Phenotype Analysis of Retinal Dystrophies in Light of the Underlying Genetic Defects: Application to Cone and Cone-Rod Dystrophies


 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Clinical and Genetic Characteristics
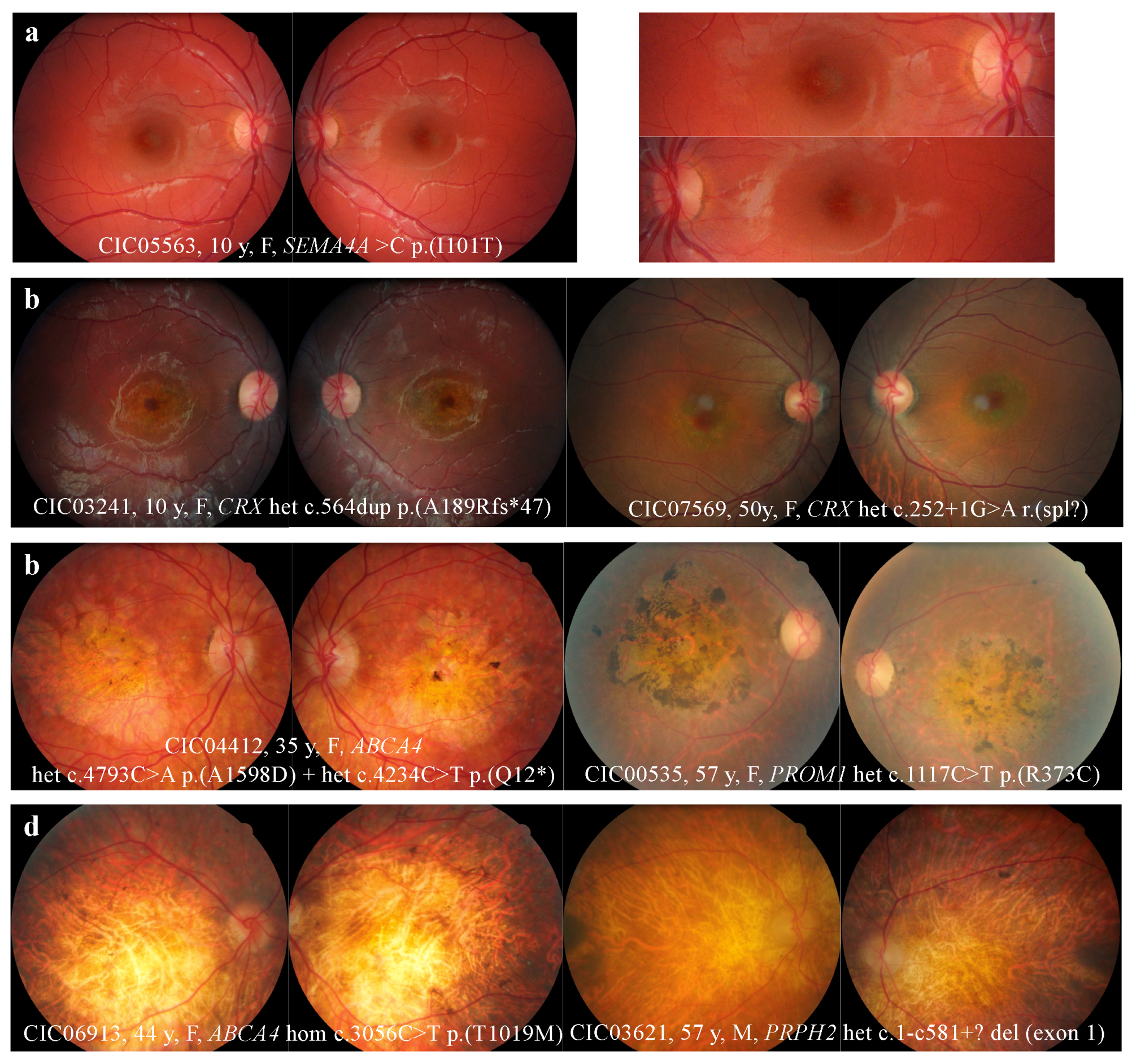
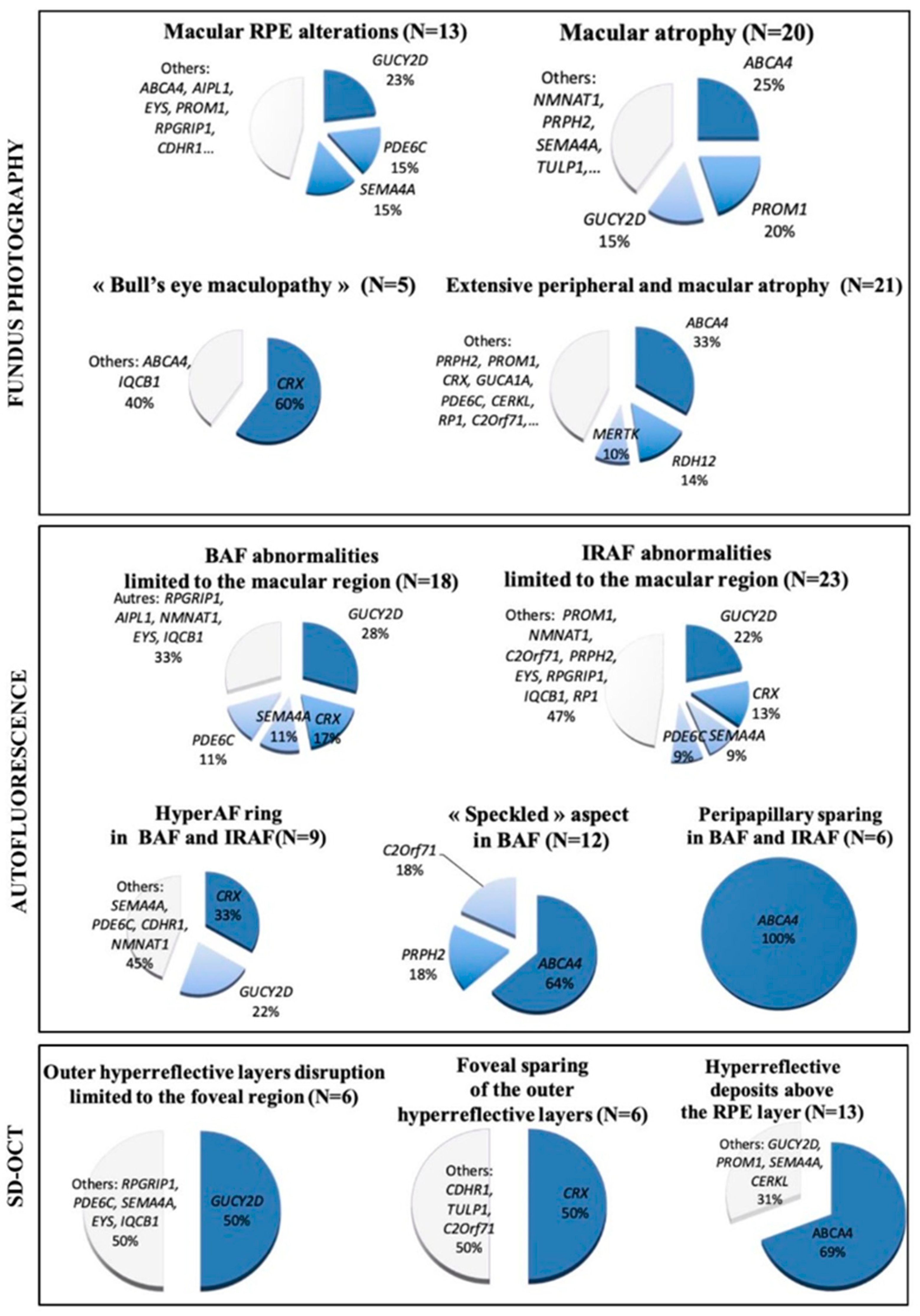
2.2. Retinophotography
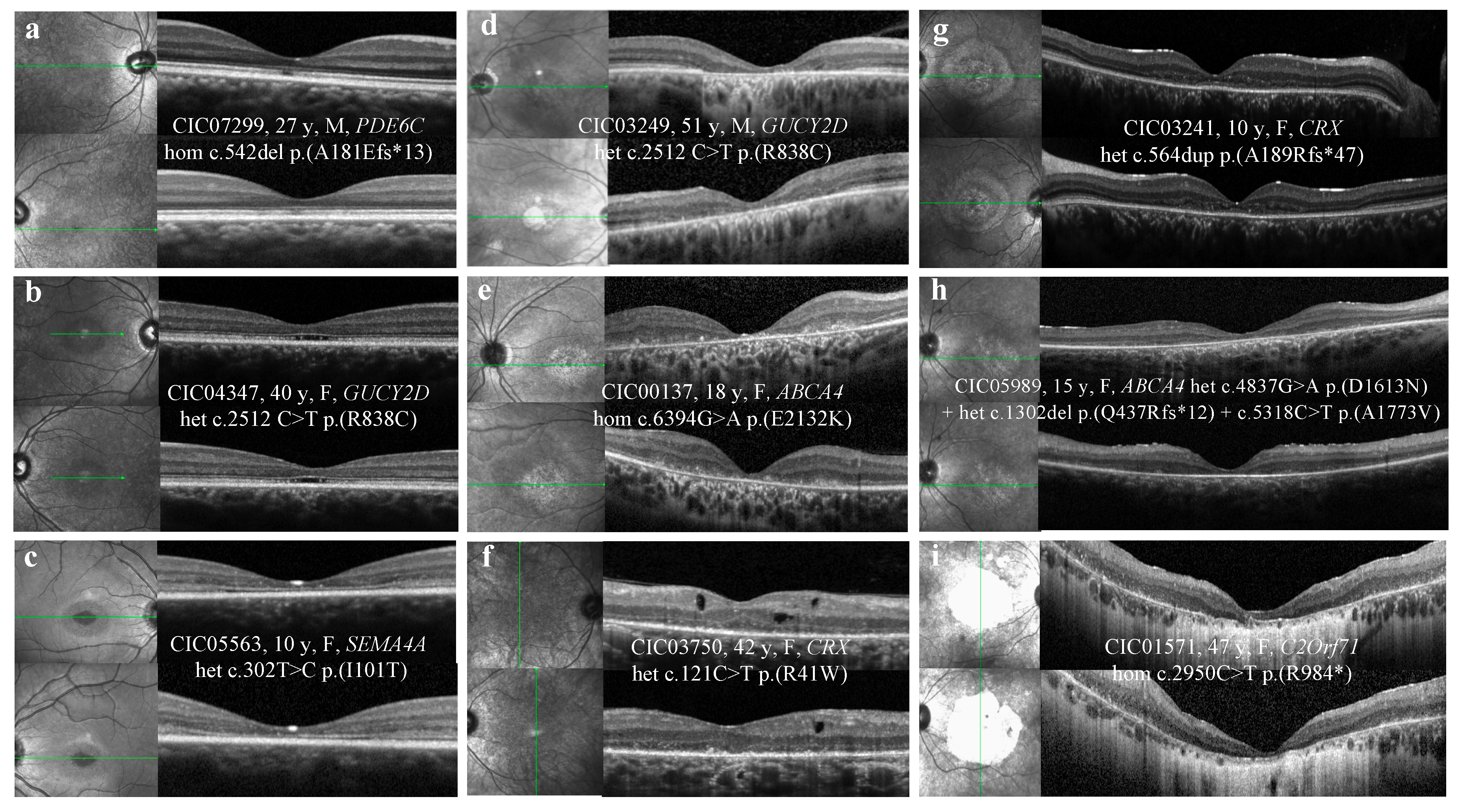
2.3. SD-OCT
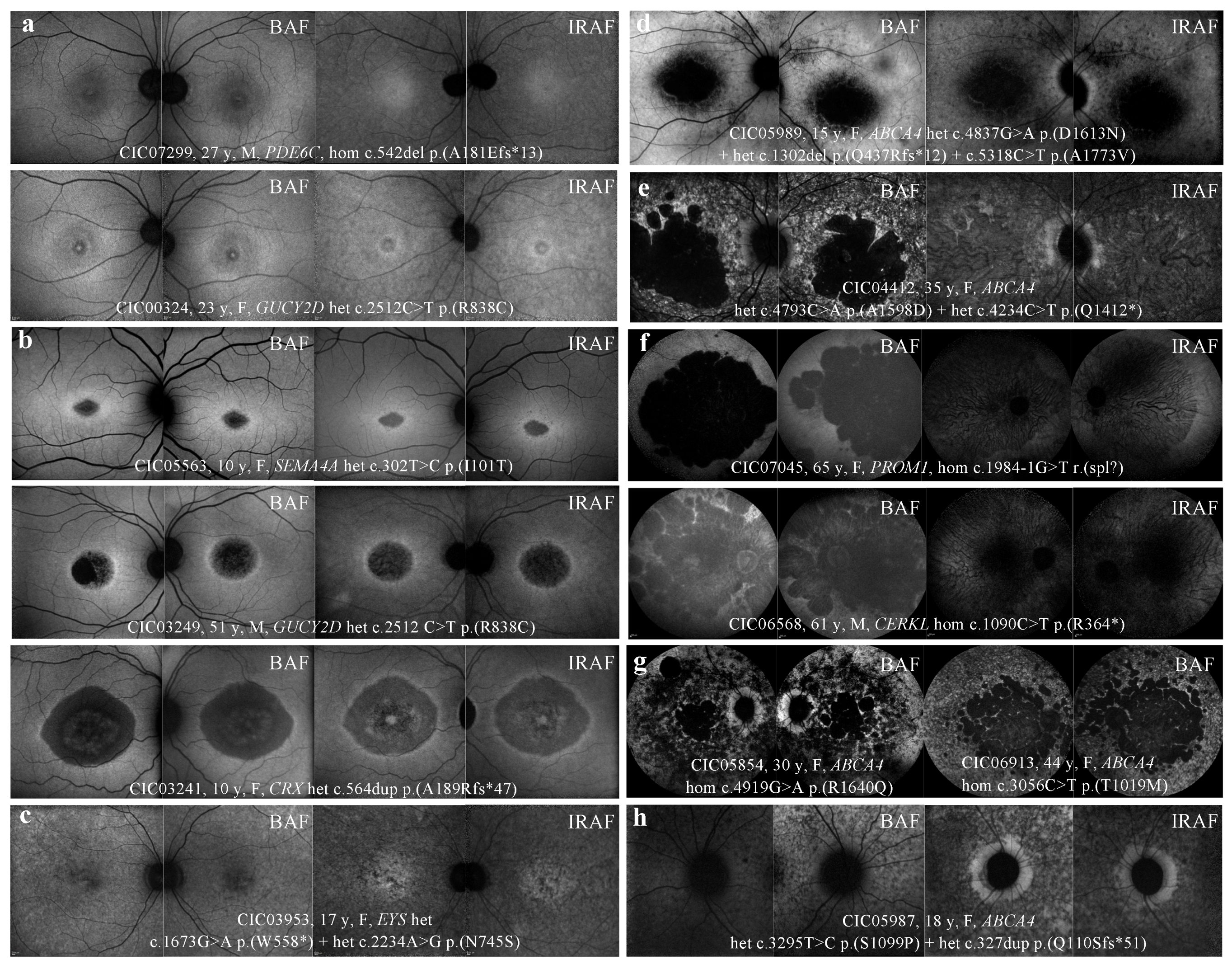
2.4. FAF
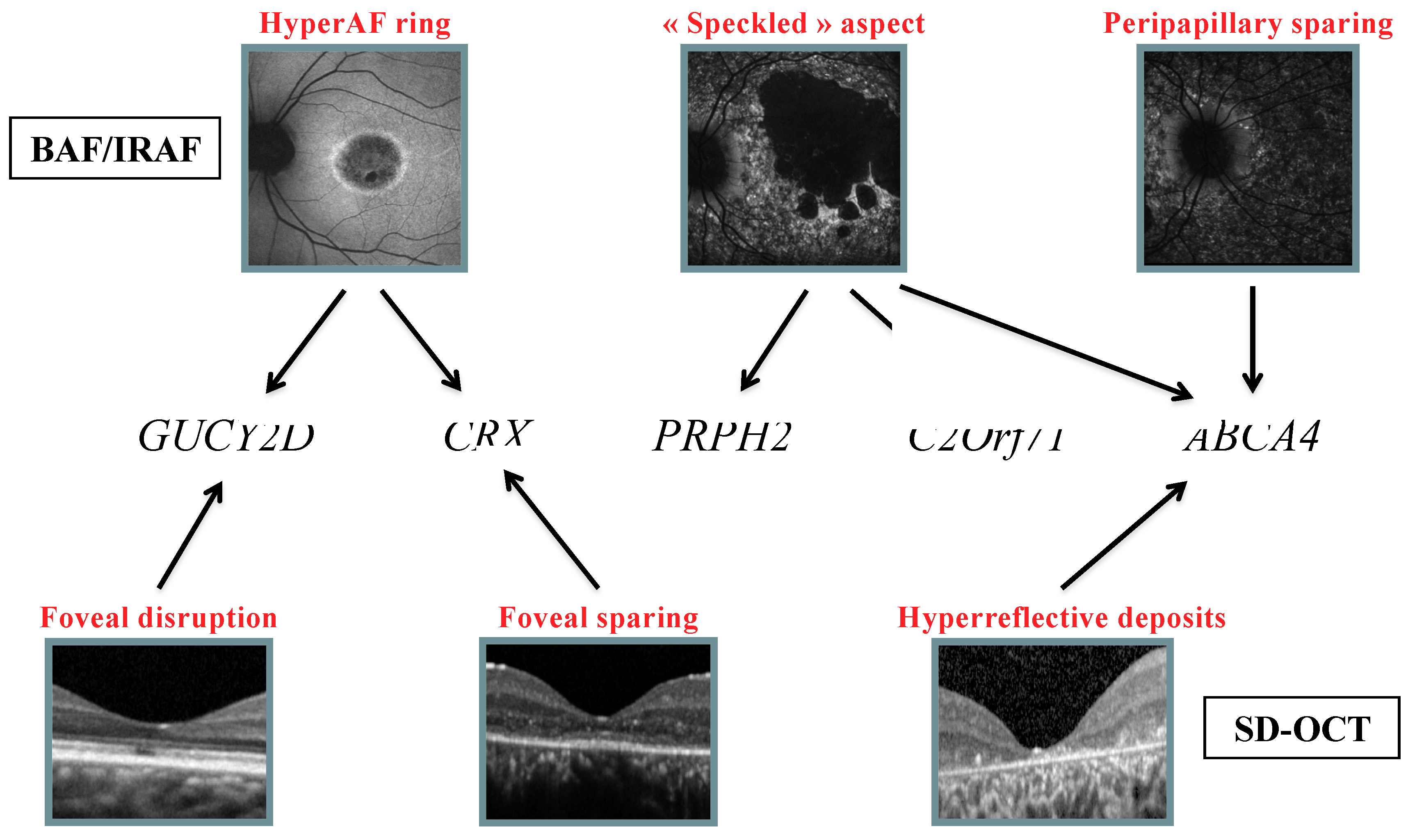
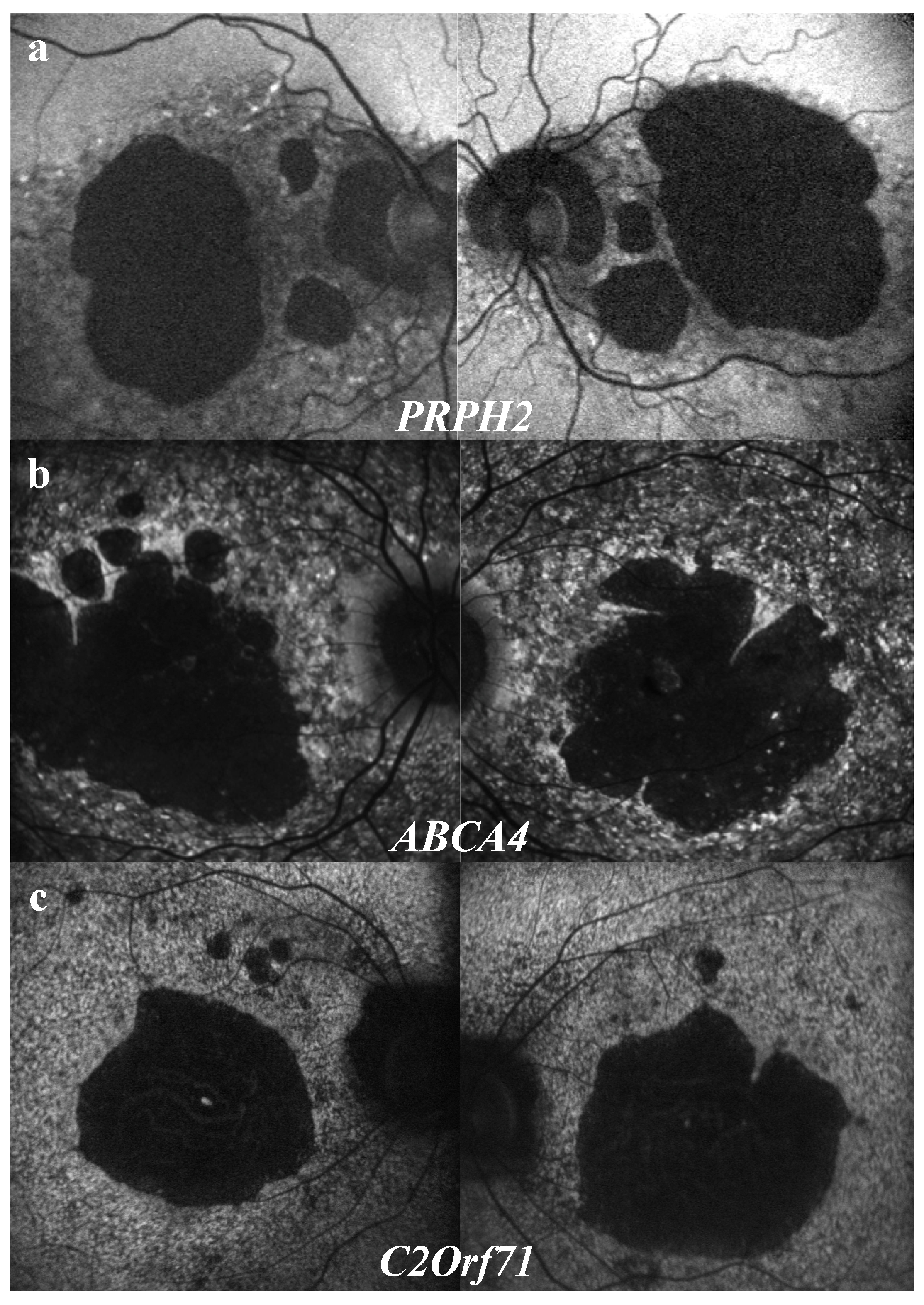
2.5. Genotype Analysis with Respect to Structural Abnormalities
3. Discussion
3.1. Retinophotography
3.2. SD-OCT
3.2.1. Outer Hyperreflective Bands
3.2.2. Hyporeflective Foveal Cavitation
3.2.3. Hyper-reflective Deposits above the RPE
3.2.4. Hyporeflective Macular Cysts
3.3. Autofluorescence
3.3.1. BAF
3.3.2. HyperAF Ring
3.3.3. “Speckled” Pattern
3.3.4. Peripapillary Sparing
3.4. IRAF
4. Patients and Methods
4.1. Study Population
4.2. Clinical Examination
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| COD/CORDs | Cone/cone-rod dystrophies |
| NGS | Next Generation Sequencing |
| SD-OCT | Spectral-Domain Optical Coherence Tomography |
| BAF/IRAF | Short-wave length /infra-red fundus autofluorescence |
| IRDs | Inherited retinal disorders |
| ERG | Electroretinogram |
| RCD | Rod-cone dystrophy |
| Ar | Autosomal recessive |
| Ad | Autosomal dominant |
| Xl | X-linked |
| ABCA4 | ATP-binding cassette, sub-family A, member 4 |
| GUCY2D | Guanylate Cyclase 2D |
| RPGR | Retinitis Pigmentosa GTPase regulator |
| SD | Standard deviation |
| BCVA | Best corrected visual acuity |
| ETDRS | Early Treatment Diabetic Retinopathy Study |
| RP | Retinitis Pigmentosa |
| MD | Macular Dystrophy |
| LCA | Leber Congenital Amaurosis |
| RPE | Retinal Pigment Epithelium |
| ELM | External limiting membrane |
| EZ | Ellipsoid zone |
| IZ | Interdigitation zone |
| HypoAF | Hypo-autofluorescence/hypo-autofluorescent |
| HyperAF | Hyper-autofluorescence/hyper-autofluorescent |
References
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.H.J.; Phan, T.M.L.; Zekveld-Vroon, R.C.; Leroy, B.P.; van den Born, L.I.; Hoyng, C.B.; Klaver, C.C.; Writing committee for the cone disorders study group consortium; Roosing, S.; Pott, J.W.; et al. Clinical Course, Genetic Etiology, and Visual Outcome in Cone and Cone-Rod Dystrophy. Ophthalmol. 2012, 119, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive cone and cone-rod dystrophies: Phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006, 51, 232–258. [Google Scholar] [CrossRef] [PubMed]
- Jalili, I.K.; Smith, N.J. A progressive cone-rod dystrophy and amelogenesis imperfecta: A new syndrome. J. Med. Genet. 1988, 25, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Cideciyan, A.V.; Volpe, N.J.; Stevanin, G.; Brice, A.; Jacobson, S.G. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp. Eye Res. 2002, 74, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Thiadens, A.A.H.J.; Hoyng, C.B.; Klaver, C.C.W.; den Hollander, A.I.; Cremers, F.P.M. Causes and consequences of inherited cone disorders. Prog. Retin. Eye Res. 2014, 42, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, A.; Klevering, B.J.; Rohrschneider, K.; Blankenagel, A.; Brunner, H.G.; Deutman, A.F.; Hoyng, C.B.; Cremers, F.P. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000, 67, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Kelsell, R.E.; Gregory-Evans, K.; Payne, A.M.; Perrault, I.; Kaplan, J.; Yang, R.B.; Garbers, D.L.; Bird, A.C.; Moore, A.T.; Hunt, D.M. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 1998, 7, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Kitiratschky, V.B.D.; Wilke, R.; Renner, A.B.; Kellner, U.; Vadalà, M.; Birch, D.G.; Wissinger, B.; Zrenner, E.; Kohl, S. Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Invest. Ophthalmol. Vis. Sci. 2008, 49, 5015–5023. [Google Scholar] [CrossRef] [PubMed]
- Demirci, F.Y.K.; Rigatti, B.W.; Wen, G.; Radak, A.L.; Mah, T.S.; Baic, C.L.; Traboulsi, E.I.; Alitalo, T.; Ramser, J.; Gorin, M.B. X-linked cone-rod dystrophy (locus COD1): Identification of mutations in RPGR exon ORF15. Am. J. Hum. Genet. 2002, 70, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Peachey, N.S.; Moshfeghi, D.M.; Thirumalaichary, S.; Chorich, L.; Shugart, Y.Y.; Fan, K.; Zhang, K. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum. Mol. Genet. 2002, 11, 605–611. [Google Scholar] [CrossRef]
- Boulanger-Scemama, E.; El Shamieh, S.; Démontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.-P.; Letexier, M.; Souied, E.; et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: Mutation spectrum and new genotype-phenotype correlation. Orphanet J. Rare Dis. 2015, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.H.; Sallum, J.M.F.; Spaide, R.F. Outer retina analysis by optical coherence tomography in cone-rod dystrophy patients. Retina 2013, 33, 1877–1880. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, O.; Mezey, J.G.; Boyko, A.R.; Gao, C.; Wang, W.; Bustamante, C.D.; Anguish, L.J.; Jordan, J.A.; Pearce-Kelling, S.E.; Aguirre, G.D.; et al. An ADAM9 mutation in canine cone-rod dystrophy 3 establishes homology with human cone-rod dystrophy 9. Mol. Vis. 2010, 16, 1549–1569. [Google Scholar] [PubMed]
- Parry, D.A.; Toomes, C.; Bida, L.; Danciger, M.; Towns, K.V.; McKibbin, M.; Jacobson, S.G.; Logan, C.V.; Ali, M.; Bond, J.; et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am. J. Hum. Genet. 2009, 84, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Inui, E.; Oishi, A.; Oishi, M.; Ogino, K.; Makiyama, Y.; Gotoh, N.; Kurimoto, M.; Yoshimura, N. Tomographic comparison of cone-rod and rod-cone retinal dystrophies. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 1065–1069. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Imamura, Y.; Margolis, R.; Slakter, J.S.; Spaide, R.F. Enhanced depth imaging optical coherence tomography of the choroid in highly myopic eyes. Am. J. Ophthalmol. 2009, 148, 445–450. [Google Scholar] [CrossRef]
- Switzer, D.W.; Mendonça, L.S.; Saito, M.; Zweifel, S.A.; Spaide, R.F. Segregation of ophthalmoscopic characteristics according to choroidal thickness in patients with early age-related macular degeneration. Retina 2012, 32, 1265–1271. [Google Scholar] [CrossRef]
- Barthelmes, D.; Sutter, F.K.; Kurz-Levin, M.M.; Bosch, M.M.; Helbig, H.; Niemeyer, G.; Fleischhauer, J.C. Quantitative analysis of OCT characteristics in patients with achromatopsia and blue-cone monochromatism. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1161–1166. [Google Scholar] [CrossRef]
- Thiadens, A.A.H.J.; Somervuo, V.; van den Born, L.I.; Roosing, S.; van Schooneveld, M.J.; Kuijpers, R.W.A.M.; van Moll-Ramirez, N.; Cremers, F.P.M.; Hoyng, C.B.; Klaver, C.C.W. Progressive loss of cones in achromatopsia: An imaging study using spectral-domain optical coherence tomography. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5952–5957. [Google Scholar] [CrossRef]
- Leng, T.; Marmor, M.F.; Kellner, U.; Thompson, D.A.; Renner, A.B.; Moore, W.; Sowden, J.C. Foveal cavitation as an optical coherence tomography finding in central cone dysfunction. Retina 2012, 32, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Duncker, T.; Tsang, S.H.; Lee, W.; Zernant, J.; Allikmets, R.; Delori, F.C.; Sparrow, J.R. Quantitative fundus autofluorescence distinguishes ABCA4-associated and non-ABCA4-associated bull’s-eye maculopathy. Ophthalmology 2015, 122, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, S.; Genead, M.A.; Fishman, G.A. The prevalence of macular cysts in patients with clinical cone-rod dystrophy determined by spectral-domain optical coherence tomography. Ophthalmic Genet. 2014, 35, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-K.; Chou, C.L.; Lima, L.H.; Cella, W.; Tosi, J.; Yannuzzi, L.A.; Tsang, S.H. Fundus autofluorescence in cone dystrophy. Doc. Ophthalmol. 2009, 119, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, M.; Oishi, A.; Ogino, K.; Makiyama, Y.; Gotoh, N.; Kurimoto, M.; Yoshimura, N. Wide-field fundus autofluorescence abnormalities and visual function in patients with cone and cone-rod dystrophies. Invest. Ophthalmol. Vis. Sci. 2014, 55, 3572–3577. [Google Scholar] [CrossRef]
- Gelman, R.; Smith, R.T.; Tsang, S.H. Diagnostic accuracy evaluation of visual acuity and fundus autofluorescence macular geographic atrophy area for the discrimination of stargardt groups. Retina 2016, 36, 1596–1606. [Google Scholar] [CrossRef]
- Robson, A.G.; Michaelides, M.; Saihan, Z.; Bird, A.C.; Webster, A.R.; Moore, A.T.; Fitzke, F.W.; Holder, G.E. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence; A review and update. Doc. Ophthalmol. 2008, 116, 79–89. [Google Scholar] [CrossRef]
- Lima, L.H.; Burke, T.; Greenstein, V.C.; Chou, C.L.; Cella, W.; Yannuzzi, L.A.; Tsang, S.H. Progressive constriction of the hyperautofluorescent ring in retinitis pigmentosa. Am. J. Ophthalmol. 2012, 153, 718–727.e2. [Google Scholar] [CrossRef]
- Nong, E.; Lee, W.; Merriam, J.E.; Allikmets, R.; Tsang, S.H. Disease progression in autosomal dominant cone-rod dystrophy caused by a novel mutation (D100G) in the GUCA1A gene. Doc. Ophthalmol. 2014, 128, 59–67. [Google Scholar] [CrossRef]
- Mukherjee, R.; Robson, A.G.; Holder, G.E.; Stockman, A.; Egan, C.A.; Moore, A.T.; Webster, A.R. A detailed phenotypic description of autosomal dominant cone dystrophy due to a de novo mutation in the GUCY2D gene. Eye 2014, 28, 481–487. [Google Scholar] [CrossRef]
- Kellner, S.; Kellner, U.; Weber, B.H.F.; Fiebig, B.; Weinitz, S.; Ruether, K. Lipofuscin-and melanin-related fundus autofluorescence in patients with ABCA4-associated retinal dystrophies. Am. J. Ophthalmol. 2009, 147, 895–902.e1. [Google Scholar] [CrossRef] [PubMed]
- Renner, A.B.; Fiebig, B.S.; Weber, B.H.F.; Wissinger, B.; Andreasson, S.; Gal, A.; Cropp, E.; Kohl, S.; Kellner, U. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am. J. Ophthalmol. 2009, 147, 518–530.e1. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.R.; Allikmets, R.; Smith, R.T.; Gouras, P.; Tsang, S.H. Loss of peripapillary sparing in non-group I Stargardt disease. Exp. Eye Res. 2010, 91, 592–600. [Google Scholar] [CrossRef] [Green Version]
- Burke, T.R.; Fishman, G.A.; Zernant, J.; Schubert, C.; Tsang, S.H.; Smith, R.T.; Ayyagari, R.; Koenekoop, R.K.; Umfress, A.; Ciccarelli, M.L.; et al. Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci. 2012, 53, 4458–4467. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.C.; Zernant, J.; Allikmets, R.; Barile, G.R.; Chang, S.; Smith, R.T. Peripapillary atrophy in Stargardt disease. Retina 2009, 29, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Jayasundera, T.; Rhoades, W.; Branham, K.; Niziol, L.M.; Musch, D.C.; Heckenlively, J.R. Peripapillary dark choroid ring as a helpful diagnostic sign in advanced stargardt disease. Am. J. Ophthalmol. 2010, 149, 656–660.e2. [Google Scholar] [CrossRef] [PubMed]
- Tschernutter, M.; Jenkins, S.A.; Waseem, N.H.; Saihan, Z.; Holder, G.E.; Bird, A.C.; Bhattacharya, S.S.; Ali, R.R.; Webster, A.R. Clinical characterisation of a family with retinal dystrophy caused by mutation in the Mertk gene. Br. J. Ophthalmol. 2006, 90, 718–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, U.; Kellner, S.; Weinitz, S. Fundus autofluorescence (488 NM) and near-infrared autofluorescence (787 NM) visualize different retinal pigment epithelium alterations in patients with age-related macular degeneration. Retina 2010, 30, 6–15. [Google Scholar] [CrossRef]
- Audo, I.; Sahel, J.-A.; Mohand-Saïd, S.; Lancelot, M.-E.; Antonio, A.; Moskova-Doumanova, V.; Nandrot, E.F.; Doumanov, J.; Barragan, I.; Antinolo, G.; et al. EYS is a major gene for rod-cone dystrophies in France. Hum. Mutat. 2010, 31, E1406–E1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaide, R.F. Questioning optical coherence tomography. Ophthalmology 2012, 119, 2203–2204.e1. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Curcio, C.A. Anatomical correlates to the bands seen in the outer retina by optical coherence tomography: Literature review and model. Retina 2011, 31, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Gaillard, M.-C.; Escher, P.; Tiab, L.; Bedell, M.; Borruat, F.-X.; Barthelmes, D.; Carmona, R.; Zhang, K.; White, E.; et al. The PROM1 mutation p.R373C causes an autosomal dominant bull’s eye maculopathy associated with rod, rod-cone, and macular dystrophy. Invest. Ophthalmol. Vis. Sci. 2010, 51, 4771–4780. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Arno, G.; Plagnol, V.; Chamney, S.; Russell-Eggitt, I.; Thompson, D.; Ramsden, S.C.; Black, G.C.; Robson, A.G.; Holder, G.E.; et al. The phenotypic variability of retinal dystrophies associated with mutations in CRX, with report of a novel macular dystrophy phenotype. Invest. Ophthalmol. Vis. Sci. 2014, 55, 6934–6944. [Google Scholar] [CrossRef] [PubMed]
- Zobor, D.; Zrenner, E.; Wissinger, B.; Kohl, S.; Jägle, H. GUCY2D- or GUCA1A-related autosomal dominant cone-rod dystrophy: Is there a phenotypic difference? Retina 2014, 34, 1576–1587. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age at the onset of symptoms (decreased central vision and/or photophobia) (median (min–max), years) | 10 (1–55) | (N = 36) * |
| Age at clinical exam (average (SD, min–max), years) | 35 (5, 7–78) | |
| Mode of inheritance | (N = 58) | |
| Autosomal recessive, n (%) | 22 (38) | |
| Autosomal dominant, n (%) | 11 (19) | |
| Sporadic, n (%) | 25 (43) | |
| Sex | (N = 58) | |
| Male, n (%) | 14 (24) | |
| BCVA (average (SD), ETDRS) | 20:400 (20:125) | (N = 53) |
| BCVA (average (SD), LogMar) | 1.3 (0.8) | |
| Spherical equivalent (average (SD), Diopters) | −2.5 (3.5) | (N = 49) |
| ID | Type | Gene | Allele Status | cDNA | Protein | References | |
|---|---|---|---|---|---|---|---|
| Known CCRD genes | |||||||
| CIC00137 | sporadic | ABCA4 | Ho | 47 | c.6394G>A | p.(E2132K) | (Boulanger-Scemama et al. 2015) |
| CIC00162 | Ar | ABCA4 | Het | 31 | c.4546_4547del | p.(Q1516Afs*38) | (Boulanger-Scemama et al. 2015) |
| ABCA4 | Het | 16 | c.2463G>A | p.(W821*) | (Boulanger-Scemama et al. 2015) | ||
| CIC00765 | Ar | ABCA4 | Ho | 47 | c.6445C>T | p.(R2149*) | (Lewis et al. 1999) |
| (rs61750654) | |||||||
| CIC03436 | Ar | ABCA4 | Ho | 42 | c.5892del | p.(G1965Efs*9) | [1] |
| CIC04412 | sporadic | ABC4A | Het | 34 | c.4793C>A | p.(A1598D) | (Maugeri et al. 2000) |
| (rs61750155) | |||||||
| ABCA4 | Het | 28 | c.4234C>T | p.(Q1412*) | (Maugeri et al. 2000) | ||
| (rs61750137) | |||||||
| CIC04645 | Ar | ABCA4 | Ho | 13 | c.1924T>C | p.(F642L) | (Boulanger-Scemama et al. 2015), but c.1924T>A p.F642I in (Jin et al. 2014) |
| CIC05087 | sporadic | ABCA4 | Ho | IVS 11 | c.1554+1G>C | r.(spl?) | (Boulanger-Scemama et al. 2015) |
| CIC05853 | sporadic | ABC4A | Ho | 22 | c.3259G>A | p.(E1087K) | (Allikmets et al. 1997) |
| (rs61751398) | |||||||
| CIC05854 | Ar | ABC4A | Ho | 35 | c.4919G>A | p.(R1640Q) | (Simonelli et al. 2000) |
| (rs61751403) | |||||||
| CIC05987 | Ar | ABC4A | Het | 22 | c.3295T>C | p.(S1099P) | (Fumagalli et al. 2001) |
| (rs61750119) | |||||||
| ABC4A | Het | 4 | c.327dup | p.(Q110Sfs*51) | (Boulanger-Scemama et al. 2015) | ||
| (rs 61748531) | |||||||
| CIC05989 | sporadic | ABC4A | Het | 34 | c.4837G>A | p.(D1613N) | (Boulanger-Scemama et al. 2015) |
| ABC4A | Het | 10 | c.1302del | p.(Q437Rfs*12) | (Boulanger-Scemama et al. 2015) | ||
| ABCA4 | Het | 38 | c.5318C>T | p.(A1773V) | (Stenirri et al. 2008) | ||
| CIC06170 | sporadic | ABC4A | Het | 44 | c.6089G>A | p.(R2030Q) | (Lewis et al. 1999) |
| (rs61750641) | |||||||
| ABC4A | Het | IVS 24 | c.3607+3A>T | r.(spl?) | (Boulanger-Scemama et al.2015) | ||
| ABCA4 | Het | 14 | c.2034G>T | p.(K678N) | (Huang et al. 2014) | ||
| CIC06694 | sporadic | ABC4A | Het | IVS36 | c.5196+1G>A | r.(spl?) | (Kitiratschky et al. 2008) |
| ABC4A | Het | 22 | c.3322C>T | p.(R1108C) | (Briggs et al. 2001) | ||
| CIC06735 | Ar | ABC4A | Ho | 42 | c.5892del | p.(G1965Efs*9) | [1] |
| CIC06913 | Ar | ABCA4 | Ho | 21 | c.3056C>T | p.(T1019M) | (Rozet et al. 1998) |
| (rs201855602) | |||||||
| CIC04239 | Ar | CDHR1 | Ho | 9 | c.838C>T | p.(R280*) | (Boulanger-Scemama et al. 2015) |
| CIC06568 | Ar | CERKL | Ho | 8 | c.1090C>T | p.(R364*) | Thesis (Sergouniotis P. 2012) [2] |
| CIC07299 | sporadic | PDE6C | Ho | 2 | c.542del | p.(A181Efs*13) | (Boulanger-Scemama et al. 2015) |
| CIC05563 | Ad | SEMA4A | Het | 4 | c.302T>C | p.(I101T) | (Boulanger-Scemama et al. 2015) |
| (rs149652495) | |||||||
| CIC07563 | sporadic | SEMA4A | Ho | 3 | c.241C>T | p.(R81*) | (Boulanger-Scemama et al. 2015) |
| CIC00324 | Ad | GUCY2D | Het | 13 | c.2512C>T | p.(R838C) | (Kelsell et al. 1998) |
| (rs61750172) | |||||||
| CIC03249 | Ad | GUCY2D | Het | 13 | c.2512C>T | p.(R838C) | (Kelsell et al. 1998) |
| (rs61750172) | |||||||
| CIC04347 | Ad | GUCY2D | Het | 13 | c.2512C>T | p.(R838C) | (Kelsell et al. 1998) |
| (rs61750172) | |||||||
| CIC04918 | Ad | GUCY2D | Het | 13 | c.2512C>T | p.(R838C) | (Kelsell et al. 1998) |
| (rs61750172) | |||||||
| CIC00597 | sporadic | GUCY2D | Het | 14 | c.2747T>C | p.(I916T) | (De Castro-Miró et al. 2014) |
| CIC06352 | sporadic | GUCA1A | Het | 3 | c.149C>T | p.(P50L) | (Downes et al. 2001) |
| (rs104893968) | |||||||
| CIC06757 | Ad | PRPH2 | Het | 1 | c.514C>T | p.(R172W) | (Wells et al. 1993) |
| (rs61755792) | |||||||
| CIC03621 | Ad | PRPH2 | Het | 1 | c.1-c581+?del | - | (Boulanger-Scemama et al. 2015) |
| CIC00535 | Ad | PROM1 | Het | 10 | c.1117C>T | p.(R373C) | (Michaelides et al. 2006) |
| (rs137853006) | |||||||
| CIC01196 | sporadic | PROM1 | Ho | 12 | c.1354dup | p.(Y452Lfs*13) | (Pras et al. 2009) |
| CIC07188 | sporadic | PROM1 | Het | 12 | c.1354dup | p.(Y452Lfs*13) | (Pras et al. 2009) |
| PROM1 | Het | IVS 12 | c.1454+2T>C | r.(spl?) | (Boulanger-Scemama et al. 2015) | ||
| CIC07045 | sporadic | PROM1 | Ho | IVS 17 | c.1984-1G>T | r.(spl?) | (Boulanger-Scemama et al. 2015) |
| (rs373680665) | |||||||
| CIC06642 | Ad | PROM1 | Het | 1 | c.7dup | p.(L3Pfs*28) | (Boulanger-Scemama et al. 2015) |
| CIC04965 | Ad | CRX | Het | 4 | c.608_609del | p.(S203Ffs*32) | (Boulanger-Scemama et al. 2015) |
| CIC03241 | sporadic | CRX | Het | 4 | c.564dup | p.(A189Rfs*47) | Not clear if same mutation as in (Stone 2007) |
| CIC3750 | sporadic | CRX | Het | 3 | c.121C>T | p.(R41W) | (Swain et al. 1997) |
| (rs104894672) | |||||||
| CIC05218 | Ar | PDE6C | Ho | IVS 10 | c.1413+3A>T | r.(spl?) | (Boulanger-Scemama et al. 2015) |
| CIC02712 | sporadic | PDE6C | Het | 10 | c.1325T>A | p.(M442K) | (Boulanger-Scemama et al. 2015) |
| PDE6C | Het | 10 | c.1375C>G | p.(Q459E) | (Boulanger-Scemama et al. 2015) | ||
| CIC06321 | sporadic | RPGRIP1 | Ho | 14 | c.2021C>A | p.(P674H) | (Boulanger-Scemama et al. 2015) |
| CIC00190 | sporadic | AIPL1 | Het | 5 | c.769C>T | p.(L257F) | (Boulanger-Scemama et al. 2015) |
| AIPL1 | Het | 5 | c.767T>G | p.(I256S) | (Boulanger-Scemama et al. 2015) | ||
| CIC04945 | sporadic | PROM1 | Het | 23 | c.2383T>C | p.(W795R) | (Boulanger-Scemama et al. 2015) |
| PROM1 | Het | IVS 13 | c.1579-1G>C | r.(spl?) | (Boulanger-Scemama et al. 2015) | ||
| CIC07569 | sporadic | CRX | Het | IVS 3 | c.252+1G>A | r.(spl?) | (Boulanger-Scemama et al. 2015) |
| Other retinal disease genes | |||||||
| CIC01571 | Ar | C2Orf71 | Ho | 1 | c.2950C>T | p.(R984*) | (Audo et al. 2011) (RP) |
| CIC00643 | Ar | C2Orf71 | Ho | 1 | c.1949G>A | p.(W650*) | (Boulanger-Scemama et al. 2015) |
| (rs371289954) | |||||||
| CIC03112 | Ar | MERTK | Ho | 17 | c.2214del | p.(C738Wfs*32) | (Tschernutter et al. 2006) (RP) |
| CIC01242 | Ar | MERTK | Ho | 3_19 | c.483-?_c.3000+?del | - | (Boulanger-Scemama et al. 2015) |
| CIC06514 | Ar | RLBP1 | Ho | 7_9 | c.526-?_c.954+?del | - | (Boulanger-Scemama et al. 2015) |
| CIC03953 | sporadic | EYS | Het | 11 | c.1673G>A | p.(W558*) | (Audo et al. 2010) |
| (RP) | |||||||
| (rs201823777) | |||||||
| EYS | Het | 14 | c.2234A>G | p.(N745S) | (Audo et al. 2010) | ||
| (RP) | |||||||
| (rs201652272) | |||||||
| CIC05012 | sporadic | NMNAT1 | Het | 5 | c.619C>T | p.(R207W) | (Perrault et al. 2012) (LCA) |
| (rs142968179) | |||||||
| NMNAT1 | Het | 5 | c.769G>A | p.(E257K) | (Chiang et al. 2012) | ||
| (LCA) | |||||||
| (rs150726175) | |||||||
| CIC06499 | sporadic | NMNAT1 | Het | 5 | c.619C>T | p.(R207W) | (Perrault et al. 2012) (LCA) |
| (rs142968179) | |||||||
| NMNAT1 | Het | 5 | c.769G>A | p.(E257K) | |||
| CIC05394 | Ar | RDH12 | Ho | 8 | c.806_810del | p.(A269Gfs*2) | (Janecke et al. 2004) |
| (LCA) | |||||||
| (rs386834261) | |||||||
| CIC07241 | Ar | RDH12 | Ho | 7 | c.464C>T | p.(T155I) | (Thompson et al. 2005) (LCA) |
| (rs121434337) | |||||||
| CIC07447 | Ar | RDH12 | Het | 8 | c.806_810del | p.(A269Gfs*2) | (Janecke et al. 2004) |
| (LCA) | |||||||
| (rs386834261)) | |||||||
| RDH12 | Het | 8 | c.403A>G | p.(K135E) | (Boulanger-Scemama et al. 2015) | ||
| CIC00953 | sporadic | IQCB1 | Het | 6 | c.424_425del | p.(F142Pfs*5) | (Otto et al. 2005) |
| (Senior-Loken/LCA) | |||||||
| IQCB1 | Het | 8 | c.686del | p.(T229Mfs*8) | (Boulanger-Scemama et al. 2015) | ||
| CIC01300 | Ar | RP1 | Ho | 4 | c.1719_1723del | p.(S574Cfs*7) | (El Shamieh et al. 2015) |
| (arRP) | |||||||
| CIC01380 | Ar | CRB1 | Ho | 11 | c.3994T>G | p.(C1332G) | (Boulanger-Scemama et al. 2015) (LCA) |
| CIC00963 | Ar | TULP1 | Ho | 11 | c.1087G>A | p.(G363R) | (Boulanger-Scemama et al. 2015) (LCA and arRP) |
| Lower confidence | |||||||
| CIC05007 | Ad | ROM1 | Het | 1 | c.339del | p.(L114Sfs*8) | (Boulanger-Scemama et al. 2015) (adRP) |
| n (%) | |
|---|---|
| OUTER RETINA | |
| EZ irregularities | 3 (6) |
| Hyperreflective layers disruption (foveal/beyond the fovea) | |
| ELM | 39 (80)/27 (55) |
| EZ | 46 (94)/30 (61) |
| IZ | 49 (100)/42 (86) |
| RPE | 0 (0)/0 (0) |
| Hyporeflective foveal cavitation | 3 (6) |
| Foveal sparing | 6 (12) |
| Hyper-reflective deposits above the RPE | 14 (28) |
| Outer retinal tubulations | 1 (2) |
| Outer nuclear layer atrophy in the macular region | 36 (73) |
| Diffuse outer retinal atrophy beyond the vascular arcades | 20 (41) |
| INNER RETINA | |
| Hyporeflective macular cysts | 2 (4) |
| “BAF” Autofluorescence (N = 56 Patients) | “IRAF” Autofluorescence (N = 52 Patients) | ||
|---|---|---|---|
| Macular abnormalities | Macular abnormalities | ||
| Minimal alterations, n (%) | 6 (10,5) | Minimal alterations, n (%) | 6 (11,5) |
| Loss of foveal hypoAF, n (%) | 1 (2) | Loss of foveal hyperAF, n (%) | 1 (2) |
| Foveal hyperAF, n (%) | 2 (3,5) | Foveal hypoAF, n (%) | 3 (5,5) |
| Perifoveal hyperAF, n (%) | 3 (5) | Perifoveal hypoAF, n (%) | 2 (4) |
| Macular hypoAF, n (%) | 11 (19,5) | Macular hypoAF, n (%) | 16 (30,5) |
| Macular hypoAF spots, n (%) | 1 (2) | Macular hypoAF spots, n (%) | 1 (2) |
| Diffuse retinal abnormalities | Diffuse retinal abnormalities | ||
| Macular hypoAF + peripheral hypoAF spots, n (%) | 20 (36) | Macular hypoAF + peripheral hypoAF spots, n (%) | 13 (25) |
| Macular hypoAF + peripheral “speckled” aspect, n (%) | 5 (9) | Macular + peripheral hypoAF, n (%) | 16 (31) |
| Macular hypoAF + peripheral confluent hypoAF patches, n (%) | 13 (23) | ||
| without “speckled” aspect, n (%) | 6 (11) | ||
| with “speckled” aspect, n (%) | 7 (12) | ||
| HyperAF ring | HyperAF ring | ||
| macular, n (%) | 9 (16) | macular, n (%) | 9 (17) |
| macular including the optic nerve, n (%) | 4 (7) | macular including the optic nerve, n (%) | 3 (6) |
| Peripapillary sparing, n (%), N = 38 | 6 (16) | Peripapillary sparing, n (%), N = 29 | 6 (21) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulanger-Scemama, E.; Mohand-Saïd, S.; El Shamieh, S.; Démontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.-P.; Letexier, M.; et al. Phenotype Analysis of Retinal Dystrophies in Light of the Underlying Genetic Defects: Application to Cone and Cone-Rod Dystrophies. Int. J. Mol. Sci. 2019, 20, 4854. https://doi.org/10.3390/ijms20194854
Boulanger-Scemama E, Mohand-Saïd S, El Shamieh S, Démontant V, Condroyer C, Antonio A, Michiels C, Boyard F, Saraiva J-P, Letexier M, et al. Phenotype Analysis of Retinal Dystrophies in Light of the Underlying Genetic Defects: Application to Cone and Cone-Rod Dystrophies. International Journal of Molecular Sciences. 2019; 20(19):4854. https://doi.org/10.3390/ijms20194854
Chicago/Turabian StyleBoulanger-Scemama, Elise, Saddek Mohand-Saïd, Said El Shamieh, Vanessa Démontant, Christel Condroyer, Aline Antonio, Christelle Michiels, Fiona Boyard, Jean-Paul Saraiva, Mélanie Letexier, and et al. 2019. "Phenotype Analysis of Retinal Dystrophies in Light of the Underlying Genetic Defects: Application to Cone and Cone-Rod Dystrophies" International Journal of Molecular Sciences 20, no. 19: 4854. https://doi.org/10.3390/ijms20194854