Nishibori, M. Soluble form of the receptor for advanced glycation end-products attenuates inflammatory pathogenesis in a rat model of lipopolysaccharide-induced lung injury. J. Pharmacol. Sci. 2016, 130, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, W.; Yuan, X.; Zhao, J.; Zhang, Y.; Hu, J.; Wang, J.; Li, J. Dietary advanced glycation end products modify gut microbial composition and partially increase colon permeability in rats. Mol. Nutr. Food Res. 2017, 61, 1700118. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Amp Hepatol. 2016, 13, 412. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Zhang, L.; Ji, G. Vitamin E Therapy in Non-Alcoholic Fatty Liver Disease. Sci. Res. 2014, 5, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Blaslov, K.; Bulum, T.; Zibar, K.; Duvnjak, L. Incretin based therapies: A novel treatment approach for non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7356–7365. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef]
- Mizrahi, M.; Shabat, Y.; Ben Ya’acov, A.; Lalazar, G.; Adar, T.; Wong, V.; Muller, B.; Rawlin, G.; Ilan, Y. Alleviation of insulin resistance and liver damage by oral administration of Imm124-E is mediated by increased Tregs and associated with increased serum GLP-1 and adiponectin: Results of a phase I/II clinical trial in NASH. J. Inflamm. Res. 2012, 5, 141–150. [Google Scholar]
- Henderson, N.C.; Sethi, T. The regulation of inflammation by galectin-3. Immunol. Rev. 2009, 230, 160–171. [Google Scholar] [CrossRef]
- Brownlee, M.; Vlassara, H.; Kooney, A.; Ulrich, P.; Cerami, A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science 1986, 232, 1629–1632. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; He, C.; Cai, W.; Hattori, M.; Steffes, M.; Vlassara, H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes/Metab. Res. Rev. 2002, 18, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C.; et al. RAGE Blockade Stabilizes Established Atherosclerosis in Diabetic Apolipoprotein E—Null Mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef] [PubMed]
- Roman, B.; Carta, L.; Martínez-González, M.A.; Serra-Majem, L. Effectiveness of the Mediterranean diet in the elderly. Clin. Interv. Aging 2008, 3, 97–109. [Google Scholar] [PubMed]
- Romagnolo, D.F.; Selmin, O.I. Mediterranean Diet and Prevention of Chronic Diseases. Nutr. Today 2017, 52, 208–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Quezada, I.; Román-Viñas, B.; Serra-Majem, L. The Mediterranean diet and nutritional adequacy: A review. Nutrients 2014, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, H.E.; Carbone, S. The antioxidant potential of the Mediterranean diet in patients at high cardiovascular risk: An in-depth review of the PREDIMED. Nutr. Diabetes 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- George, E.S.; Kucianski, T.; Mayr, H.L.; Moschonis, G.; Tierney, A.C.; Itsiopoulos, C. A Mediterranean Diet Model in Australia: Strategies for Translating the Traditional Mediterranean Diet into a Multicultural Setting. Nutrients 2018, 10, 465. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Mechanisms of Disease: Advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat. Clin. Pract. Endocrinol. Amp Metab. 2008, 4, 285. [Google Scholar] [CrossRef]
- Monnier, V.M.; Bautista, O.; Kenny, D.; Sell, D.R.; Fogarty, J.; Dahms, W.; Cleary, P.A.; Lachin, J.; Genuth, S. Skin collagen glycation, glycoxidation, and crosslinking are lower in subjects with long-term intensive versus conventional therapy of type 1 diabetes: Relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes 1999, 48, 870–880. [Google Scholar] [PubMed]
- Rahbar, S.; Natarajan, R.; Yerneni, K.; Scott, S.; Gonzales, N.; Nadler, J.L. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin. Chim. Acta 2000, 301, 65–77. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Engelen, L.; Stehouwer, C.D.A.; Schalkwijk, C.G. Current therapeutic interventions in the glycation pathway: Evidence from clinical studies. Diabetes Obes. Metab. 2013, 15, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Borg, D.J.; Forbes, J.M. Targeting advanced glycation with pharmaceutical agents: Where are we now? Glycoconj. J. 2016, 33, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.; Ruggiero-Lopez, D. Metformin inhibition of glycation processes. Diabetes Metab. 2003, 29, S95–S103. [Google Scholar] [CrossRef]
- Bhatwadekar, A.; Glenn, J.V.; Figarola, J.L.; Scott, S.; Gardiner, T.A.; Rahbar, S.; Stitt, A.W. A new advanced glycation inhibitor, LR-90, prevents experimental diabetic retinopathy in rats. Br. J. Ophthalmol. 2008, 92, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Ning, Z.; Chen, L.; Wei, Q.; Yuan, E.; Yang, J.; Ren, J. Intracellular Antioxidant Detoxifying Effects of Diosmetin on 2,2-Azobis(2-amidinopropane) Dihydrochloride (AAPH)-Induced Oxidative Stress through Inhibition of Reactive Oxygen Species Generation. J. Agric. Food Chem. 2014, 62, 8648–8654. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Kurokawa, K.; van Ypersele de Strihou, C. Advanced Glycation and Lipoxidation End Products. Role React. Carbonyl Compd Gener. Carbohydr. Lipid Metab. 2000, 11, 1744–1752. [Google Scholar]
- Koyama, H.; Yamamoto, H.; Nishizawa, Y. RAGE and soluble RAGE: Potential therapeutic targets for cardiovascular diseases. Mol. Med. 2007, 13, 625–635. [Google Scholar] [CrossRef]
- Monteiro, F.A.; Cardoso, I.; Sousa, M.M.; Saraiva, M.J. In vitro inhibition of transthyretin aggregate-induced cytotoxicity by full and peptide derived forms of the soluble receptor for advanced glycation end products (RAGE). FEBS Lett. 2006, 580, 3451–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, T.; Yan, S.F.; Yan, S.D.; Belov, D.; Rong, L.L.; Sousa, M.; Andrassy, M.; Marso, S.P.; Duda, S.; Arnold, B.; et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Investig. 2003, 111, 959–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE–amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Goova, M.T.; Li, J.; Kislinger, T.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Nowygrod, S.; Wolf, B.M.; Caliste, X.; Yan, S.F.; et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am. J. Pathol. 2001, 159, 513–525. [Google Scholar] [CrossRef]
- Barile, G.R.; Pachydaki, S.I.; Tari, S.R.; Lee, S.E.; Donmoyer, C.M.; Ma, W.; Rong, L.L.; Buciarelli, L.G.; Wendt, T.; Hörig, H.; et al. The RAGE Axis in Early Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Koyama, H.; Morioka, T.; Tanaka, S.; Kizu, A.; Motoyama, K.; Mori, K.; Fukumoto, S.; Shioi, A.; Shimogaito, N.; et al. Receptor for Advanced Glycation End Products Is Involved in Impaired Angiogenic Response in Diabetes. Diabetes 2006, 55, 2245–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernando, D.H.; Forbes, J.M.; Angus, P.W.; Herath, C.B. Development and Progression of Non-Alcoholic Fatty Liver Disease: The Role of Advanced Glycation End Products. Int. J. Mol. Sci. 2019, 20, 5037. https://doi.org/10.3390/ijms20205037
Fernando DH, Forbes JM, Angus PW, Herath CB. Development and Progression of Non-Alcoholic Fatty Liver Disease: The Role of Advanced Glycation End Products. International Journal of Molecular Sciences. 2019; 20(20):5037. https://doi.org/10.3390/ijms20205037
Chicago/Turabian StyleFernando, Dinali H., Josephine M. Forbes, Peter W. Angus, and Chandana B. Herath. 2019. "Development and Progression of Non-Alcoholic Fatty Liver Disease: The Role of Advanced Glycation End Products" International Journal of Molecular Sciences 20, no. 20: 5037. https://doi.org/10.3390/ijms20205037