Regulation of Brain Cholesterol: What Role Do Liver X Receptors Play in Neurodegenerative Diseases?

 and
and
Abstract
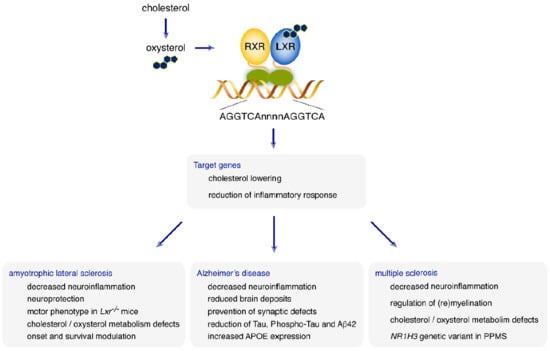
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
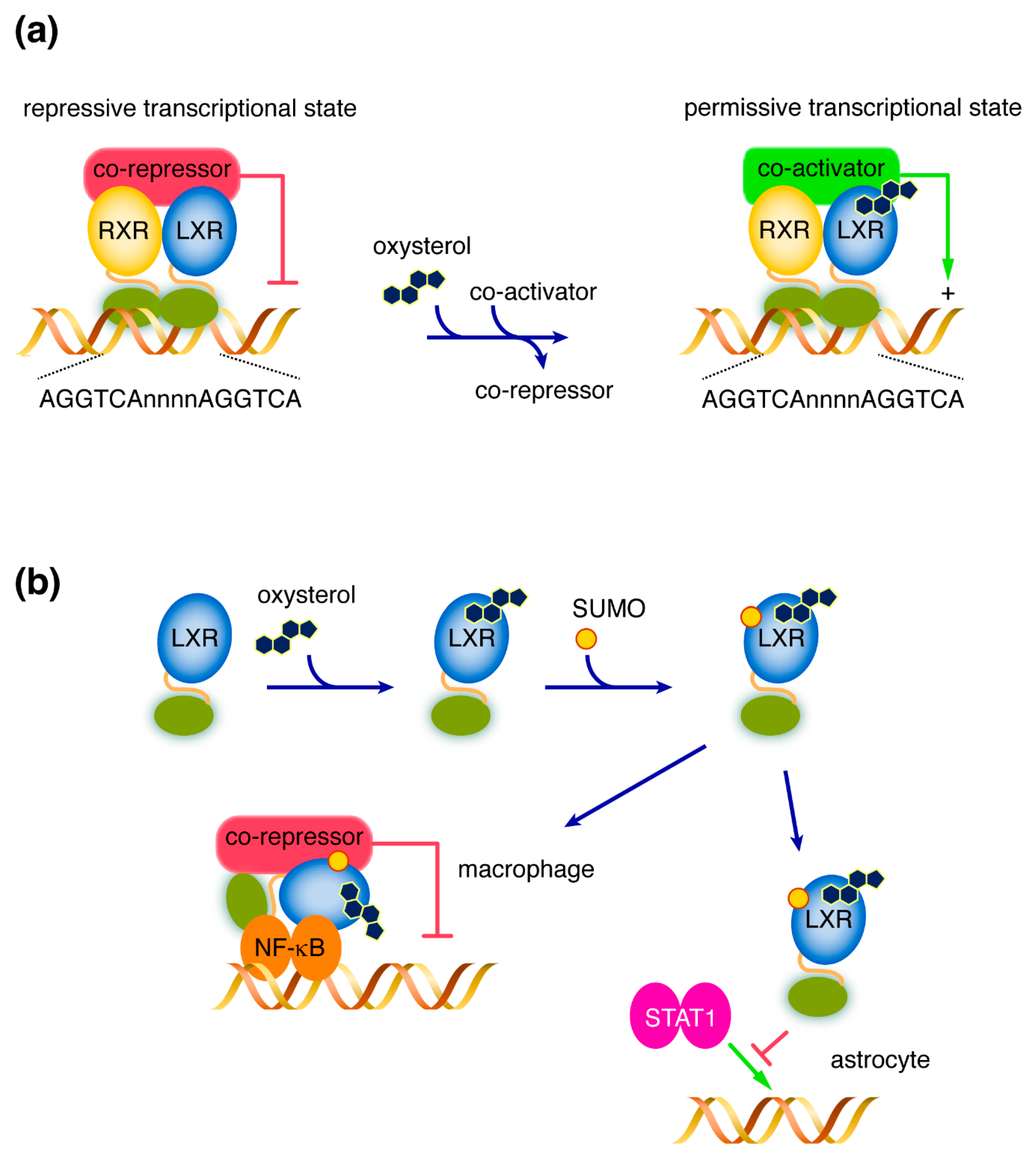
2. LXRs: Structure and Mode of Action
3. LXRs Ligands
4. LXRs: Two Central Regulators of Cholesterol Metabolism
5. Cholesterol and Oxysterol in the CNS: Which Place for LXRs in the Regulation of Brain Homeostasis?
6. What Role for LXRs in Neurodegenerative Diseases?
6.1. Amyotrophic Lateral Sclerosis
6.2. Alzheimer’s Disease
6.3. Multiple Sclerosis
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| APP | Amyloid precursor protein |
| BBB | Blood brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DBD | DNA-binding domain |
| FALS | Familial ALS |
| FDR | False discovery rate |
| FTD | Frontotemporal dementia |
| IDOL | Inducible degrader of LDLR |
| IL | Interleukin |
| LDLR | LDL receptor |
| LXR | Liver X Receptor |
| LXRE | LXR Responsive Element |
| MN | Motor neuron |
| MS | Multiple sclerosis |
| NLS | Nuclear localization signal |
| PPMS | Primary progressive MS |
| RRMS | Relapsing remitting MS |
| RXR | Retinoid X Receptor |
| SPMS | Secondary Progressive MS |
| STAT1 | Signal transducer and activator of transcription 1 |
| SUMO | Small ubiquitin-like modifier |
| TNF | Tumor necrosis factor |
References
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the X-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Shinar, D.M.; Endo, N.; Rutledge, S.J.; Vogel, R.; Rodan, G.A.; Schmidt, A. NER, a new member of the gene family encoding the human steroid hormone nuclear receptor. Gene 1994, 147, 273–276. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Maqdasy, S.; Trousson, A.; Tauveron, I.; Volle, D.H.; Baron, S.; Lobaccaro, J.-M.A. Once and for all, LXRα and LXRβ are gatekeepers of the endocrine system. Mol. Aspects Med. 2016, 49, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Dallel, S.; Tauveron, I.; Brugnon, F.; Baron, S.; Lobaccaro, J.M.A.; Maqdasy, S. Liver X Receptors: A Possible Link between Lipid Disorders and Female Infertility. Int. J. Mol. Sci. 2018, 19, 2177. [Google Scholar] [CrossRef] [PubMed]
- El-Hajjaji, F.Z.; Oumeddour, A.; Pommier, A.J.; Ouvrier, A.; Viennois, E.; Dufour, J.; Caira, F.; Drevet, J.R.; Volle, D.H.; Baron, S.; et al. Liver X receptors, lipids and their reproductive secrets in the male. Biochim. Biophys. Acta 2011, 1812, 974–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessler, M.B. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef]
- De Boussac, H.; Alioui, A.; Viennois, E.; Dufour, J.; Trousson, A.; Vega, A.; Guy, L.; Volle, D.H.; Lobaccaro, J.-M.A.; Baron, S. Oxysterol receptors and their therapeutic applications in cancer conditions. Expert Opin. Ther. Targets 2013, 17, 1029–1038. [Google Scholar] [CrossRef]
- Courtney, R.; Landreth, G.E. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol. Metab. 2016, 27, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Volle, D.H.; Repa, J.J.; Mazur, A.; Cummins, C.L.; Val, P.; Henry-Berger, J.; Caira, F.; Veyssiere, G.; Mangelsdorf, D.J.; Lobaccaro, J.M. Regulation of the aldo-keto reductase gene akr1b7 by the nuclear oxysterol receptor LXRalpha (liver X receptor-alpha) in the mouse intestine: Putative role of LXRs in lipid detoxification processes. Mol. Endocrinol. 2004, 18, 888–898. [Google Scholar] [CrossRef]
- Henry-Berger, J.; Mouzat, K.; Baron, S.; Bernabeu, C.; Marceau, G.; Saru, J.P.; Sapin, V.; Lobaccaro, J.M.; Caira, F. Endoglin (CD105) expression is regulated by the liver X receptor alpha (NR1H3) in human trophoblast cell line JAR. Biol. Reprod. 2008, 78, 968–975. [Google Scholar] [CrossRef]
- Horlein, A.J.; Naar, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Soderstrom, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef]
- Chen, J.D.; Evans, R.M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [Google Scholar] [CrossRef]
- Viennois, E.; Mouzat, K.; Dufour, J.; Morel, L.; Lobaccaro, J.M.; Baron, S. Selective liver X receptor modulators (SLiMs): What use in human health? Mol. Cell Endocrinol. 2012, 351, 129–141. [Google Scholar] [CrossRef]
- Hu, X.; Lazar, M.A. Transcriptional repression by nuclear hormone receptors. Trends Endocrinol. Metab. 2000, 11, 6–10. [Google Scholar] [CrossRef]
- Lazar, M.A. Nuclear receptor corepressors. Nucl. Recept Signal 2003, 1, e001. [Google Scholar] [CrossRef]
- McKenna, N.J.; O’Malley, B.W. Minireview: Nuclear receptor coactivators—An update. Endocrinology 2002, 143, 2461–2465. [Google Scholar] [CrossRef]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- Landis, M.S.; Patel, H.V.; Capone, J.P. Oxysterol activators of liver X receptor and 9-cis-retinoic acid promote sequential steps in the synthesis and secretion of tumor necrosis factor-alpha from human monocytes. J. Biol. Chem. 2002, 277, 4713–4721. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Ghisletti, S.; Huang, W.; Ogawa, S.; Pascual, G.; Lin, M.E.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol. Cell 2007, 25, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.M.; Kim, O.S.; Lee, C.S.; Woo, J.H.; Park, S.J.; Joe, E.; Jou, I. Differential SUMOylation of LXRalpha and LXRbeta mediates transrepression of STAT1 inflammatory signaling in IFN-gamma-stimulated brain astrocytes. Mol. Cell 2009, 35, 806–817. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 2015, 4, e08009. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.G.; Doran, A.C.; Fotakis, P.; Westerterp, M.; Antonson, P.; Jiang, H.; Jiang, X.-C.; Gustafsson, J.-Å.; Tabas, I.; Tall, A.R. LXR Suppresses Inflammatory Gene Expression and Neutrophil Migration through cis-Repression and Cholesterol Efflux. Cell Rep. 2018, 25, 3774.e4–3785.e4. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Schroepfer, G.J., Jr. Oxysterols: Modulators of cholesterol metabolism and other processes. Physiol. Rev. 2000, 80, 361–554. [Google Scholar] [CrossRef]
- Mutemberezi, V.; Guillemot-Legris, O.; Muccioli, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169. [Google Scholar] [CrossRef]
- Fakheri, R.J.; Javitt, N.B. 27-Hydroxycholesterol, does it exist? On the nomenclature and stereochemistry of 26-hydroxylated sterols. Steroids 2012, 77, 575–577. [Google Scholar] [CrossRef]
- Muse, E.D.; Yu, S.; Edillor, C.R.; Tao, J.; Spann, N.J.; Troutman, T.D.; Seidman, J.S.; Henke, A.; Roland, J.T.; Ozeki, K.A.; et al. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, E4680–E4689. [Google Scholar] [CrossRef] [Green Version]
- Jansen, M.; Wang, W.; Greco, D.; Bellenchi, G.C.; di Porzio, U.; Brown, A.J.; Ikonen, E. What dictates the accumulation of desmosterol in the developing brain? FASEB J. 2013, 27, 865–870. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Mauch, D.H.; Nägler, K.; Schumacher, S.; Göritz, C.; Müller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Tassew, N.G.; Mothe, A.J.; Shabanzadeh, A.P.; Banerjee, P.; Koeberle, P.D.; Bremner, R.; Tator, C.H.; Monnier, P.P. Modifying lipid rafts promotes regeneration and functional recovery. Cell Rep. 2014, 8, 1146–1159. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Abdel-Khalik, J.; Hearn, T.; Yutuc, E.; Morgan, A.H.; Wang, Y. Current trends in oxysterol research. Biochem. Soc. Trans. 2016, 44, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Gosselet, F.; Saint-Pol, J.; Fenart, L. Effects of oxysterols on the blood-brain barrier: Implications for Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2014, 446, 687–691. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. Oxysterols as biomarkers in neurodegenerative diseases. Chem. Phys. Lipids 2011, 164, 515–524. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. Potential diagnostic applications of side chain oxysterols analysis in plasma and cerebrospinal fluid. Biochem. Pharmacol. 2013, 86, 26–36. [Google Scholar] [CrossRef]
- Bezine, M.; Namsi, A.; Sghaier, R.; Ben Khalifa, R.; Hamdouni, H.; Brahmi, F.; Badreddine, I.; Mihoubi, W.; Nury, T.; Vejux, A.; et al. The effect of oxysterols on nerve impulses. Biochimie 2018, 153, 46–51. [Google Scholar] [CrossRef]
- Theofilopoulos, S.; Abreu de Oliveira, W.A.; Yang, S.; Yutuc, E.; Saeed, A.; Abdel-Khalik, J.; Ullgren, A.; Cedazo-Minguez, A.; Björkhem, I.; Wang, Y.; et al. 24(S),25-Epoxycholesterol and cholesterol 24S-hydroxylase (CYP46A1) overexpression promote midbrain dopaminergic neurogenesis in vivo. J. Biol. Chem. 2019, 294, 4169–4176. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Chen, G.; Head, D.L.; Mangelsdorf, D.J.; Russell, D.W. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 2007, 5, 73–79. [Google Scholar] [CrossRef]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar] [CrossRef]
- Yu, L.; York, J.; von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8. J. Biol. Chem. 2003, 278, 15565–15570. [Google Scholar] [CrossRef]
- Alberti, S.; Schuster, G.; Parini, P.; Feltkamp, D.; Diczfalusy, U.; Rudling, M.; Angelin, B.; Bjorkhem, I.; Pettersson, S.; Gustafsson, J.A. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice. J. Clin. Investig. 2001, 107, 565–573. [Google Scholar] [CrossRef]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Rogers, P.M.; Su, C.; Varga, G.; Stayrook, K.R.; Burris, T.P. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J. Biol. Chem. 2008, 283, 26332–26339. [Google Scholar] [CrossRef]
- Tontonoz, P.; Mangelsdorf, D.J. Liver X receptor signaling pathways in cardiovascular disease. Mol. Endocrinol. 2003, 17, 985–993. [Google Scholar] [CrossRef]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Venkateswaran, A.; Tarr, P.T.; Xenarios, I.; Kudoh, J.; Shimizu, N.; Edwards, P.A. Characterization of the human ABCG1 gene: Liver X receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J. Biol. Chem. 2001, 276, 39438–39447. [Google Scholar] [CrossRef]
- Engel, T.; Lorkowski, S.; Lueken, A.; Rust, S.; Schluter, B.; Berger, G.; Cullen, P.; Assmann, G. The human ABCG4 gene is regulated by oxysterols and retinoids in monocyte-derived macrophages. Biochem. Biophys. Res. Commun. 2001, 288, 483–488. [Google Scholar] [CrossRef]
- Laffitte, B.A.; Repa, J.J.; Joseph, S.B.; Wilpitz, D.C.; Kast, H.R.; Mangelsdorf, D.J.; Tontonoz, P. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 507–512. [Google Scholar] [CrossRef]
- Mak, P.A.; Laffitte, B.A.; Desrumaux, C.; Joseph, S.B.; Curtiss, L.K.; Mangelsdorf, D.J.; Tontonoz, P.; Edwards, P.A. Regulated expression of the apolipoprotein E/C-I/C-IV/C-II gene cluster in murine and human macrophages. A critical role for nuclear liver X receptors alpha and beta. J. Biol. Chem. 2002, 277, 31900–31908. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef]
- Björkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef]
- Goritz, C.; Mauch, D.H.; Pfrieger, F.W. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell. Neurosci. 2005, 29, 190–201. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Donizetti, A.; Iannotta, L.; Aliperti, V.; Cupidi, C.; Bruni, A.C.; Cigliano, L. Brain-derived neurotrophic factor modulates cholesterol homeostasis and Apolipoprotein E synthesis in human cell models of astrocytes and neurons. J. Cell. Physiol. 2018, 233, 6925–6943. [Google Scholar] [CrossRef]
- Mouzat, K.; Raoul, C.; Polge, A.; Kantar, J.; Camu, W.; Lumbroso, S. Liver X receptors: From cholesterol regulation to neuroprotection—A new barrier against neurodegeneration in amyotrophic lateral sclerosis? Cell. Mol. Life Sci. 2016, 73, 3801–3808. [Google Scholar] [CrossRef]
- Eckert, G.P.; Vardanian, L.; Rebeck, G.W.; Burns, M.P. Regulation of central nervous system cholesterol homeostasis by the liver X receptor agonist TO-901317. Neurosci. Lett. 2007, 423, 47–52. [Google Scholar] [CrossRef]
- Abildayeva, K.; Jansen, P.J.; Hirsch-Reinshagen, V.; Bloks, V.W.; Bakker, A.H.F.; Ramaekers, F.C.S.; de Vente, J.; Groen, A.K.; Wellington, C.L.; Kuipers, F.; et al. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J. Biol. Chem. 2006, 281, 12799–12808. [Google Scholar] [CrossRef]
- Meffre, D.; Shackleford, G.; Hichor, M.; Gorgievski, V.; Tzavara, E.T.; Trousson, A.; Ghoumari, A.M.; Deboux, C.; Nait Oumesmar, B.; Liere, P.; et al. Liver X receptors alpha and beta promote myelination and remyelination in the cerebellum. Proc. Natl. Acad. Sci. USA 2015, 112, 7587–7592. [Google Scholar] [CrossRef] [Green Version]
- Makoukji, J.; Shackleford, G.; Meffre, D.; Grenier, J.; Liere, P.; Lobaccaro, J.-M.A.; Schumacher, M.; Massaad, C. Interplay between LXR and Wnt/β-catenin signaling in the negative regulation of peripheral myelin genes by oxysterols. J. Neurosci. 2011, 31, 9620–9629. [Google Scholar] [CrossRef]
- Wu, C.-H.; Chen, C.-C.; Lai, C.-Y.; Hung, T.-H.; Lin, C.-C.; Chao, M.; Chen, S.-F. Treatment with TO901317, a synthetic liver X receptor agonist, reduces brain damage and attenuates neuroinflammation in experimental intracerebral hemorrhage. J. Neuroinflamm. 2016, 13, 62. [Google Scholar] [CrossRef]
- Haukedal, H.; Freude, K. Implications of Microglia in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. J. Mol. Biol. 2019, 431, 1818–1829. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.S.; Lee, C.S.; Joe, E.; Jou, I. Oxidized low density lipoprotein suppresses lipopolysaccharide-induced inflammatory responses in microglia: Oxidative stress acts through control of inflammation. Biochem. Biophys. Res. Commun. 2006, 342, 9–18. [Google Scholar] [CrossRef]
- Zhang-Gandhi, C.X.; Drew, P.D. Liver X receptor and retinoid X receptor agonists inhibit inflammatory responses of microglia and astrocytes. J. Neuroimmunol. 2007, 183, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Secor McVoy, J.R.; Oughli, H.A.; Oh, U. Liver X receptor-dependent inhibition of microglial nitric oxide synthase 2. J. Neuroinflamm. 2015, 12, 27. [Google Scholar] [CrossRef]
- Paterniti, I.; Campolo, M.; Siracusa, R.; Cordaro, M.; Di Paola, R.; Calabrese, V.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Liver X receptors activation, through TO901317 binding, reduces neuroinflammation in Parkinson’s disease. PLoS ONE 2017, 12, e0174470. [Google Scholar] [CrossRef]
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef]
- Talbot, K. Motor neuron disease: The bare essentials. Pract. Neurol. 2009, 9, 303–309. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primer 2017, 3, 17071. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Eurals Consortium Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2009, 10, 310–323. [Google Scholar]
- Van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8+ T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef]
- Aebischer, J.; Bernard-Marissal, N.; Pettmann, B.; Raoul, C. Death Receptors in the Selective Degeneration of Motoneurons in Amyotrophic Lateral Sclerosis. J. Neurodegener. Dis. 2013, 2013, 746845. [Google Scholar] [CrossRef]
- Schmitt, F.; Hussain, G.; Dupuis, L.; Loeffler, J.-P.; Henriques, A. A plural role for lipids in motor neuron diseases: Energy, signaling and structure. Front. Cell. Neurosci. 2014, 8, 25. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Irish, M.; Piguet, O.; Halliday, G.M.; Ittner, L.M.; Farooqi, S.; Hodges, J.R.; Kiernan, M.C. Amyotrophic lateral sclerosis and frontotemporal dementia: Distinct and overlapping changes in eating behaviour and metabolism. Lancet Neurol. 2016, 15, 332–342. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.-L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Abdel-Khalik, J.; Yutuc, E.; Crick, P.J.; Gustafsson, J.-Å.; Warner, M.; Roman, G.; Talbot, K.; Gray, E.; Griffiths, W.J.; Turner, M.R.; et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J. Lipid Res. 2017, 58, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-M.; Noh, M.-Y.; Kim, H.; Cheon, S.-Y.; Lee, K.M.; Lee, J.; Cha, E.; Park, K.S.; Lee, K.-W.; Sung, J.-J.; et al. 25-Hydroxycholesterol is involved in the pathogenesis of amyotrophic lateral sclerosis. Oncotarget 2017, 8, 11855–11867. [Google Scholar] [CrossRef]
- Kang, J.; Rivest, S. Lipid metabolism and neuroinflammation in Alzheimer’s disease: A role for liver X receptors. Endocr. Rev. 2012, 33, 715–746. [Google Scholar] [CrossRef]
- Andersson, S.; Gustafsson, N.; Warner, M.; Gustafsson, J.A. Inactivation of liver X receptor beta leads to adult-onset motor neuron degeneration in male mice. Proc. Natl. Acad. Sci. USA 2005, 102, 3857–3862. [Google Scholar] [CrossRef]
- Bigini, P.; Steffensen, K.R.; Ferrario, A.; Diomede, L.; Ferrara, G.; Barbera, S.; Salzano, S.; Fumagalli, E.; Ghezzi, P.; Mennini, T.; et al. Neuropathologic and biochemical changes during disease progression in liver X receptor beta-/- mice, a model of adult neuron disease. J. Neuropathol. Exp. Neurol. 2010, 69, 593–605. [Google Scholar] [CrossRef]
- Kim, H.J.; Fan, X.; Gabbi, C.; Yakimchuk, K.; Parini, P.; Warner, M.; Gustafsson, J.A. Liver X receptor beta (LXRbeta): A link between beta-sitosterol and amyotrophic lateral sclerosis-Parkinson’s dementia. Proc. Natl. Acad. Sci. USA 2008, 105, 2094–2099. [Google Scholar] [CrossRef]
- Wilson, J.M.B.; Shaw, C.A. Late appearance of glutamate transporter defects in a murine model of ALS-parkinsonism dementia complex. Neurochem. Int. 2007, 50, 1067–1077. [Google Scholar] [CrossRef]
- Xu, Z.; Lee, A.; Nouwens, A.; Henderson, R.D.; McCombe, P.A. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 362–376. [Google Scholar] [CrossRef]
- Mouzat, K.; Molinari, N.; Kantar, J.; Polge, A.; Corcia, P.; Couratier, P.; Clavelou, P.; Juntas-Morales, R.; Pageot, N.; Lobaccaro, J.-M.A.; et al. Liver X Receptor Genes Variants Modulate ALS Phenotype. Mol. Neurobiol. 2018, 55, 1959–1965. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Robinson, M.; Lee, B.Y.; Hane, F.T. Recent Progress in Alzheimer’s Disease Research, Part 2: Genetics and Epidemiology. J. Alzheimers Dis. 2017, 57, 317–330. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Jick, H.; Zornberg, G.L.; Jick, S.S.; Seshadri, S.; Drachman, D.A. Statins and the risk of dementia. Lancet 2000, 356, 1627–1631. [Google Scholar] [CrossRef]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef]
- Koldamova, R.P.; Lefterov, I.M.; Staufenbiel, M.; Wolfe, D.; Huang, S.; Glorioso, J.C.; Walter, M.; Roth, M.G.; Lazo, J.S. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem. 2005, 280, 4079–4088. [Google Scholar] [CrossRef]
- Zelcer, N.; Khanlou, N.; Clare, R.; Jiang, Q.; Reed-Geaghan, E.G.; Landreth, G.E.; Vinters, H.V.; Tontonoz, P. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10601–10606. [Google Scholar] [CrossRef]
- Sandoval-Hernández, A.G.; Buitrago, L.; Moreno, H.; Cardona-Gómez, G.P.; Arboleda, G. Role of Liver X Receptor in AD Pathophysiology. PLoS ONE 2015, 10, e0145467. [Google Scholar] [CrossRef]
- Vanmierlo, T.; Rutten, K.; Dederen, J.; Bloks, V.W.; van Vark-van der Zee, L.C.; Kuipers, F.; Kiliaan, A.; Blokland, A.; Sijbrands, E.J.G.; Steinbusch, H.; et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 2011, 32, 1262–1272. [Google Scholar] [CrossRef]
- Cui, W.; Sun, Y.; Wang, Z.; Xu, C.; Peng, Y.; Li, R. Liver X receptor activation attenuates inflammatory response and protects cholinergic neurons in APP/PS1 transgenic mice. Neuroscience 2012, 210, 200–210. [Google Scholar] [CrossRef]
- Báez-Becerra, C.; Filipello, F.; Sandoval-Hernández, A.; Arboleda, H.; Arboleda, G. Liver X Receptor Agonist GW3965 Regulates Synaptic Function upon Amyloid Beta Exposure in Hippocampal Neurons. Neurotox. Res. 2018, 33, 569–579. [Google Scholar] [CrossRef]
- Sharpe, L.J.; Wong, J.; Garner, B.; Halliday, G.M.; Brown, A.J. Is seladin-1 really a selective Alzheimer’s disease indicator? J. Alzheimers Dis. 2012, 30, 35–39. [Google Scholar] [CrossRef]
- Wisniewski, T.; Newman, K.; Javitt, N.B. Alzheimer’s disease: Brain desmosterol levels. J. Alzheimers Dis. 2013, 33, 881–888. [Google Scholar] [CrossRef]
- Adighibe, O.; Arepalli, S.; Duckworth, J.; Hardy, J.; Wavrant-De Vrièze, F. Genetic variability at the LXR gene (NR1H2) may contribute to the risk of Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1431–1434. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, E.; Sánchez-Juan, P.; Mateo, I.; Infante, J.; Sánchez-Quintana, C.; García-Gorostiaga, I.; Berciano, J.; Combarros, O. Interaction between CD14 and LXRbeta genes modulates Alzheimer’s disease risk. J. Neurol. Sci. 2008, 264, 97–99. [Google Scholar] [CrossRef]
- Natunen, T.; Helisalmi, S.; Vepsäläinen, S.; Sarajärvi, T.; Antikainen, L.; Mäkinen, P.; Herukka, S.-K.; Koivisto, A.M.; Haapasalo, A.; Soininen, H.; et al. Genetic Analysis of Genes Involved in Amyloid-β Degradation and Clearance in Alzheimer’s Disease. J. Alzheimers Dis. 2012, 28, 553–559. [Google Scholar] [CrossRef]
- Natunen, T.; Martiskainen, H.; Sarajärvi, T.; Helisalmi, S.; Pursiheimo, J.-P.; Viswanathan, J.; Laitinen, M.; Mäkinen, P.; Kauppinen, T.; Rauramaa, T.; et al. Effects of NR1H3 genetic variation on the expression of liver X receptor α and the progression of Alzheimer’s disease. PLoS ONE 2013, 8, e80700. [Google Scholar] [CrossRef]
- Nylander, A.; Hafler, D.A. Multiple sclerosis. J. Clin. Investig. 2012, 122, 1180–1188. [Google Scholar] [CrossRef]
- Browne, P.; Chandraratna, D.; Angood, C.; Tremlett, H.; Baker, C.; Taylor, B.V.; Thompson, A.J. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology 2014, 83, 1022–1024. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, R.; Rashid, W. Multiple sclerosis. Am. Fam. Phys. 2013, 87, 712–714. [Google Scholar]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shahi, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar]
- Björkhem, I.; Cedazo-Minguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Aspects Med. 2009, 30, 171–179. [Google Scholar] [CrossRef]
- Xu, P.; Li, D.; Tang, X.; Bao, X.; Huang, J.; Tang, Y.; Yang, Y.; Xu, H.; Fan, X. LXR agonists: New potential therapeutic drug for neurodegenerative diseases. Mol. Neurobiol. 2013, 48, 715–728. [Google Scholar] [CrossRef]
- Leoni, V.; Masterman, T.; Diczfalusy, U.; De Luca, G.; Hillert, J.; Björkhem, I. Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci. Lett. 2002, 331, 163–166. [Google Scholar] [CrossRef]
- Fellows Maxwell, K.; Bhattacharya, S.; Bodziak, M.L.; Jakimovski, D.; Hagemeier, J.; Browne, R.W.; Weinstock-Guttman, B.; Zivadinov, R.; Ramanathan, M. Oxysterols and apolipoproteins in multiple sclerosis: A 5 year follow-up study. J. Lipid Res. 2019, 60, 1190–1198. [Google Scholar] [CrossRef]
- Crick, P.J.; Griffiths, W.J.; Zhang, J.; Beibel, M.; Abdel-Khalik, J.; Kuhle, J.; Sailer, A.W.; Wang, Y. Reduced Plasma Levels of 25-Hydroxycholesterol and Increased Cerebrospinal Fluid Levels of Bile Acid Precursors in Multiple Sclerosis Patients. Mol. Neurobiol. 2017, 54, 8009–8020. [Google Scholar] [CrossRef]
- Shackleford, G.G.; Grenier, J.; Abi Habib, W.; Massaad, C.; Meffre, D. Liver X Receptors differentially modulate central myelin gene mRNA levels in a region-, age- and isoform-specific manner. J. Steroid Biochem. Mol. Biol. 2017, 169, 61–68. [Google Scholar] [CrossRef]
- Wang, Z.; Sadovnick, A.D.; Traboulsee, A.L.; Ross, J.P.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; de Lemos, M.; Greenwood, T.; Lee, J.D.; et al. Nuclear Receptor NR1H3 in Familial Multiple Sclerosis. Neuron 2016, 90, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sadovnick, A.D.; Traboulsee, A.L.; Ross, J.P.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; de Lemos, M.; Greenwood, T.; Lee, J.D.; et al. Editorial Note to:Nuclear Receptor NR1H3 in Familial Multiple Sclerosis. Neuron 2016, 92, 331–332. [Google Scholar] [CrossRef]
- Antel, J.; Ban, M.; Baranzini, S.; Barcellos, L.; Barizzone, N.; Beecham, A.; Berge, T.; Bernardinelli, L.; Booth, D.; Bos, S.; et al. NR1H3 p.Arg415Gln Is Not Associated to Multiple Sclerosis Risk. Neuron 2016, 92, 333–335. [Google Scholar] [CrossRef] [Green Version]
- Minikel, E.V.; MacArthur, D.G. Publicly Available Data Provide Evidence against NR1H3 R415Q Causing Multiple Sclerosis. Neuron 2016, 92, 336–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sadovnick, A.D.; Traboulsee, A.L.; Ross, J.P.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; de Lemos, M.; Greenwood, T.; Lee, J.D.; et al. Case-Control Studies Are Not Familial Studies. Neuron 2016, 92, 339–341. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouzat, K.; Chudinova, A.; Polge, A.; Kantar, J.; Camu, W.; Raoul, C.; Lumbroso, S. Regulation of Brain Cholesterol: What Role Do Liver X Receptors Play in Neurodegenerative Diseases? Int. J. Mol. Sci. 2019, 20, 3858. https://doi.org/10.3390/ijms20163858
Mouzat K, Chudinova A, Polge A, Kantar J, Camu W, Raoul C, Lumbroso S. Regulation of Brain Cholesterol: What Role Do Liver X Receptors Play in Neurodegenerative Diseases? International Journal of Molecular Sciences. 2019; 20(16):3858. https://doi.org/10.3390/ijms20163858
Chicago/Turabian StyleMouzat, Kevin, Aleksandra Chudinova, Anne Polge, Jovana Kantar, William Camu, Cédric Raoul, and Serge Lumbroso. 2019. "Regulation of Brain Cholesterol: What Role Do Liver X Receptors Play in Neurodegenerative Diseases?" International Journal of Molecular Sciences 20, no. 16: 3858. https://doi.org/10.3390/ijms20163858