Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy


Institute of Pharmacology, University of Bern, 3010 Bern, Switzerland
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(15), 3624; https://doi.org/10.3390/ijms20153624
Submission received: 28 June 2019
/
Revised: 22 July 2019
/
Accepted: 22 July 2019
/
Published: 24 July 2019
(This article belongs to the Special Issue Lipid as a Cancer Therapeutic Target)
Abstract
:The deregulation of cancer cell metabolic networks is now recognized as one of the hallmarks of cancer. Abnormal lipid synthesis and extracellular lipid uptake are advantageous modifications fueling the needs of uncontrolled cancer cell proliferation. Fatty acids are placed at the crossroads of anabolic and catabolic pathways, as they are implicated in the synthesis of phospholipids and triacylglycerols, or they can undergo β-oxidation. Key players to these decisions are the long-chain acyl-CoA synthetases, which are enzymes that catalyze the activation of long-chain fatty acids of 12–22 carbons. Importantly, the long-chain acyl-CoA synthetases are deregulated in many types of tumors, providing a rationale for anti-tumor therapeutic opportunities. The purpose of this review is to summarize the last up-to-date findings regarding their role in cancer, and to discuss the related emerging tumor targeting opportunities.
Keywords:
ACSL1; ACSL3; ACSL4; ACSL5; ACSL6; cancer; lipid metabolism; fatty acid; cancer therapy; therapy target1. Introduction
1.1. Fatty Acid Metabolism
Lipid metabolism is a wide and intertwined network that mediates multiple functions, such as energy production [1], thermoregulation [2] and the synthesis of signaling molecules [3], while studies have demonstrated that abnormal levels of lipids contribute to numerous pathologies such as cardiovascular diseases, metabolic diseases and cancer [4]. Fatty acid imbalances have a prognostic value, as their levels in serum can act as tumor biomarkers, or they can predict responses to therapy in multiple cancer types, including leukemia [5], non-small cell lung cancer [6,7], breast cancer [8] and colorectal cancer [9,10].
Alterations of main metabolic pathways [11], especially those involving glucose and lipids [12], are among the most striking metabolic features that span many different cancer types [13]. Moreover, it is now clear that different oncogenic drivers induce different metabolic phenotypes in cancer cells. Indeed, the lipid metabolic network remodeling can embrace multiple aspects; some tumors tend to increase the intake of extracellular fatty acids derived from the diet [14,15], while others tend to rely more on de novo fatty acid synthesis [1]. Most enzymes involved in this fatty acid synthesis are under the transcriptional control of the sterol regulatory element binding proteins (SREBPs). The serine/threonine kinase Akt (also known as protein kinase B (PKB)), through the mammalian target of rapamycin complex 1 (mTORC1), induces the expression of SREBP target genes in order to control de novo lipid synthesis [16]. In de novo lipid synthesis, the acetyl-CoA is the building block for fatty acids and can be generated from citrate or acetate. Citrate comes from either glycolysis followed by the tricarboxylic acid (TCA) cycle or from glutaminolysis followed by reductive carboxylation. Citrate is cleaved by ATP-citrate lyase (ACLY) to form acetyl-CoA and oxaloacetate. Next, acetyl-CoA is carboxylated by acetyl-CoA carboxylase (ACC) to form malonyl-CoA, and this latter produces palmitate in a reaction catalyzed by fatty acid synthase (FASN) [1]. Palmitate is a completely saturated fatty acid that is subjected to desaturation by desaturases. The stearoyl-CoA desaturase (SCD1), an oxygen-depend fatty-acyl desaturase, catalyzes the biosynthesis of Δ9 monounsaturated fatty acids (MUFA), oleic and palmitoleic acid, from stearic and palmitic acid. Interestingly, SCD1 inhibitors are shown to suppress the formation of tumors that rely on de novo fatty acid synthesis, and are more potent in suppressing cancer cell proliferation compared to ACC or FASN inhibitors [17,18]. However, recent evidence suggests that sapienate biosynthesis, through fatty acid desaturase 2 (FADS2), enables cancer cells to bypass the requirement for SCD1 [19]. Although these results suggest some new emerging targeting opportunities, they also predict a resistance to SCD1 inhibitors. On the other hand, some tumors, for instance those driven by oncogenic RAS, depend upon extracellularly-derived unsaturated fatty acids, and therefore, they are inherently resistant to SCD1 inhibition [15]. Indeed, the suppression of extracellularly-derived fatty acid activation is sufficient to trigger anti-tumor effects in KRAS-driven lung cancer [20]. Interestingly, fatty acid activation catalyzed by acyl-CoA synthetases (ACSs), is essential for the metabolism of both extracellularly-derived and de novo-synthesized fatty acids, suggesting that the development of pharmacological inhibitors against ACSs may trigger a potent anti-tumor response by overcoming a compensatory activation of one of the two pathways. Thus, the use ACSs inhibitors may be particularly relevant in cases of cancer cells that normally rely on de novo fatty acid synthesis, but in a hypoxic tumor environment, where the oxygen-dependent SCD1 is impaired, cells are forced to switch to extracellular fatty acid uptake for survival [15,21].
1.2. Acyl-CoA-Synthetases
Free fatty acids, depending on their chain length, can either freely pass the plasma membrane or be transported by specialized proteins [22,23], but in order to be channeled towards anabolic or catabolic pathways, they require a two-step activation with the following ATP-dependent reaction catalyzed by acyl-CoA synthetases (ACSs):
Fatty Acid + ATP ![Ijms 20 03624 i001]() Acyl-AMP + PPi
Acyl-AMP + PPi
 Acyl-AMP + PPi
Acyl-AMP + PPi
Acyl-AMP + CoASH ![Ijms 20 03624 i002]() Acyl-CoA + AMP
Acyl-CoA + AMP
 Acyl-CoA + AMP
Acyl-CoA + AMP
ACSs, depending upon the fatty chain length of their preferred substrate, are divided into five enzyme families: Short-chain (C2–C4), medium-chain (C4–C12), long-chain (C12–C22), bubblegum (C14–C24) and very long-chain acyl-CoA (C18–C26) [3]. The synthesis of fatty acyl-CoAs is required for different physiological processes, among which: Proliferation and migration [24,25,26], energy fueling [20,27,28], steroid synthesis, reduction of pro-apoptotic free fatty acids [29] and glucose tolerance [30].
The acyl-CoA synthetase long-chain family (ACSLs) comprises five isoenzymes: ACSL1, ACSL3, ACSL4, ACSL5 and ACSL6 [31]. The five ACSL isoenzymes were identified, cloned and characterized by Yamamoto and his colleagues [32,33,34,35]. ACSLs catalyze substrates with a backbone of 12 to 22 carbons, but the isoenzymes differ in their substrate preference. For instance, ACSL1 has a marked preference for oleate and linoleate [36], ACSL3 for myristate, palmitate, arachidonate and eicosapentaenoate [3], ACSL4 for arachidonate [37], ACSL5 is reported to prefer palmitate, palmitoleate, oleate and linoleate [37] and ACSL6 has an equal preference for saturated and polyunsaturated fatty acids with a backbone of C16–C20 [38]. Furthermore, evidence supports the fact that their substrate preference is more complex, and is affected by many factors including membrane quality, coactivators, inhibitors, interactions with other enzymes, specific ACSL cellular location, specific ACSL tissue expression and posttranslational modifications [37,39].
Long-chain acyl-CoAs have multiple metabolic fates, including incorporation into ether lipids by fatty acyl-CoA reductases, incorporation into complex lipids and acylated proteins by acyl-CoA acyltransferases, convertion into acylcarnitines that enter the mitochondria for β-oxidation by carnitine palmitoyltransferase 1 (CPT1), or synthesis of bioactive lipids [40].
Moreover, an important aspect of ACSLs is their participation in lipid droplet biogenesis and the formation of triacylglycerols (TAG) and cholesteryl esters (CE), neutral lipids that constitute the cargo of lipid droplets [3,41,42]. This is in particular relevant in cancer because mounting evidence supports the fact that the number and size of lipid droplets are associated with cancer aggressiveness and resistance to therapy [43,44,45,46].
The ACSL isoforms differ also in their tissue and subcellular distribution. ACSL1 is localized in mitochondria, lipid droplets and microsomes of liver, heart, white and brown adipose tissue and skeletal muscle [3,47]. ACSL3 localizes to the periphery of the lipid droplets of lipogenic cells (for instance, hepatocytes) and thanks to its hydrophobic hairpin-like N-terminus, it is also present on the cytoplasmic face of the endoplasmatic reticulum (ER) [41,42,48]. It is controversial whether ACSL3 also plays a role in the trans-Golgi network, as it is implicated in the post-Golgi trafficking to the plasma membrane [49,50]. ACSL4 is reported to be present at the endosomes [51], peroxisomes [52], in the secretory pathway [49], at the plasma membrane [53] and at ER regions in contact with the mitochondria, named mitochondrial associated membranes [52]. The mechanism of ACSL4 membrane anchoring is not fully understood yet, but it has been proposed that specific splicing variants could express an analogous hydrophobic N-terminal region, as in the case for ACSL3 [52]. High levels of ACSL4 are mostly found in the adrenal gland, ovary, testis and brain [32]. ACSL5 is mainly located at the mitochondria, and it is highly expressed in intestinal mucosa, lung, liver, adrenal gland, kidney and brown adipose tissue [34,54]. ACSL6 is highly expressed in brain tissue [55], but its subcellular localization is still unclear. An extensive description about the subcellular localization and the specific function of ACSL isoforms can be found in the review by Tang and colleagues [4].
2. ACSLs in Cancer
The following chapter brings together the existing literature about the differential expression and, when available, the role of ACSLs in different types of cancers.
2.1. Colorectal Cancer
Three main ACSL isoforms have been found to be upregulated in colorectal cancer, ACSL1, ACSL4 and ACSL6 [28]. Importantly, high levels of ACSL1 and/or ACSL4 expression in patient tumor samples correlates with a worse prognosis [24,28,56]. Overexpression experiments performed in colorectal cancer cell lines evidenced that these isoforms mediate different aspects of carcinogenesis. ACSL1 overexpression elicits an upregulation of the epithelial to mesenchymal transition (EMT) markers N-cadherin and Slug, but is not sufficient for a complete EMT that is, in fact, only achieved by cells upon a combined overexpression of ACSL1, ACSL4 and SCD1 [56]. However, overexpression of either ACSL1 or ACSL4 in colorectal cancer cells is sufficient to increase wound healing, cell invasion [56] and proliferation [24] compared to empty vector control cells. Moreover, in colorectal cancer cells, ACSL4 overexpression and, to a lesser extent ACSL1, cause a shift of energy metabolism towards glucose utilization [24]. Conversely, the shRNA-mediated knockdown of ACSL1 or ACSL4 reduces cell proliferation in colorectal cancer cell lines, with a more marked effect elicited by the ACSL4 knockdown [24]. In support of these findings, another recent report evidenced that ACSL1 knockdown suppresses anchorage-independent growth and reduces cell migration [28].
Another interesting relationship between colorectal cancer and ACSLs involves the regulation of ACSL1 and ACSL4 by non-canonical micro-RNAs (miRNAs). Cruz-Gil and colleagues have shown that ACSL1, ACSL4 and SCD1 are all targets of miR-19b-1. miR-19b-1 is shown to reduce the neutral lipid content of colorectal cancer cells, and interestingly, its expression correlates with a better prognosis for stage II/III colorectal cancer patients. This suggests that miR-19b-1 can act as a prognostic marker in colorectal cancer [57].
Reports suggest different expression levels of ACSL5 in human colorectal cancer. In the study of Gassler et al. [58] it is suggested that ACSL5 expression and enzymatic activity is enhanced in adenomas and adenocarcinomas, while a more recent report from Hartmann et al. [59] describes a more complex picture. According to the latter study, ACSL5 expression levels vary dramatically among colorectal cancer patients. Out of the 72 patient samples assessed by standard immunohistochemical staining, 41 were scored as low-ACSL5 while 31 as high-ACSL5. However, regardless of the differences in ACSL5 status, the survival rate of those patients was similar.
In a bioinformatic study in colorectal cancer using the Oncomine database, it was found that ACSL6 is upregulated compared to healthy tissue [28]. However, due to the lack of experimental evidence, its functional significance remains unknown.
2.2. Breast Cancer
Breast cancer shows a dysregulated lipid metabolism and alterations in fatty acid β-oxidation (FAO) levels. The main characteristic is the progressive increase of lipid droplets, a signature that is strongly associated with the transformation from normal tissue to invasive carcinoma [60]. Some breast cancer-relevant oncogenes have been reported to participate in this lipid network dysregulation in triple-negative breast cancer, on top of them Src [27] and Myc [61]. Moreover, in the context of hypoxia, lipid droplets have been shown to contribute to breast cancer cell survival upon hypoxia re-oxygenation. This is mediated by a hypoxia-inducible factor-1a (HIF-1a)-driven upregulation of the fatty acid binding protein 3 (FABP3) and FABP7 that are responsible for increased fatty acid uptake [46].
ACSL1 results to be transcriptionally upregulated in both estrogen receptor (ER)-negative [62], ER-positive [63] and HER2-positive [62] breast cancer subtypes, and its mRNA levels correlate with poor patient survival [28], but the specific mechanisms and functional significance of their upregulation are unclear.
ACSL3, although found to be upregulated in women with ER-negative breast cancer [64], has also been shown to be downregulated in triple-negative breast cancer [26]. In fact, in triple-negative breast cancer, ACSL3 interacts with the pro-metastatic protein CUB domain-containing protein 1 (CDCP1). CDCP1 inhibits the activity of ACSL3, resulting in a decreased fatty acid activation, and concomitantly, increased FAO. Indeed, in this context, high FAO/low lipid droplet abundance serve as a prognostic marker of triple-negative breast cancer metastatic potential [26].
ACSL4 is upregulated in quadruple-negative breast cancer (triple-negative breast cancer tumors that lack androgen receptor expression) [65]. In this setting, the expression level of ACSL4 could be used as a diagnostic and prognostic biomarker. ACSL4 overexpression results in higher proliferation rates, higher invasion and anchorage independent growth in vitro and increased tumor burden in vivo [65]. Moreover, ACSL4 was shown to confer chemotherapy resistance in ER-positive breast cancer cell lines by upregulating the transcription of ATP-binding cassette transporters (ABC transporters), which are responsible for drug efflux [66].
ACSL5 is upregulated in ER-negative, basal and normal-like subtypes. One of the interesting aspects of ACSL5 expression is its association with estrogen and progesterone receptor expression, but the mechanism of this association remains elusive [62].
Taken together, this evidence highlights the complexity behind the expression of the different ACSL isoenzymes in breast cancer.
2.3. Prostate Cancer
ACSL3 is expressed in both androgen-sensitive and castration-resistant prostate cancer, but in the latter its expression is dramatically higher [28,67]. Interestingly, ACSL3 overexpression leads to the upregulation of enzymes involved in steroids synthesis, one of the mechanisms underlying the development of castration-resistant prostate cancer [67]. In the castration-resistant prostate cancer context, ACSL3 promotes androgen synthesis through adrenal androgen dehydroepiandrosterone sulfate as a substrate, suggesting that ACSL3 may select for castration-resistant cancer cell populations.
Of note, the current standard for the treatment of prostate cancer is androgen-deprivation treatment. However, after androgen-deprivation treatment, most patients develop castration-resistant prostate cancer after 2–3 years. Thus, it would be interesting to assess whether the increased ACSL3 expression is concomitant to the cessation of the efficacy of the androgen-deprivation treatment and the development of castration-resistant prostate cancer. This would suggest that ACSL3-mediated steroidogenesis is an alternative way that cells utilize to cope with the androgen receptor signaling deficiency.
2.4. Melanoma
ACSL3 is upregulated in melanoma, and its levels correlate with a worse patient prognosis, but the molecular mechanism regarding its role is unknown [28]. A signature of dysregulated lipid biosynthesis has been already observed by several reports [68]. Melanoma cells show high fatty acid synthase and acetyl-CoA carboxylase (key enzymes in the de novo pathway of fatty acid synthesis) compared to benign nevi, suggesting an increase in de novo fatty acid synthesis in melanoma patients [68]. High de novo fatty acid synthesis is accompanied by increased lipid droplet content, and altogether has been associated with increased therapy resistance and poor patient survival [69,70]. Because ACSL3 regulates lipid droplet biogenesis and maintenance [41], it would be interesting to investigate whether the lipid droplet increase and therapy resistance in melanoma patients is mediated by ACSL3.
2.5. Liver Cancer
Lipid metabolism occupies a central role in liver function. In liver, lipoproteins are assembled, and the regulation of the bodily distribution of various diet-derived lipid species has to pass through this physiologic and metabolic bottleneck. Hepatoma cell models show that ACSL3 is required for the assembly of very low density lipoproteins (VLDLs) [71] while ACSL1 spans a broader plethora of functions, among which are ER, peroxisome and mitochondrial lipid metabolism [72].
In a study of Nwosu et al. [73], an extensive analysis of publicly available patient-derived hepatocellular carcinoma expression data has been performed. The data were analyzed with a pathway finder algorithm, and the alteration of several pathways was assessed. Interestingly, lipid metabolism is altered in carcinoma samples, and among the ACSL family members, ACSL3 and ACSL4 expression results being up-regulated compared to a healthy liver. In parallel, ACSL1 and ACSL5 are down-regulated. These data are in agreement with the results of Chen et al. [28] and Liang et al. [74]. Finally, ACSL6 contributes to the accumulation of lipid droplets in fatty liver disease [75], with the latter being involved in the etiology of hepatocellular carcinoma [76]. Thus, it is tempting to speculate that the ACSL6-dependent lipid droplet increase could be required for the early stages of hepatocellular carcinoma development.
2.6. Lung Cancer
ACSL1 is reported to be downregulated in non-small cell lung cancer. Moreover, in vitro experiments show that RNA interference-mediated ACSL1 knockdown enhances the proliferation and invasiveness of non-small cell lung cancer cell lines [28]. These data indicate an anti-tumor role of ACSL1 in non-small cell lung cancer. On the other hand, bioinformatic analyses [28] and tumor tissue microarray (TMA) staining [20] showed that ACSL3 is up-regulated in lung cancer compared to the healthy lung tissue. Moreover, the H-score of ACSL3 (which takes into consideration the staining intensity in conjunction with the percentage of positive cells) appears overall reduced, with the ACSL3 levels being inversely proportional to the tumor stage. These results support a role of ACSL3 in tumor initiation, but not maintenance. This is additionally corroborated by a Kras-driven mouse model of lung tumorigenesis where the loss of Acsl3 suppressed the tumor onset [20]. Interestingly, in this context, oncogenic KRAS induces the ACSL3 promoter activity through the mammalian target of rapamycin complex 1 (mTORC1) regulation [20]. Whether this is a conserved mechanism in all KRAS-mutant tumors remains to be addressed.
2.7. Soft Tissue Cancers
The expression levels of ACSLs in soft tissue cancers differ by isoenzyme and cancer type. Immunohistochemical staining revealed that ACSL3 is moderately to highly expressed in human fibrosarcomas, leiomyosarcomas and rhabdomyosarcomas [77]. On the other hand, low intensity staining was obtained for liposarcoma samples and a very scattered expression pattern was evidenced in dermatofibrosarcoma. Regarding ACSL4, a less defined pattern can be found; in fact, its expression is less homogeneous among the same tumor type. Notably, ACSL4 is highly expressed in some leiomyosarcomas and rhabdomyosarcomas, but is more consistently overexpressed in fibrosarcomas [77].
2.8. Blood Cancers
ACSL1 and ACSL6 are reported to be downregulated in a pan-leukemia dataset [28]. Accordingly, low levels of ACSL6 significantly correlate with poor patient survival in acute myelogenous leukemia, suggesting a tumor suppressing role in this context [28]. On the other hand, although it is known that lipid metabolism is dysregulated in acute myeloid leukemia [13], a small body of literature is available regarding the status and the role of ACSL3, ACSL4 and ACSL5 in blood cancers. Enhanced pathways are fatty acid synthesis [78] and FAO [78,79]. Therefore, it could be hypothesized that ACSL3, ACSL4 and ACSL5 are involved in the aforementioned metabolic re-shaping of cancer cells.
3. Pharmacological Targeting of ACSLs
A few compounds are known for their ability to target ACSLs, but none are selective. The main issue about that is the conserved catalytic domain that makes the five ACSL isoenzymes have redundant catalytic functions [3]. The lead compound that is able to directly inhibit ACSL1, ACSL3 and ACSL4, by competing with fatty acids for their catalytic domain, is the fungal metabolite, Triacsin C [80]. At a higher dose, Triacsin C becomes also a competitive inhibitor of ACSL5 [81]. However, Triacsin C is toxic to cells at high concentrations and its polyunsaturated chain would make the pharmacokinetics and cell penetrance properties challenging.
Attempts to increase the activity of Triacsin C have been performed without a substantial success. In one study, the authors generated a library of six compounds by modifying the alkylidene terminal moiety of Triacsin C, and tested their activity in inhibiting the infective and replicative potential of Rotavirus using the lipid droplets content as a readout [82]. Interestingly, one compound named 1e showed a 50% lower median effective dose (ED50) than did Triacsin C. Another follow up study from Prior et al. [83] aimed to expand the previously synthetized library, and to test their inhibitory activity in a direct manner. Twelve different compounds were synthetized this time, and their capacity to inhibit the ACSL isoenzymes was tested either in the protein lysate or in living cells. Although some compounds showed a decrease in 14C-palmitate incorporation, none of the synthetized compounds showed any higher inhibition rate than Triacsin C. Another competitive ACSL inhibitor is 2-fluoropalmitic acid, a modified structure of palmitic acid with a higher affinity for ACSLs than the natural fatty acid [84]. The limitation of this drug is the high IC50. Moreover, no data are available regarding its affinity for any ACSL isoenzyme over others. Since the preference of ACSL3 and ACSL5 for palmitic acid is high, one could speculate that these may be the isoenzymes majorly inhibited by this molecule. Still more experiments have to be performed in order to understand which pharmacophore groups could confer specificity. In this way, the scientific community would finally be able to design more efficient and isoenzyme-selective analogs of Triacsin C.
Alternatively, an approach that could be used to interfere with ACSLs is the use of the antagonists of the peroxisome proliferator-activated receptors (PPARs), which transcriptionally regulate ACSLs [85,86]. Obviously, one drawback of this indirect inhibition is the high presence of off-target effects due to the various PPAR-controlled functions.
4. ACSLs as Therapeutic Targets in Cancer
Here, we report the cancer-specific effects of targeting individual ACSL isoenzymes (summarized in Table 1) and we discuss the emerging therapeutic opportunities (summarized in Figure 1).
4.1. ACSL1
The shRNA-mediated knockdown of ACSL1 in colorectal cancer shows reduced cell proliferation and migration [28]. This suggests a role of ACSL1 as a direct therapeutic target for colorectal cancer. Moreover, given the involvement of ACSL1 in favoring glycolysis [24], a potentially interesting treatment opportunity may arise from the combination of ACSL1 and glycolysis intermediates inhibition in colorectal cancer.
As suggested by Cruz-Gill and colleagues [57], the inhibitory effect of miR-19b-1 on ACSL1, ACSL4 and SCD1 in colorectal carcinoma could be exploited as a therapeutic strategy. An in silico functional analysis performed in colorectal cancer cell lines revealed an effect of miR-19b-1 on focal adhesion formation and cytoskeleton remodeling which caused a reduction in cancer cell invasion and abolished their proliferation. This confirms the potential role of miR-19b-1 in ACSL1/4 axis targeting.
As previously stated, ACSL1 is upregulated in breast cancer, and its expression correlates with poor survival. RNAi experiments show that ACSL1 targeting reduces proliferation, colony formation and cell viability in breast cancer cell lines [28,62], thus rendering ACSL1 an attractive therapeutic target for breast, as well as colorectal cancer.
4.2. ACSL3
ACSL3 is found to be upregulated in fibrosarcomas, ER-negative, prostate cancer, melanoma, hepatocellular carcinomas and lung cancer, suggesting that targeting ACSL3 in this context may be clinically relevant for many cancers. Recent evidence in non-small cell lung cancer suggests that ACSL3 is important in maintaining the channeling of extracellularly-derived lipids to FAO [20]. Thus, FAO inhibitors such as Etomoxir, a carnitine palmitoyltransferase I (CPT1) inhibitor, could have an antitumor effect in lung cancer. On the other hand, in triple-negative breast cancer, ACSL3 is significantly reduced, resulting in high FAO/low lipid droplet abundance, and serves as a poor prognosis marker of the triple-negative breast cancer metastatic potential. However, also in this case, an inhibition of FAO could reduce cancer cell viability and suppress invasiveness.
We previously showed that KRAS drives the transcriptional upregulation of ACSL3 and ACSL4 in non-small cell lung cancer, and that this can be prevented by the inhibition of the mammalian target of rapamycin complex 1 (mTORC1) [20]. mTORC1 is a regulator of the sterol regulatory element-binding protein-1 (SREBP-1) prolipogenic transcription factor, which promotes aberrant proliferation in cancer cells [20]. Interestingly, both the ACSL3 and ACSL4, but not the ACSL1 and ACSL6 promoters, contain SREBP-binding sites. Moreover, mTORC1 inhibition abrogates the ACSL4-mediated chemotherapy resistance in breast cancer cells [66]. Thus, these results strongly support the idea that KRAS controls ACSL3 and ACSL4 through the mTORC1-SREBP signaling axis.
Some recent reports link ACSL3 and an unfolded protein response in cancer [91]. Thapsigargin-induced ER stress results in the downregulation of ACSL3, ACSL4 and solute carrier family 27 member 2 (SLC27A2) in lung cancer. Moreover, in a prostate cancer cell model, ACSL3 overexpression conferred resistance to tunicamycin-sensitive cancer cells [67]. These findings suggest that ACSL3 supports ER function to confer a survival advantage to cancer cells. ER stress triggers cancer cell death in many contexts [92,93,94,95], therefore ER stress inducers together with ACSL3 inhibition could result in enhanced antitumor response.
Finally, ACSL3 is able to drive steroidogenesis in castration-resistant prostatic cancer [67]. Thus, in this context, ACSL3 may be an attractive therapeutic target to prevent prostate cancer relapse.
4.3. ACSL4
Similar to ACSL1, ACSL4 is shown to have the potential for colorectal carcinoma treatment. Specific ACSL4 knockdown shows a reduced colorectal cancer cell proliferation [56]. On the other hand, the inhibition of ACSL4 with a non-toxic dose of Triacsin C does not have a significant impact on the cell viability of colorectal cancer cell lines. This is dramatically ameliorated by co-treatment with the SCD1 inhibitor, A939572 [56]. Accordingly, in vivo experiments performed in a xenograft model of colorectal carcinoma show that the treatment with Triacsin C is not effective in reducing the tumor burden. However, Triacsin C is sufficient to abolish the lipid droplet-mediated therapy resistance to the chemotherapeutic drugs, 5-fluorouracyl and oxaliplatin [90].
In breast cancer, another ACSL4-mediated resistance mechanism has been shown. The ABC transporters, ABCG2, ABCC4 and ABCC8 were identified as ACSL4-responsive genes involved in paclitaxel, cisplatin and doxorubicin resistance [66]. Indeed, combined ACSL4 inhibition with Triacsin C and treatment with the aforementioned chemotherapeutic drugs result in remarkable synergistic antitumor effects. In the light of these data, ACSL4 targeting could increase the efficacy of chemotherapy.
ACSL4 has been shown to be a ferroptosis-sensitizing factor [87]. In the study of Doll et al., the authors showed in mouse embryonic fibroblasts (MEFs) that ACSL4 mediates the incorporation of polyunsaturated fatty acids in phosphatidylethanolamine (PE) and phosphatidylinositol (PI). As a result, the shaping of the phospholipids landscape causes cells to become more sensitive to the ferroptotic inducers [96]. This occurs due to the intrinsic higher lipid peroxidation potential of lipids conjugated with unsaturated fatty acids [97,98]. Moreover, ACSL4 knockout MEFs are resistant to ferroptotic stimuli and the expression of ACSL4 or arachidonic acid supplementation is able to completely restore sensitivity [96]. Interestingly, the role of ACSL4 in sensitizing to ferroptosis has also been confirmed in basal-like breast cancer cell lines [96]. Additionally, in a study assessing the sensitivity of different colorectal carcinoma cell lines to Bromelain treatment (a mixture of proteolytic enzymes derived from pineapple stems), it was shown that ferroptosis was accompanied by higher levels of ACSL4 and lower levels of miRNAs targeting, among others, ACSL4 [25]. Intriguingly, the sensitivity to ferroptosis could be abrogated by RNAi-mediated ACSL4 knockdown or treatment with thiazolidinediones, a class of antidiabetic compounds, that among others, target ACSL4 and ameliorate the tissue demise in a murine model of ferroptosis. Altogether, these results suggest that ACSL4 inhibition is a viable therapeutic approach to prevent ferroptosis-related diseases. Interestingly, triggering ferroptosis could represent a therapeutic strategy for the treatment of many neoplasias, alone or in combination with chemoterapeutic agents [99]. Thus, it could be speculated that ACSL4 may represent a double-edged sword target, since both enhances cell proliferation and invasion and sensitizes cancer cells to ferroptotic stimuli.
The work of Liang and colleagues [74] shows that ACSL4 knockdown decreases the proliferation rates of hepatocellular carcinoma cell lines. Moreover, the expression of ACSL4 is driven by the p38 MAPK signaling pathway, and it can be inhibited by bromo-cAMP or SB203508 (a p38 MAPK inhibitor) treatment [74]. This suggests that ACSL4 may be indirectly targeted with p38 MAPK inhibitors.
4.4. ACSL5
ACSL5 is upregulated in several cancers such as colorectal cancer, ER-negative, basal and normal-like breast cancer subtypes, suggesting that the identification of specific ACSL5 inhibitors is of paramount importance. Moreover, ACSL5 is frequently overexpressed in malignant gliomas [89]. Using gain- and loss-of-function experiments, Mashima et al. showed that ACSL5 selectively promotes human glioma cell survival under conditions of extracellular acidosis [88]. Moreover, Triacsin C enhances the antitumor efficacy of the chemotherapeutic drug etoposide [89]. These results suggest that ACSL5 may be critical for the malignant progression and metastatic dissemination of gliomas, and support the fact that targeting ACSL5 in this context may be an effective therapeutic strategy.
4.5. ACSL6
The data available for ACSL6 suggest that this isoenzyme has opposite roles in different cancers. In colorectal cancer it has been shown to be overexpressed, while in acute myelogenous, leukemia is downregulated, and is proposed to act as a tumor suppressor in this context. However, the lack of experimental evidence precludes further conclusions. Future investigations are needed in order to understand the role of this isoenzyme in different cancers.
5. Conclusions
Here we collected and summarized evidence that ACSLs are potential therapeutic targets in multiple cancer types (Figure 1 and Table 1). The individual ACSL isoenzymes, due to their different fatty acid chain length preference, tissue distribution and subcellular location, are able to channel specific fatty acids toward distinct metabolic fates. In cancer, the expression of most of the isoenzymes is amplified, and this feature can be used to drive rationalized therapeutic approaches.
Validation of the whole-body mouse knockout of some isoenzymes such as ACSL3 [20], ACSL5 [100] and ACSL6 [101] does not result in lethality or any overt dysfunction which renders them cancer cell specific vulnerabilities. Nevertheless, still a great effort is needed towards developing the necessary medicinal chemistry in order to select adequate and selective inhibitors for this purpose.
There is significant caution in the field of metabolic targets progressing through clinical development due to extrinsic supply nullifying any effect of target inhibition [102]. For instance, it was shown that monoacylglycerol lipase (MAGL), by liberating free fatty acids from neutral lipid stores, controls the free fatty acid levels in cancer cells. Indeed, MAGL knockdown suppresses tumorigenesis in mice. However, the suppressed tumor growth that was observed after MAGL knockdown could be rescued by the high-fat diet in mice, indicating that exogenous sources of fatty acids can contribute to malignancy in cancers lacking MAGL activity. Since ACSLs are able to activate fatty acids deriving both from de novo and exogenous sources, a combined inhibition of MAGL and a tissue-relevant ACSL isoenzyme may result in a significant anti-tumor response. Further studies are warranted to assess combination therapies involving ACSL isoenzymes in cancer.
Author Contributions
M.R.S. wrote the review and prepared the figure/table, G.K. wrote the review.
Funding
Funding was provided by the Swiss National Science Foundation (SNSF) professorship (PP00P3_163929) and Novartis Foundation for medical-biological research (16C190).
Acknowledgments
We thank Maria Saliakoura (Institute of Pharmacology, University of Bern) for critically reading the review.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| HCC | Hepatocellular carcinoma |
| ER | Estrogen receptor |
| CRC | Colorectal Carcinoma |
| ACSL1 | Acyl CoA long chain synthetase 1 |
| ACSL3 | Acyl CoA long chain synthetase 3 |
| ACSL4 | Acyl CoA long chain synthetase 4 |
| ACSL5 | Acyl CoA long chain synthetase 5 |
| ACSL6 | Acyl CoA long chain synthetase 6 |
| EMT | Epithelial to Mesenchymal transition |
| SCD1 | Stearoyl-CoA Desaturase 1 |
| CRPC | Castration-resistant prostate cancer |
| BC | Breast cancer |
| FAO | Fatty Acid Oxidation |
| NSCLC | Non-small cell lung cancer |
References
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell. Metab. 2013, 18, 153–161. [Google Scholar] [PubMed]
- Ellis, J.M.; Li, L.O.; Wu, P.C.; Koves, T.R.; Ilkayeva, O.; Stevens, R.D.; Watkins, S.M.; Muoio, D.M.; Coleman, R.A. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell. Metab. 2010, 12, 53–64. [Google Scholar] [PubMed]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [PubMed]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.M.; Lu, G.D. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [PubMed]
- Khalid, A.; Siddiqui, A.J.; Huang, J.H.; Shamsi, T.; Musharraf, S.G. Alteration of serum free fatty acids are indicators for progression of pre-leukaemia diseases to leukaemia. Sci. Rep. 2018, 8, 14883. [Google Scholar] [PubMed]
- Ren, J.; Zhang, D.; Liu, Y.; Zhang, R.; Fang, H.; Guo, S.; Zhou, D.; Zhang, M.; Xu, Y.; Qiu, L.; et al. Simultaneous quantification of serum nonesterified and esterified fatty acids as potential biomarkers to differentiate benign lung diseases from lung cancer. Sci. Rep. 2016, 6, 34201. [Google Scholar] [PubMed]
- Zhang, Y.; He, C.; Qiu, L.; Wang, Y.; Zhang, L.; Qin, X.; Liu, Y.; Zhang, D.; Li, Z. Serum unsaturated free fatty acids: Potential biomarkers for early detection and disease progression monitoring of non-small cell lung cancer. J. Cancer 2014, 5, 706–714. [Google Scholar]
- Shaikh, S.; Channa, N.A.; Talpur, F.N.; Younis, M.; Tabassum, N. Radiotherapy improves serum fatty acids and lipid profile in breast cancer. Lipids Health Dis. 2017, 16, 92. [Google Scholar]
- Zhang, Y.; He, C.; Qiu, L.; Wang, Y.; Qin, X.; Liu, Y.; Li, Z. Serum unsaturated free fatty acids: A potential biomarker panel for early-stage detection of colorectal cancer. J. Cancer 2016, 7, 477–483. [Google Scholar]
- Iemoto, T.; Nishiumi, S.; Kobayashi, T.; Fujigaki, S.; Hamaguchi, T.; Kato, K.; Shoji, H.; Matsumura, Y.; Honda, K.; Yoshida, M. Serum level of octanoic acid predicts the efficacy of chemotherapy for colorectal cancer. Oncol. Lett. 2019, 17, 831–842. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [PubMed]
- Balaban, S.; Nassar, Z.D.; Zhang, A.Y.; Hosseini-Beheshti, E.; Centenera, M.M.; Schreuder, M.; Lin, H.M.; Aishah, A.; Varney, B.; Liu-Fu, F.; et al. Extracellular fatty acids are the major contributor to lipid synthesis in prostate cancer. Mol. Cancer Res. 2019, 17, 949–962. [Google Scholar] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [PubMed]
- Porstmann, T.; Griffiths, B.; Chung, Y.L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. Pkb/akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of srebp. Oncogene 2005, 24, 6465–6481. [Google Scholar]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.P.; Wang, B.; Cohen, A.; et al. Scd1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 2012, 7, e33823. [Google Scholar]
- Peck, B.; Schulze, A. Lipid desaturation—The next step in targeting lipogenesis in cancer? FEBS J. 2016, 283, 2767–2778. [Google Scholar]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant kras lung tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mtorc1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [PubMed]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 1994, 79, 427–436. [Google Scholar] [PubMed]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot Essent Fatty Acids 2010, 82, 149–154. [Google Scholar] [PubMed]
- Sanchez-Martinez, R.; Cruz-Gil, S.; Garcia-Alvarez, M.S.; Reglero, G.; Ramirez de Molina, A. Complementary acsl isoforms contribute to a non-warburg advantageous energetic status characterizing invasive colon cancer cells. Sci. Rep. 2017, 7, 11143. [Google Scholar] [PubMed]
- Park, S.; Oh, J.; Kim, M.; Jin, E.J. Bromelain effectively suppresses kras-mutant colorectal cancer by stimulating ferroptosis. Anim. Cells Syst. (Seoul) 2018, 22, 334–340. [Google Scholar] [PubMed]
- Wright, H.J.; Hou, J.; Xu, B.; Cortez, M.; Potma, E.O.; Tromberg, B.J.; Razorenova, O.V. Cdcp1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2017, 114, E6556–E6565. [Google Scholar] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [PubMed]
- Chen, W.C.; Wang, C.Y.; Hung, Y.H.; Weng, T.Y.; Yen, M.C.; Lai, M.D. Systematic analysis of gene expression alterations and clinical outcomes for long-chain acyl-coenzyme a synthetase family in cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar]
- Li, L.O.; Klett, E.L.; Coleman, R.A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 246–251. [Google Scholar] [Green Version]
- Bauer, P.V.; Duca, F.A.; Waise, T.M.Z.; Dranse, H.J.; Rasmussen, B.A.; Puri, A.; Rasti, M.; O’Brien, C.A.; Lam, T.K.T. Lactobacillus gasseri in the upper small intestine impacts an acsl3-dependent fatty acid-sensing pathway regulating whole-body glucose homeostasis. Cell Metab. 2018, 27, 572–587. [Google Scholar]
- Ellis, J.M.; Frahm, J.L.; Li, L.O.; Coleman, R.A. Acyl-coenzyme a synthetases in metabolic control. Curr. Opin. Lipidol. 2010, 21, 212–217. [Google Scholar] [PubMed]
- Kang, M.J.; Fujino, T.; Sasano, H.; Minekura, H.; Yabuki, N.; Nagura, H.; Iijima, H.; Yamamoto, T.T. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. USA 1997, 94, 2880–2884. [Google Scholar] [PubMed] [Green Version]
- Fujino, T.; Kang, M.J.; Suzuki, H.; Iijima, H.; Yamamoto, T. Molecular characterization and expression of rat acyl-CoA synthetase 3. J. Biol. Chem. 1996, 271, 16748–16752. [Google Scholar] [PubMed]
- Oikawa, E.; Iijima, H.; Suzuki, T.; Sasano, H.; Sato, H.; Kamataki, A.; Nagura, H.; Kang, M.J.; Fujino, T.; Suzuki, H.; et al. A novel acyl-CoA synthetase, acs5, expressed in intestinal epithelial cells and proliferating preadipocytes. J. Biochem. 1998, 124, 679–685. [Google Scholar] [PubMed]
- Mashek, D.G.; Bornfeldt, K.E.; Coleman, R.A.; Berger, J.; Bernlohr, D.A.; Black, P.; DiRusso, C.C.; Farber, S.A.; Guo, W.; Hashimoto, N.; et al. Revised nomenclature for the mammalian long-chain acyl-CoA synthetase gene family. J. Lipid Res. 2004, 45, 1958–1961. [Google Scholar] [PubMed] [Green Version]
- Kanter, J.E.; Tang, C.; Oram, J.F.; Bornfeldt, K.E. Acyl-CoA synthetase 1 is required for oleate and linoleate mediated inhibition of cholesterol efflux through atp-binding cassette transporter a1 in macrophages. Biochim. Biophys. Acta. 2012, 1821, 358–364. [Google Scholar]
- Klett, E.L.; Chen, S.; Yechoor, A.; Lih, F.B.; Coleman, R.A. Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J. Lipid Res. 2017, 58, 884–894. [Google Scholar] [Green Version]
- Lopes-Marques, M.; Cunha, I.; Reis-Henriques, M.A.; Santos, M.M.; Castro, L.F. Diversity and history of the long-chain acyl-CoA synthetase (acsl) gene family in vertebrates. BMC Evol. Biol. 2013, 13, 271. [Google Scholar]
- Spector, A.A.; Yorek, M.A. Membrane lipid composition and cellular function. J. Lipid Res. 1985, 26, 1015–1035. [Google Scholar]
- Yamashita, A.; Hayashi, Y.; Nemoto-Sasaki, Y.; Ito, M.; Oka, S.; Tanikawa, T.; Waku, K.; Sugiura, T. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid Res. 2014, 53, 18–81. [Google Scholar]
- Kassan, A.; Herms, A.; Fernandez-Vidal, A.; Bosch, M.; Schieber, N.L.; Reddy, B.J.; Fajardo, A.; Gelabert-Baldrich, M.; Tebar, F.; Enrich, C.; et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in er microdomains. J. Cell Biol. 2013, 203, 985–1001. [Google Scholar] [PubMed]
- Fujimoto, Y.; Itabe, H.; Kinoshita, T.; Homma, K.J.; Onoduka, J.; Mori, M.; Yamaguchi, S.; Makita, M.; Higashi, Y.; Yamashita, A.; et al. Involvement of acsl in local synthesis of neutral lipids in cytoplasmic lipid droplets in human hepatocyte huh7. J. Lipid Res. 2007, 48, 1280–1292. [Google Scholar] [PubMed]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by pten loss and PI3k/Akt activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [PubMed]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules 2018, 23, 1941. [Google Scholar]
- Ackerman, D.; Tumanov, S.; Qiu, B.; Michalopoulou, E.; Spata, M.; Azzam, A.; Xie, H.; Simon, M.C.; Kamphorst, J.J. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 2018, 24, 2596–2605.e5. [Google Scholar] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by hif-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [PubMed]
- Durgan, D.J.; Smith, J.K.; Hotze, M.A.; Egbejimi, O.; Cuthbert, K.D.; Zaha, V.G.; Dyck, J.R.; Abel, E.D.; Young, M.E. Distinct transcriptional regulation of long-chain acyl-CoA synthetase isoforms and cytosolic thioesterase 1 in the rodent heart by fatty acids and insulin. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2480–H2497. [Google Scholar] [PubMed]
- Poppelreuther, M.; Rudolph, B.; Du, C.; Grossmann, R.; Becker, M.; Thiele, C.; Ehehalt, R.; Fullekrug, J. The n-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J. Lipid Res. 2012, 53, 888–900. [Google Scholar]
- Ansari, I.H.; Longacre, M.J.; Stoker, S.W.; Kendrick, M.A.; O’Neill, L.M.; Zitur, L.J.; Fernandez, L.A.; Ntambi, J.M.; MacDonald, M.J. Characterization of acyl-CoA synthetase isoforms in pancreatic beta cells: Gene silencing shows participation of acsl3 and acsl4 in insulin secretion. Arch. Biochem. Biophys. 2017, 618, 32–43. [Google Scholar]
- Obata, Y.; Fukumoto, Y.; Nakayama, Y.; Kuga, T.; Dohmae, N.; Yamaguchi, N. The lyn kinase c-lobe mediates golgi export of lyn through conformation-dependent acsl3 association. J. Cell Sci. 2010, 123, 2649–2662. [Google Scholar]
- Liu, Z.; Huang, Y.; Hu, W.; Huang, S.; Wang, Q.; Han, J.; Zhang, Y.Q. Dacsl, the drosophila ortholog of acyl-CoA synthetase long-chain family member 3 and 4, inhibits synapse growth by attenuating bone morphogenetic protein signaling via endocytic recycling. J. Neurosci. 2014, 34, 2785–2796. [Google Scholar]
- Lewin, T.M.; Kim, J.H.; Granger, D.A.; Vance, J.E.; Coleman, R.A. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 2001, 276, 24674–24679. [Google Scholar]
- Kuch, E.M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brugger, B.; Sreemmel, W.; Ehehalt, R.; Poppelreuther, M.; Fullekrug, J. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 2014, 1841, 227–239. [Google Scholar]
- Meller, N.; Morgan, M.E.; Wong, W.P.; Altemus, J.B.; Sehayek, E. Targeting of acyl-CoA synthetase 5 decreases jejunal fatty acid activation with no effect on dietary long-chain fatty acid absorption. Lipids Health Dis. 2013, 12, 88. [Google Scholar]
- Fujino, T.; Yamamoto, T. Cloning and functional expression of a novel long-chain acyl-CoA synthetase expressed in brain. J. Biochem. 1992, 111, 197–203. [Google Scholar]
- Sanchez-Martinez, R.; Cruz-Gil, S.; Gomez de Cedron, M.; Alvarez-Fernandez, M.; Vargas, T.; Molina, S.; Garcia, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar]
- Cruz-Gil, S.; Sanchez-Martinez, R.; Gomez de Cedron, M.; Martin-Hernandez, R.; Vargas, T.; Molina, S.; Herranz, J.; Davalos, A.; Reglero, G.; Ramirez de Molina, A. Targeting the lipid metabolic axis acsl/scd in colorectal cancer progression by therapeutic mirnas: Mir-19b-1 role. J. Lipid Res. 2018, 59, 14–24. [Google Scholar]
- Gassler, N.; Herr, I.; Schneider, A.; Penzel, R.; Langbein, L.; Schirmacher, P.; Kopitz, J. Impaired expression of acyl-CoA synthetase 5 in sporadic colorectal adenocarcinomas. J. Pathol. 2005, 207, 295–300. [Google Scholar]
- Hartmann, F.; Sparla, D.; Tute, E.; Tamm, M.; Schneider, U.; Jeon, M.K.; Kasperk, R.; Gassler, N.; Kaemmerer, E. Low acyl-CoA synthetase 5 expression in colorectal carcinomas is prognostic for early tumour recurrence. Pathol. Res. Pract. 2017, 213, 261–266. [Google Scholar]
- Wilson, K.E.; Bachawal, S.V.; Tian, L.; Willmann, J.K. Multiparametric spectroscopic photoacoustic imaging of breast cancer development in a transgenic mouse model. Theranostics 2014, 4, 1062–1071. [Google Scholar]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for myc-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar]
- Yen, M.C.; Kan, J.Y.; Hsieh, C.J.; Kuo, P.L.; Hou, M.F.; Hsu, Y.L. Association of long-chain acyl-coenzyme a synthetase 5 expression in human breast cancer by estrogen receptor status and its clinical significance. Oncol. Rep. 2017, 37, 3253–3260. [Google Scholar]
- Wang, Y.; Cai, X.; Zhang, S.; Cui, M.; Liu, F.; Sun, B.; Zhang, W.; Zhang, X.; Ye, L. Hbxip up-regulates acsl1 through activating transcriptional factor sp1 in breast cancer. Biochem. Biophys. Res. Commun. 2017, 484, 565–571. [Google Scholar]
- Wang, J.; Scholtens, D.; Holko, M.; Ivancic, D.; Lee, O.; Hu, H.; Chatterton, R.T., Jr.; Sullivan, M.E.; Hansen, N.; Bethke, K.; et al. Lipid metabolism genes in contralateral unaffected breast and estrogen receptor status of breast cancer. Cancer Prev. Res. (Phila) 2013, 6, 321–330. [Google Scholar]
- Wu, X.; Li, Y.; Wang, J.; Wen, X.; Marcus, M.T.; Daniels, G.; Zhang, D.Y.; Ye, F.; Wang, L.H.; Du, X.; et al. Long chain fatty acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS ONE 2013, 8, e77060. [Google Scholar]
- Orlando, U.D.; Castillo, A.F.; Medrano, M.A.R.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4 is implicated in drug resistance in breast cancer cell lines involving the regulation of energy-dependent transporter expression. Biochem. Pharmacol. 2019, 159, 52–63. [Google Scholar]
- Migita, T.; Takayama, K.I.; Urano, T.; Obinata, D.; Ikeda, K.; Soga, T.; Takahashi, S.; Inoue, S. Acsl3 promotes intratumoral steroidogenesis in prostate cancer cells. Cancer Sci. 2017, 108, 2011–2021. [Google Scholar]
- Saab, J.; Santos-Zabala, M.L.; Loda, M.; Stack, E.C.; Hollmann, T.J. Fatty acid synthase and acetyl-CoA carboxylase are expressed in nodal metastatic melanoma but not in benign intracapsular nodal nevi. Am. J. Dermatopathol. 2018, 40, 259–264. [Google Scholar]
- Giampietri, C.; Petrungaro, S.; Cordella, M.; Tabolacci, C.; Tomaipitinca, L.; Facchiano, A.; Eramo, A.; Filippini, A.; Facchiano, F.; Ziparo, E. Lipid storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 2017, 18, 1271. [Google Scholar]
- Shyu, P., Jr.; Wong, X.F.A.; Crasta, K.; Thibault, G. Dropping in on lipid droplets: Insights into cellular stress and cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Yao, H.; Ye, J. Long chain acyl-CoA synthetase 3-mediated phosphatidylcholine synthesis is required for assembly of very low density lipoproteins in human hepatoma huh7 cells. J. Biol. Chem. 2008, 283, 849–854. [Google Scholar]
- Young, P.A.; Senkal, C.E.; Suchanek, A.L.; Grevengoed, T.J.; Lin, D.D.; Zhao, L.; Crunk, A.E.; Klett, E.L.; Fullekrug, J.; Obeid, L.M.; et al. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J. Biol. Chem. 2018, 293, 16724–16740. [Google Scholar] [Green Version]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the consistently altered metabolic targets in human hepatocellular carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323. [Google Scholar]
- Liang, Y.C.; Wu, C.H.; Chu, J.S.; Wang, C.K.; Hung, L.F.; Wang, Y.J.; Ho, Y.S.; Chang, J.G.; Lin, S.Y. Involvement of fatty acid-CoA ligase 4 in hepatocellular carcinoma growth: Roles of cyclic amp and p38 mitogen-activated protein kinase. World J. Gastroenterol. 2005, 11, 2557–2563. [Google Scholar]
- Xu, C.; Wang, G.; Hao, Y.; Zhi, J.; Zhang, L.; Chang, C. Correlation analysis between gene expression profile of rat liver tissues and high-fat emulsion-induced nonalcoholic fatty liver. Dig. Dis. Sci. 2011, 56, 2299–2308. [Google Scholar]
- Issa, D.; Alkhouri, N. Nonalcoholic fatty liver disease and hepatocellular carcinoma: New insights on presentation and natural history. Hepatobiliary Surg. Nutr. 2017, 6, 401–403. [Google Scholar]
- Radif, Y.; Ndiaye, H.; Kalantzi, V.; Jacobs, R.; Hall, A.; Minogue, S.; Waugh, M.G. The endogenous subcellular localisations of the long chain fatty acid-activating enzymes acsl3 and acsl4 in sarcoma and breast cancer cells. Mol. Cell. Biochem. 2018, 448, 275–286. [Google Scholar]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable isotope labeling highlights enhanced fatty acid and lipid metabolism in human acute myeloid leukemia. Int. J. Mol. Sci. 2018, 19, 3325. [Google Scholar]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone marrow adipocytes facilitate fatty acid oxidation activating ampk and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar]
- Tomoda, H.; Igarashi, K.; Cyong, J.C.; Omura, S. Evidence for an essential role of long chain acyl-CoA synthetase in animal cell proliferation. Inhibition of long chain acyl-CoA synthetase by triacsins caused inhibition of raji cell proliferation. J. Biol Chem 1991, 266, 4214–4219. [Google Scholar]
- Kaemmerer, E.; Peuscher, A.; Reinartz, A.; Liedtke, C.; Weiskirchen, R.; Kopitz, J.; Gassler, N. Human intestinal acyl-CoA synthetase 5 is sensitive to the inhibitor triacsin c. World J. Gastroenterol 2011, 17, 4883–4889. [Google Scholar] [PubMed]
- Kim, Y.; George, D.; Prior, A.M.; Prasain, K.; Hao, S.; Le, D.D.; Hua, D.H.; Chang, K.O. Novel triacsin c analogs as potential antivirals against rotavirus infections. Eur. J. Med. Chem. 2012, 50, 311–318. [Google Scholar] [PubMed]
- Prior, A.M.; Zhang, M.; Blakeman, N.; Datta, P.; Pham, H.; Chen, Q.; Young, L.H.; Weis, M.T.; Hua, D.H. Inhibition of long chain fatty acyl-CoA synthetase (acsl) and ischemia reperfusion injury. Bioorg. Med. Chem. Lett. 2014, 24, 1057–1061. [Google Scholar] [PubMed]
- Recuero-Checa, M.A.; Sharma, M.; Lau, C.; Watkins, P.A.; Gaydos, C.A.; Dean, D. Chlamydia trachomatis growth and development requires the activity of host long-chain acyl-CoA synthetases (acsls). Sci. Rep. 2016, 6, 23148. [Google Scholar] [PubMed]
- Cao, A.; Li, H.; Zhou, Y.; Wu, M.; Liu, J. Long chain acyl-CoA synthetase-3 is a molecular target for peroxisome proliferator-activated receptor delta in hepg2 hepatoma cells. J. Biol. Chem. 2010, 285, 16664–16674. [Google Scholar]
- Burton, J.D.; Goldenberg, D.M.; Blumenthal, R.D. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Res. 2008, 2008, 494161. [Google Scholar] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of acsl4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar]
- Mashima, T.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Promotion of glioma cell survival by acyl-CoA synthetase 5 under extracellular acidosis conditions. Oncogene 2009, 28, 9–19. [Google Scholar]
- Mashima, T.; Sato, S.; Okabe, S.; Miyata, S.; Matsuura, M.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Acyl-CoA synthetase as a cancer survival factor: Its inhibition enhances the efficacy of etoposide. Cancer Sci. 2009, 100, 1556–1562. [Google Scholar]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangere, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar]
- Liu, K.T.; Yeh, I.J.; Chou, S.K.; Yen, M.C.; Kuo, P.L. Regulatory mechanism of fatty acidcoa metabolic enzymes under endoplasmic reticulum stress in lung cancer. Oncol. Rep. 2018, 40, 2674–2682. [Google Scholar] [PubMed]
- Guha, P.; Kaptan, E.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mtorc1. Oncotarget 2017, 8, 68191–68207. [Google Scholar] [PubMed]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates er stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting n-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [PubMed]
- Wang, X.; Xiong, W.; Tang, Y. Tunicamycin suppresses breast cancer cell growth and metastasis via regulation of the protein kinase b/nuclear factor-kappab signaling pathway. Oncol. Lett. 2018, 15, 4137–4142. [Google Scholar] [PubMed]
- Ramadori, G.; Konstantinidou, G.; Venkateswaran, N.; Biscotti, T.; Morlock, L.; Galie, M.; Williams, N.S.; Luchetti, M.; Santinelli, A.; Scaglioni, P.P.; et al. Diet-induced unresolved er stress hinders kras-driven lung tumorigenesis. Cell Metab. 2015, 21, 117–125. [Google Scholar] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. Acsl4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell. Biol. 2016, 26, 165–176. [Google Scholar]
- Taylor, W.R.; Fedorka, S.R.; Gad, I.; Shah, R.; Alqahtani, H.D.; Koranne, R.; Kuganesan, N.; Dlamini, S.; Rogers, T.; Al-Hamashi, A.; et al. Small-molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci. Rep. 2019, 9, 5926. [Google Scholar]
- Bowman, T.A.; O’Keeffe, K.R.; D’Aquila, T.; Yan, Q.W.; Griffin, J.D.; Killion, E.A.; Salter, D.M.; Mashek, D.G.; Buhman, K.K.; Greenberg, A.S. Acyl coa synthetase 5 (acsl5) ablation in mice increases energy expenditure and insulin sensitivity and delays fat absorption. Mol. Metab. 2016, 5, 210–220. [Google Scholar]
- Fernandez, R.F.; Kim, S.Q.; Zhao, Y.; Foguth, R.M.; Weera, M.M.; Counihan, J.L.; Nomura, D.K.; Chester, J.A.; Cannon, J.R.; Ellis, J.M. Acyl-CoA synthetase 6 enriches the neuroprotective omega-3 fatty acid dha in the brain. Proc. Natl. Acad. Sci. USA 2018, 115, 12525–12530. [Google Scholar] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [PubMed]
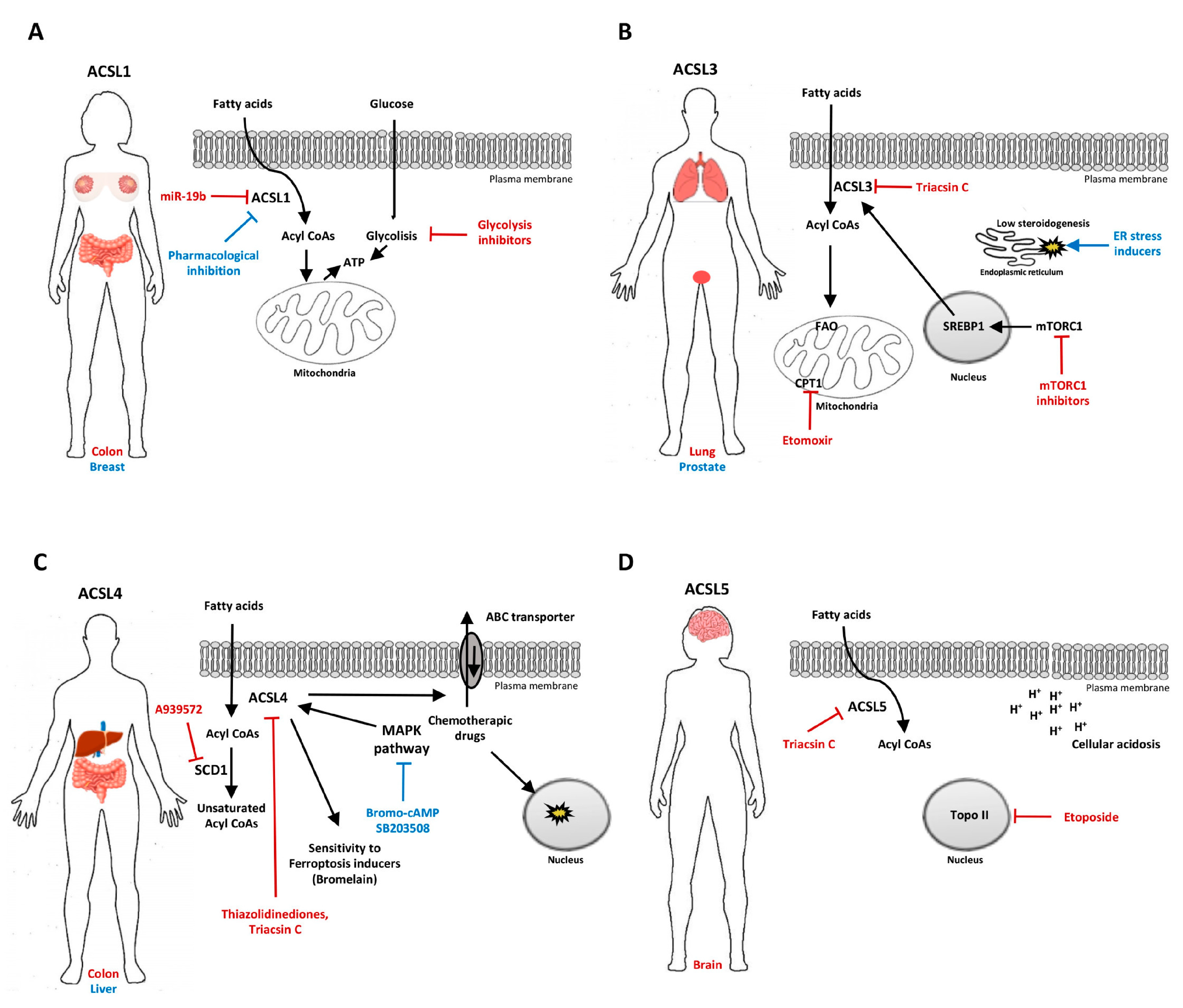
Figure 1.
Proposed targeting approaches involving ACSL isoenzymes in cancer. (A) ACSL1 and glycolytic pathway co-targeting in colon cancer (red), and ACSL1 single targeting breast cancer (blue). (B) ACSL3, CPT1 and the mammalian target of rapamycin complex 1 (mTORC1) co-targeting in lung cancer (red) and the combination of ACSL3 inhibition with ER stress inducers in lung and prostate cancer. (C) Combined ACSL4 and SCD1 inhibition in colon cancer (red) and ACSL4 indirect targeting via the p38 MAPK pathway inhibition in liver cancer (blue). (D) ACSL5 and Topoisomerase II (Topo II) co-targeting with a potential therapeutic benefit in glioma.
Figure 1.
Proposed targeting approaches involving ACSL isoenzymes in cancer. (A) ACSL1 and glycolytic pathway co-targeting in colon cancer (red), and ACSL1 single targeting breast cancer (blue). (B) ACSL3, CPT1 and the mammalian target of rapamycin complex 1 (mTORC1) co-targeting in lung cancer (red) and the combination of ACSL3 inhibition with ER stress inducers in lung and prostate cancer. (C) Combined ACSL4 and SCD1 inhibition in colon cancer (red) and ACSL4 indirect targeting via the p38 MAPK pathway inhibition in liver cancer (blue). (D) ACSL5 and Topoisomerase II (Topo II) co-targeting with a potential therapeutic benefit in glioma.


{kind=link}
Table 1.
Impact of the deregulation of ACSL isoenzymes in cancer models.
| Target Isoenzyme | Cancer Type | Effect | Reference |
|---|---|---|---|
| ACSL1 | CRC cell line | Overexpression: enhanced wound healing, invasion, proliferation, glycolysis and EMT (this latter, only when combined with ACSL4 and stearoyl-CoA desaturase (SCD1) overexpression). Knockdown: decreased cell proliferation, migration, and anchorage-independent growth. miR-19b-1 expression: decreased invasion and proliferation. | [24,28,56,57] |
| BC cell lines | Knockdown: decreased proliferation, colony formation and cell viability. | [28] | |
| NSCLC cell lines | Knockdown: enhanced proliferation and invasiveness of non-small cell lung cancer cell lines. | [28] | |
| ACSL3 | BC cell lines | Knockdown: decreased proliferation and viability, increased FAO/decreased lipid droplets content. | [26] |
| PC cell lines | Overexpression: protection against ER stress inducers. Increased intracellular steroidogenesis. | [67] | |
| NSCLC cell lines | Knockdown: decreased cell proliferation, colony formation and reduced FAO. | [20] | |
| HCC | Knockdown: decreased very low density lipoprotein (VLDL) secretion. | [72] | |
| KRAS NSCLC GEM mouse model | Total mouse knockout: reduced tumor initiation/tumor burden. | [20] | |
| ACSL4 | CRC cell line | Overexpression: enhanced wound healing, invasion, proliferation and glycolysis. Knockdown: decreased cell proliferation and glycolysis, insensitivity to ferroptosis inducers. miR-19b-1 expression: decreased invasion and proliferation. | [24,56,57,87] |
| HCC cell lines | Knockdown: reduced proliferation. | [74] | |
| ER+ BC cell lines | Overexpression: enhanced cell proliferation, increased drug efflux and chemotherapy resistance. | [65,66] | |
| Quadruple-negative BC cell lines | Overexpression: Increased invasion and anchorage independent growth in vitro and increased tumor burden in vivo. | [65,66] | |
| ACSL5 | Glioma cell lines | Overexpression: enhanced cell survival under extracellular acidosis. Knockdown: reduced cell survival under extracellular acidosis. | [88] |
| ACSL6 | NA | NA | NA |
| ACSL1, 3, 4, (5) | ER+, ER− and BC cell lines | Triacsin C: decreased proliferation and decreased chemotherapy resistance. | [62,65] |
| CRC cell lines | Triacsin C: enhanced sensitization to stearoyl-CoA desaturase (SCD1) inhibition treatment. | [56] | |
| Glioma xenograft mouse model | Triacsin C: sensitization to low dose etoposide treatment. | [89] | |
| CRC xenograft mouse model | Triacsin C: decreased lipid droplets content and increased sensitivity to chemotherapy. | [90] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rossi Sebastiano, M.; Konstantinidou, G. Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 3624. https://doi.org/10.3390/ijms20153624
AMA Style
Rossi Sebastiano M, Konstantinidou G. Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. International Journal of Molecular Sciences. 2019; 20(15):3624. https://doi.org/10.3390/ijms20153624
Chicago/Turabian StyleRossi Sebastiano, Matteo, and Georgia Konstantinidou. 2019. "Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy" International Journal of Molecular Sciences 20, no. 15: 3624. https://doi.org/10.3390/ijms20153624
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.