Association between Lysosomal Dysfunction and Obesity-Related Pathology: A Key Knowledge to Prevent Metabolic Syndrome

 , , , and
, , , and
Abstract
:1. Introduction
2. Overview of Lysosomes and Lysosome-Associated Diseases
3. Lysosomal Proteases
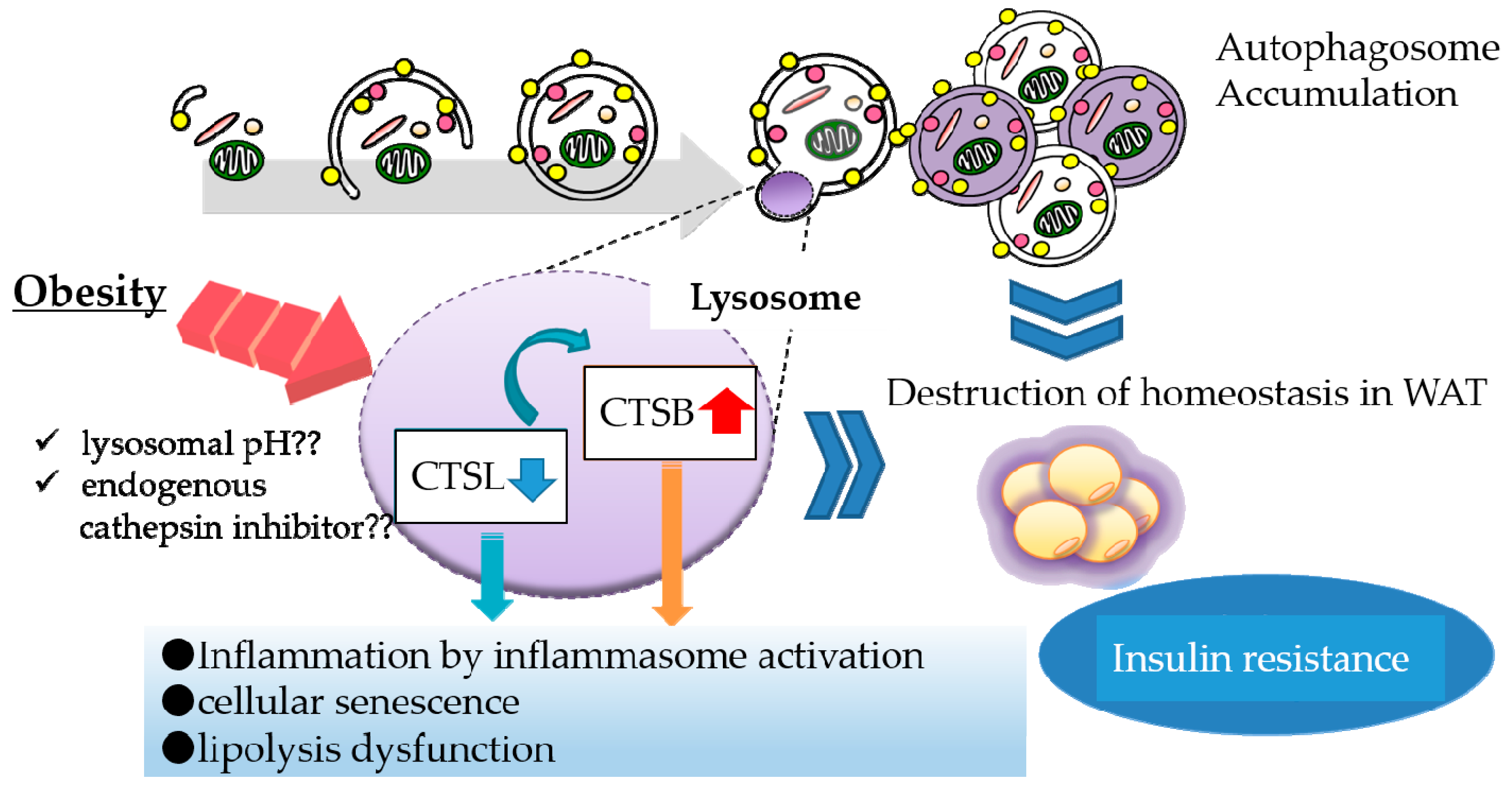
4. Lysosomal Dysfunction in Obese Adipose Tissue
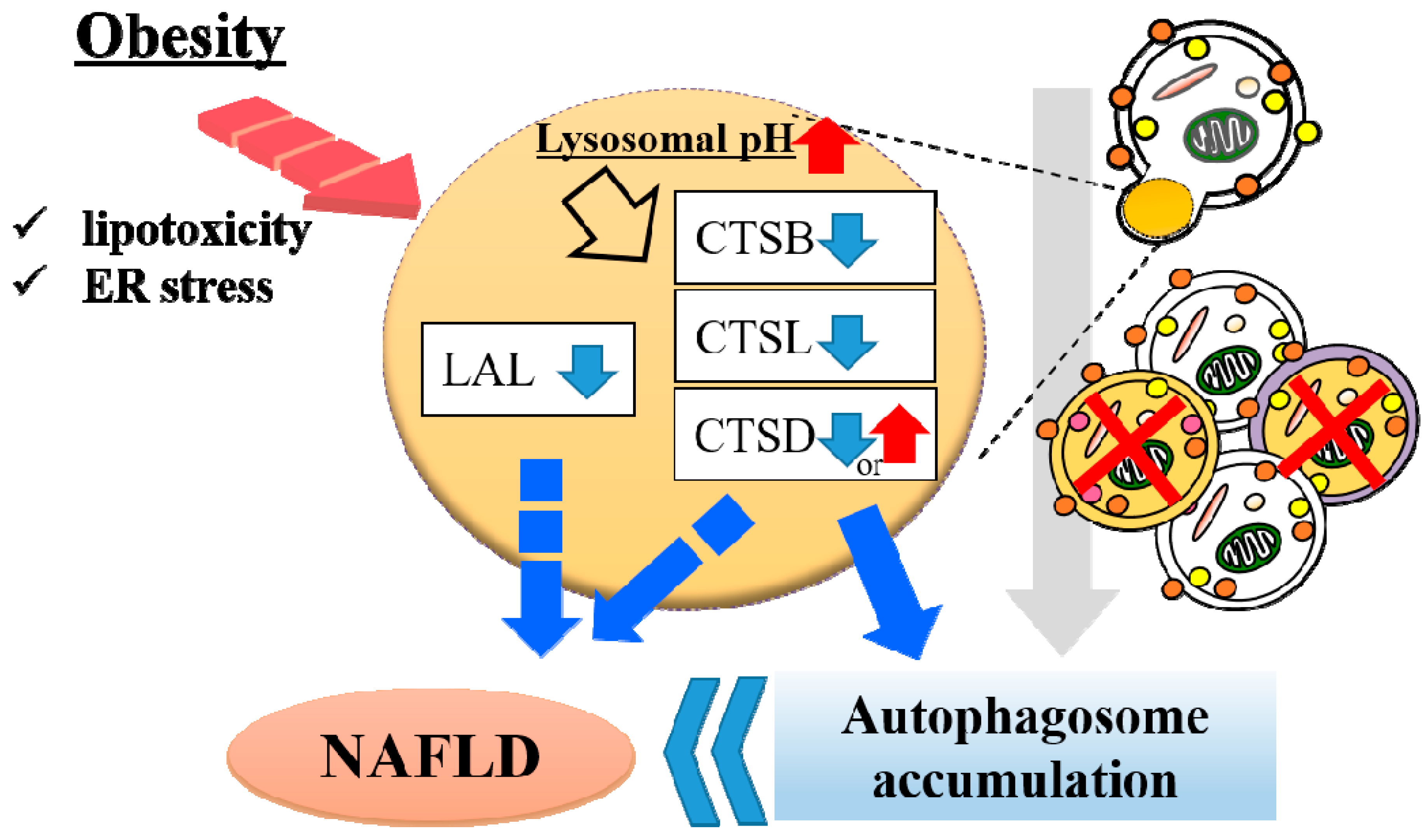
5. Lysosomal Dysfunction in Obese Liver
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Gaal, L.F.; Mertens, I.L.; de Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell Biol. 2015, 208, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. The adipose organ. Prostaglandins Leukot. Essent. Fatty Acids. 2005, 73, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Sindhu, S.; Thomas, R.; Shihab, P.; Sriraman, D.; Behbehani, K.; Ahmad, R. Obesity Is a Positive Modulator of IL-6R and IL-6 Expression in the Subcutaneous Adipose Tissue: Significance for Metabolic Inflammation. PLoS ONE 2015, 10, e0133494. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Alessi, M.C.; Poggi, M.; Juhan-Vague, I. Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Curr. Opin. Lipidol. 2007, 18, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, Y.; Shimano, H. CREBH Regulates Systemic Glucose and Lipid Metabolism. Int. J. Mol. Sci 2018, 19, 1396. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Williams, R. Global challenges in liver disease. Hepatology 2006, 44, 3. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. GI Epideniology: Nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Sherif, Z.A.; Saeed, A.; Ghavimi, S.; Nouraie, S.M.; Laiyemo, A.O.; Brim, H.; Ashktorab, H. Global Epidemiology of Nonalcoholic Fatty Liver Disease and Perspectives on US Minority Populations. Dig. Dis. Sci. 2016, 61, 1214–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Schwake, M.; Schröder, B.; Saftig, P. Lysosomal membrane proteins and their central role in physiology. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. (Lausanne) 2019, 9, 778. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef]
- Ozawa, T.; Koide, R.; Nakata, Y.; Saitsu, H.; Matsumoto, N.; Takahashi, K.; Nakano, I.; Orimo, S. A novel WDR45 mutation in a patient with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA). Am. J. Med. Genet. A 2014, 164A, 2388–2390. [Google Scholar] [CrossRef]
- Cox, T.M.; Cachón-González, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2012, 226, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Kohler, L.; Puertollano, R.; Raben, N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics 2018, 15, 928–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, G.; Yao, Y.; Liu, J.; Yu, Z.; Cheung, S.; Xie, A.; Liang, X.; Bi, X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am. J. Pathol. 2007, 171, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Scita, G.; Di Fiore, P.P. The endocytic matrix. Nature 2010, 463, 464–473. [Google Scholar] [CrossRef]
- Zhao, G.N.; Zhang, P.; Gong, J.; Zhang, X.J.; Wang, P.X.; Yin, M.; Jiang, Z.; Shen, L.J.; Ji, Y.X.; Tong, J.; et al. Tmbim1 is a multivesicular body regulator that protects against non-alcoholic fatty liver disease in mice and monkeys by targeting the lysosomal degradation of Tlr4. Nat. Med. 2017, 23, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Li, F.Y.; Lam, K.S.; Li, H.; Jia, W.; Wang, Y.; Man, K.; Lo, C.M.; Li, X.; Xu, A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 2012, 61, 1058–1067. [Google Scholar] [CrossRef] [Green Version]
- Dall, E.; Brandstetter, H. Structure and function of legumain in health and disease. Biochimie 2016, 122, 126–150. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Kon, Y.; Konno, A.; Iwanaga, T. Cellular distribution of napsin (kidney-derived aspartic protease-like protein, KAP) mRNA in the kidney, lung and lymphatic organs of adult and developing mice. Arch. Histol. Cytol. 2001, 64, 319–327. [Google Scholar] [CrossRef]
- Ketterer, S.; Gomez-Auli, A.; Hillebrand, L.E.; Petrera, A.; Ketscher, A.; Reinheckel, T. Inherited diseases caused by mutations in cathepsin protease genes. FEBS J. 2017, 284, 1437–1454. [Google Scholar] [CrossRef] [Green Version]
- Turk, V.; Turk, B.; Turk, D. Lysosomal cysteine proteases: Facts and opportunities. EMBO J. 2001, 20, 4629–4633. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Katunuma, N. Posttranslational processing and modification of cathepsins and cystatins. J. Signal. Transduct. 2010, 2010, 375345. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 2012, 1824, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, V.; Zhivotovsky, B. Proteases in autophagy. Biochim. Biophys. Acta 2012, 1824, 44–50. [Google Scholar] [CrossRef]
- Turk, D.; Janjić, V.; Stern, I.; Podobnik, M.; Lamba, D.; Dahl, S.W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. Structure of human dipeptidyl peptidase I (cathepsin C): Exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001, 20, 6570–6582. [Google Scholar] [CrossRef]
- Katunuma, N.; Kominami, E. Structures and functions of lysosomal thiol proteinases and their endogenous inhibitor. Curr. Top. Cell. Regul. 1983, 22, 71–101. [Google Scholar]
- Towatari, T.; Katunuma, N. Amino acid sequence of rat liver cathepsin L. FEBS Lett. 1988, 236, 57–61. [Google Scholar] [CrossRef]
- Ishidoh, K.; Kominami, E.; Suzuki, K.; Katunuma, N. Gene structure and 5’-upstream sequence of rat cathepsin L. FEBS Lett. 1989, 259, 71–74. [Google Scholar] [CrossRef]
- Takio, K.; Towatari, T.; Katunuma, N.; Titani, K. Primary structure study of rat liver cathepsin B—A striking resemblance to papain. Biochem. Biophys. Res. Commun. 1980, 97, 340–346. [Google Scholar] [CrossRef]
- Towatari, T.; Katunuma, N. Crystallization and amino acid composition of cathepsin B from rat liver lysosomes. Biochem. Biophys. Res. Commun. 1978, 83, 513–520. [Google Scholar] [CrossRef]
- Gelb, B.D.; Shi, G.P.; Heller, M.; Weremowicz, S.; Morton, C.; Desnick, R.J.; Chapman, H.A. Structure and chromosomal assignment of the human cathepsin K gene. Genomics 1997, 41, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef]
- Turk, B.; Bieth, J.G.; Björk, I.; Dolenc, I.; Turk, D.; Cimerman, N.; Kos, J.; Colic, A.; Stoka, V.; Turk, V. Regulation of the activity of lysosomal cysteine proteinases by pH-induced inactivation and/or endogenous protein inhibitors, cystatins. Biol. Chem. Hoppe Seyler 1995, 376, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Potts, W.; Bowyer, J.; Jones, H.; Tucker, D.; Freemont, A.J.; Millest, A.; Martin, C.; Vernon, W.; Neerunjun, D.; Slynn, G.; et al. Cathepsin L-deficient mice exhibit abnormal skin and bone development and show increased resistance to osteoporosis following ovariectomy. Int J. Exp. Pathol. 2004, 85, 85–96. [Google Scholar] [CrossRef]
- Stypmann, J.; Gläser, K.; Roth, W.; Tobin, D.J.; Petermann, I.; Matthias, R.; Mönnig, G.; Haverkamp, W.; Breithardt, G.; Schmahl, W.; et al. Dilated cardiomyopathy in mice deficient for the lysosomal cysteine peptidase cathepsin L. Proc. Natl. Acad. Sci. USA 2002, 99, 6234–6239. [Google Scholar] [CrossRef]
- Petermann, I.; Mayer, C.; Stypmann, J.; Biniossek, M.L.; Tobin, D.J.; Engelen, M.A.; Dandekar, T.; Grune, T.; Schild, L.; Peters, C.; et al. Lysosomal, cytoskeletal, and metabolic alterations in cardiomyopathy of cathepsin L knockout mice. FASEB J. 2006, 20, 1266–1268. [Google Scholar] [CrossRef]
- Spira, D.; Stypmann, J.; Tobin, D.J.; Petermann, I.; Mayer, C.; Hagemann, S.; Vasiljeva, O.; Günther, T.; Schüle, R.; Peters, C.; et al. Cell type-specific functions of the lysosomal protease cathepsin L in the heart. J. Biol. Chem. 2007, 282, 37045–37052. [Google Scholar] [CrossRef]
- Felbor, U.; Kessler, B.; Mothes, W.; Goebel, H.H.; Ploegh, H.L.; Bronson, R.T.; Olsen, B.R. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc. Natl. Acad. Sci. USA 2002, 99, 7883–7888. [Google Scholar] [CrossRef] [PubMed]
- Cocchiaro, P.; De Pasquale, V.; Della Morte, R.; Tafuri, S.; Avallone, L.; Pizard, A.; Moles, A.; Pavone, L.M. The Multifaceted Role of the Lysosomal Protease Cathepsins in Kidney Disease. Front. Cell Dev. Biol. 2017, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Fang, J.; Ao, G.Z. Cathepsin B and L inhibitors: A patent review (2010 - present). Expert Opin. Ther. Pat. 2017, 27, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Gornicka, A.; Fettig, J.; Eguchi, A.; Berk, M.P.; Thapaliya, S.; Dixon, L.J.; Feldstein, A.E. Adipocyte hypertrophy is associated with lysosomal permeability both in vivo and in vitro: Role in adipose tissue inflammation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E597–E606. [Google Scholar] [CrossRef]
- Hannaford, J.; Guo, H.; Chen, X. Involvement of cathepsins B and L in inflammation and cholesterol trafficking protein NPC2 secretion in macrophages. Obesity (Silver Spring) 2013, 21, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Soussi, H.; Reggio, S.; Alili, R.; Prado, C.; Mutel, S.; Pini, M.; Rouault, C.; Clément, K.; Dugail, I. DAPK2 Downregulation Associates with Attenuated Adipocyte Autophagic Clearance in Human Obesity. Diabetes 2015, 64, 3452–3463. [Google Scholar] [CrossRef]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef] [Green Version]
- Ju, L.; Han, J.; Zhang, X.; Deng, Y.; Yan, H.; Wang, C.; Li, X.; Chen, S.; Alimujiang, M.; Li, X.; et al. Obesity-associated inflammation triggers an autophagy-lysosomal response in adipocytes and causes degradation of perilipin 1. Cell Death Dis. 2019, 10, 121. [Google Scholar] [CrossRef]
- Inami, Y.; Yamashina, S.; Izumi, K.; Ueno, T.; Tanida, I.; Ikejima, K.; Watanabe, S. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem. Biophys. Res. Commun. 2011, 412, 618–625. [Google Scholar] [CrossRef]
- Fukuo, Y.; Yamashina, S.; Sonoue, H.; Arakawa, A.; Nakadera, E.; Aoyama, T.; Uchiyama, A.; Kon, K.; Ikejima, K.; Watanabe, S. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol. Res. 2014, 44, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Chu, E.S.H.; Chen, X.; Kang, W.; Wu, F.; To, K.F.; Wong, V.W.S.; Chan, H.L.Y.; Chan, M.T.V.; et al. Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J. 2018, 32, 37–51. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Kusunoki, C.; Kondo, M.; Yasuda, M.; Kume, S.; Morino, K.; Sekine, O.; Ugi, S.; Uzu, T.; Nishio, Y.; et al. Autophagy regulates inflammation in adipocytes. Biochem. Biophys. Res. Commun. 2012, 417, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Mikami, K.; Okita, N.; Tokunaga, Y.; Ichikawa, T.; Okazaki, T.; Takemoto, K.; Nagai, W.; Matsushima, S.; Higami, Y. Autophagosomes accumulate in differentiated and hypertrophic adipocytes in a p53-independent manner. Biochem. Biophys. Res. Commun. 2012, 427, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Naour, N.; Fellahi, S.; Renucci, J.F.; Poitou, C.; Rouault, C.; Basdevant, A.; Dutour, A.; Alessi, M.C.; Bastard, J.P.; Clément, K.; et al. Potential contribution of adipose tissue to elevated serum cystatin C in human obesity. Obesity (Silver Spring) 2009, 17, 2121–2126. [Google Scholar] [CrossRef] [PubMed]
- Bevec, T.; Stoka, V.; Pungercic, G.; Dolenc, I.; Turk, V. Major histocompatibility complex class II-associated p41 invariant chain fragment is a strong inhibitor of lysosomal cathepsin L. J. Exp. Med. 1996, 183, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Bruchard, M.; Mignot, G.; Derangère, V.; Chalmin, F.; Chevriaux, A.; Végran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walenbergh, S.M.; Houben, T.; Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Vreugdenhil, A.C.; Adriaanse, M.P.; Buurman, W.A.; Hofker, M.H.; Mosca, A.; et al. Plasma cathepsin D levels: A novel tool to predict pediatric hepatic inflammation. Am. J. Gastroenterol. 2015, 110, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Walenbergh, S.M.; Houben, T.; Rensen, S.S.; Bieghs, V.; Hendrikx, T.; van Gorp, P.J.; Oligschlaeger, Y.; Jeurissen, M.L.; Gijbels, M.J.; Buurman, W.A.; et al. Plasma cathepsin D correlates with histological classifications of fatty liver disease in adults and responds to intervention. Sci. Rep. 2016, 6, 38278. [Google Scholar] [CrossRef] [PubMed]
- Houben, T.; Oligschlaeger, Y.; Hendrikx, T.; Bitorina, A.V.; Walenbergh, S.M.A.; van Gorp, P.J.; Gijbels, M.J.J.; Friedrichs, S.; Plat, J.; Schaap, F.G.; et al. Cathepsin D regulates lipid metabolism in murine steatohepatitis. Sci. Rep. 2017, 7, 3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svegliati-Baroni, G.; Pierantonelli, I.; Torquato, P.; Marinelli, R.; Ferreri, C.; Chatgilialoglu, C.; Bartolini, D.; Galli, F. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Werneburg, N.W.; Li, Z.; Bronk, S.F.; Gores, G.J. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1339–G1346. [Google Scholar] [CrossRef] [Green Version]
- Fucho, R.; Martínez, L.; Baulies, A.; Torres, S.; Tarrats, N.; Fernandez, A.; Ribas, V.; Astudillo, A.M.; Balsinde, J.; Garcia-Rovés, P.; et al. ASMase regulates autophagy and lysosomal membrane permeabilization and its inhibition prevents early stage non-alcoholic steatohepatitis. J. Hepatol. 2014, 61, 1126–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, K.; Oe, S.; Honma, Y.; Izumi, H.; Baba, R.; Harada, M. Lipid-Induced Endoplasmic Reticulum Stress Impairs Selective Autophagy at the Step of Autophagosome-Lysosome Fusion in Hepatocytes. Am. J. Pathol. 2016, 186, 1861–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, Ž.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Jones, S.; Ćorić, M.; Calandra, S.; Hamilton, J.; et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Gomaraschi, M.; Bonacina, F.; Norata, G.D. Lysosomal Acid Lipase: From Cellular Lipid Handler to Immunometabolic Target. Trends Pharmacol. Sci. 2019, 40, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Deegan, P.B.; Enns, G.M.; Guardamagna, O.; Horslen, S.; Hovingh, G.K.; Lobritto, S.J.; Malinova, V.; McLin, V.A.; Raiman, J.; et al. Clinical Features of Lysosomal Acid Lipase Deficiency. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pericleous, M.; Kelly, C.; Wang, T.; Livingstone, C.; Ala, A. Wolman’s disease and cholesteryl ester storage disorder: The phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol. Hepatol. 2017, 2, 670–679. [Google Scholar] [CrossRef]
- Du, H.; Heur, M.; Duanmu, M.; Grabowski, G.A.; Hui, D.Y.; Witte, D.P.; Mishra, J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 2001, 42, 489–500. [Google Scholar]
- Leopold, C.; Duta-Mare, M.; Sachdev, V.; Goeritzer, M.; Maresch, L.K.; Kolb, D.; Reicher, H.; Wagner, B.; Stojakovic, T.; Ruelicke, T.; et al. Hepatocyte-specific lysosomal acid lipase deficiency protects mice from diet-induced obesity but promotes hepatic inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 500–511. [Google Scholar] [CrossRef]
- Cahova, M.; Dankova, H.; Palenickova, E.; Papackova, Z.; Komers, R.; Zdychova, J.; Sticova, E.; Kazdova, L. The increased activity of liver lysosomal lipase in nonalcoholic Fatty liver disease contributes to the development of hepatic insulin resistance. Biochem. Res. Int. 2012, 2012, 135723. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Del Ben, M.; Polimeni, L.; Labbadia, G.; Di Santo, S.; Piemonte, F.; Tozzi, G.; Violi, F.; Angelico, F. Reduced Lysosomal Acid Lipase Activity in Adult Patients with Non-alcoholic Fatty Liver Disease. EBioMedicine 2015, 2, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Tovoli, F.; Napoli, L.; Negrini, G.; D’Addato, S.; Tozzi, G.; D’Amico, J.; Piscaglia, F.; Bolondi, L. A Relative Deficiency of Lysosomal Acid Lypase Activity Characterizes Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2017, 18, 1134. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Zhang, Z.; Li, M.; Savage, K.; Cheng, D.; Rauckhorst, A.J.; Ankrum, J.A.; Taylor, E.B.; Ding, W.X.; Xiao, Y.; et al. Hepatic Lysosomal iNOS Activity Impairs Autophagy in Obesity. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 95–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Metabolic Organ | Characteristics | Results | Reference |
|---|---|---|---|
| Adipose tissue (WAT) | Upregulation of CTSB | Increased lysosomal permeablization in adipocytes and contribution to cell death | Gornicka et al., 2012 [75] |
| Upregulation of CTSB | Involvement in mediating the inflammatory response in cholesterol trafficking | Hannaford et al., 2013 [76] | |
| Downregulation of DAPK2 | Modulation of Lysosome-Mediated Remodeling | Soussi et al., 2015 [77] | |
| Downregulation of CTSL | Autophagosome accumulation | Mizunoe et al., 2017 [78] | |
| Complementary upregulation of CTSB | Inflammasome activation in obese WAT | ||
| Upregulation of CTSB | Contribution to the pathogenesis of obesity-related inflammation | Ju et al., 2019 [79] | |
| Liver | Downregulation of CTSB | Autophagosome accumulation in liver from ob/ob mice | Inami et al., 2011 [80] |
| Downregulation of CTSL | |||
| Downregulation of CTSL, CTSB | patients with NAFLD | Fukuo et al., 2014 [81] | |
| Reduced lysosomal acidity Dysfunction of CTSD maturation | Defective lysosomal clearance of autophagosomes | Wang et al., 2018 [82] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizunoe, Y.; Kobayashi, M.; Tagawa, R.; Nakagawa, Y.; Shimano, H.; Higami, Y. Association between Lysosomal Dysfunction and Obesity-Related Pathology: A Key Knowledge to Prevent Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 3688. https://doi.org/10.3390/ijms20153688
Mizunoe Y, Kobayashi M, Tagawa R, Nakagawa Y, Shimano H, Higami Y. Association between Lysosomal Dysfunction and Obesity-Related Pathology: A Key Knowledge to Prevent Metabolic Syndrome. International Journal of Molecular Sciences. 2019; 20(15):3688. https://doi.org/10.3390/ijms20153688
Chicago/Turabian StyleMizunoe, Yuhei, Masaki Kobayashi, Ryoma Tagawa, Yoshimi Nakagawa, Hitoshi Shimano, and Yoshikazu Higami. 2019. "Association between Lysosomal Dysfunction and Obesity-Related Pathology: A Key Knowledge to Prevent Metabolic Syndrome" International Journal of Molecular Sciences 20, no. 15: 3688. https://doi.org/10.3390/ijms20153688