Transcriptome Analysis of Clinopodium chinense (Benth.) O. Kuntze and Identification of Genes Involved in Triterpenoid Saponin Biosynthesis

Abstract
:
1. Introduction
2. Results
2.1. Total Saponin Content in C. chinense Samples
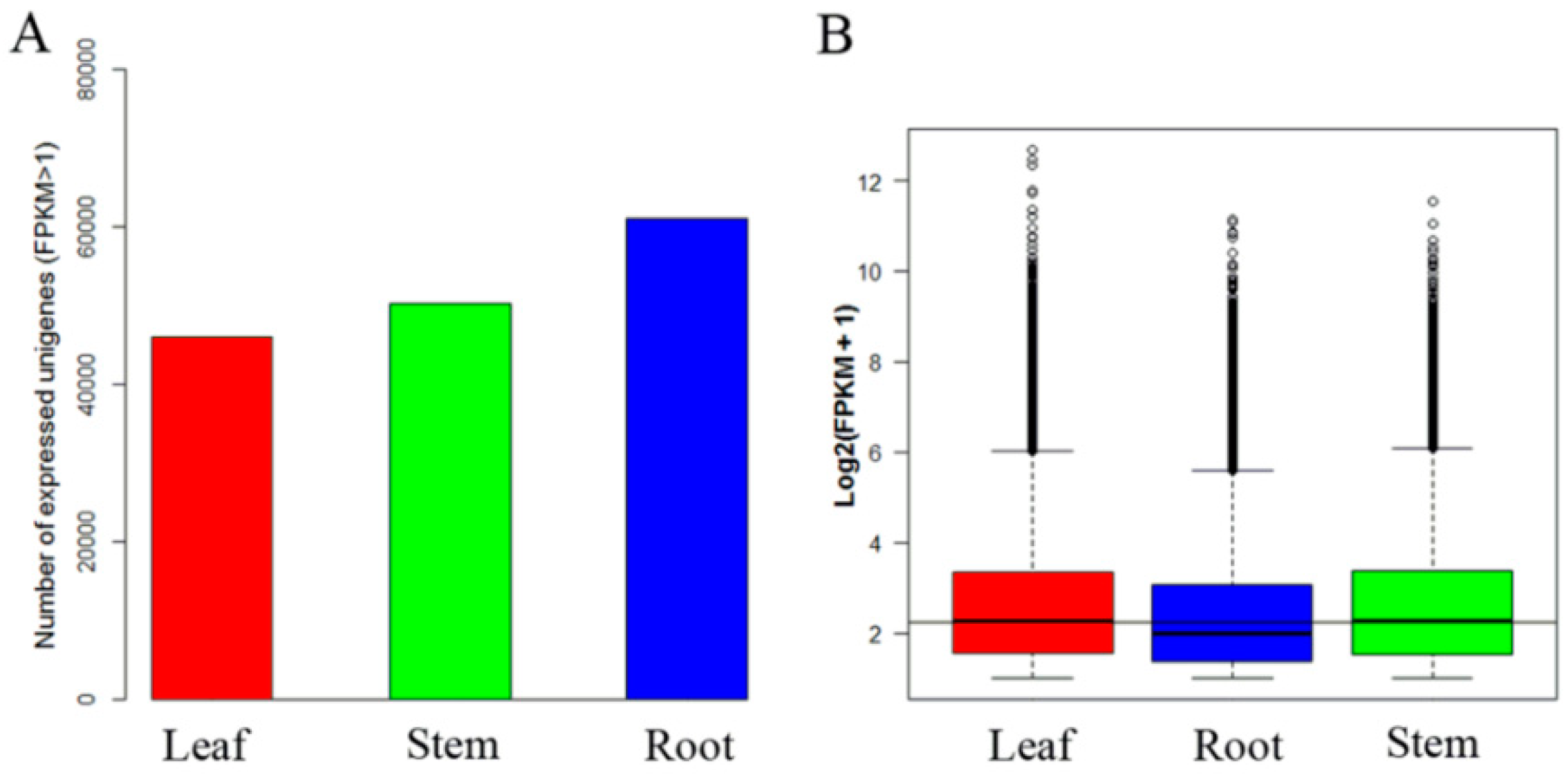
2.2. Sequencing and de novo Assembly
2.3. Unigene Functional Annotation and Overview of Unigene Expression
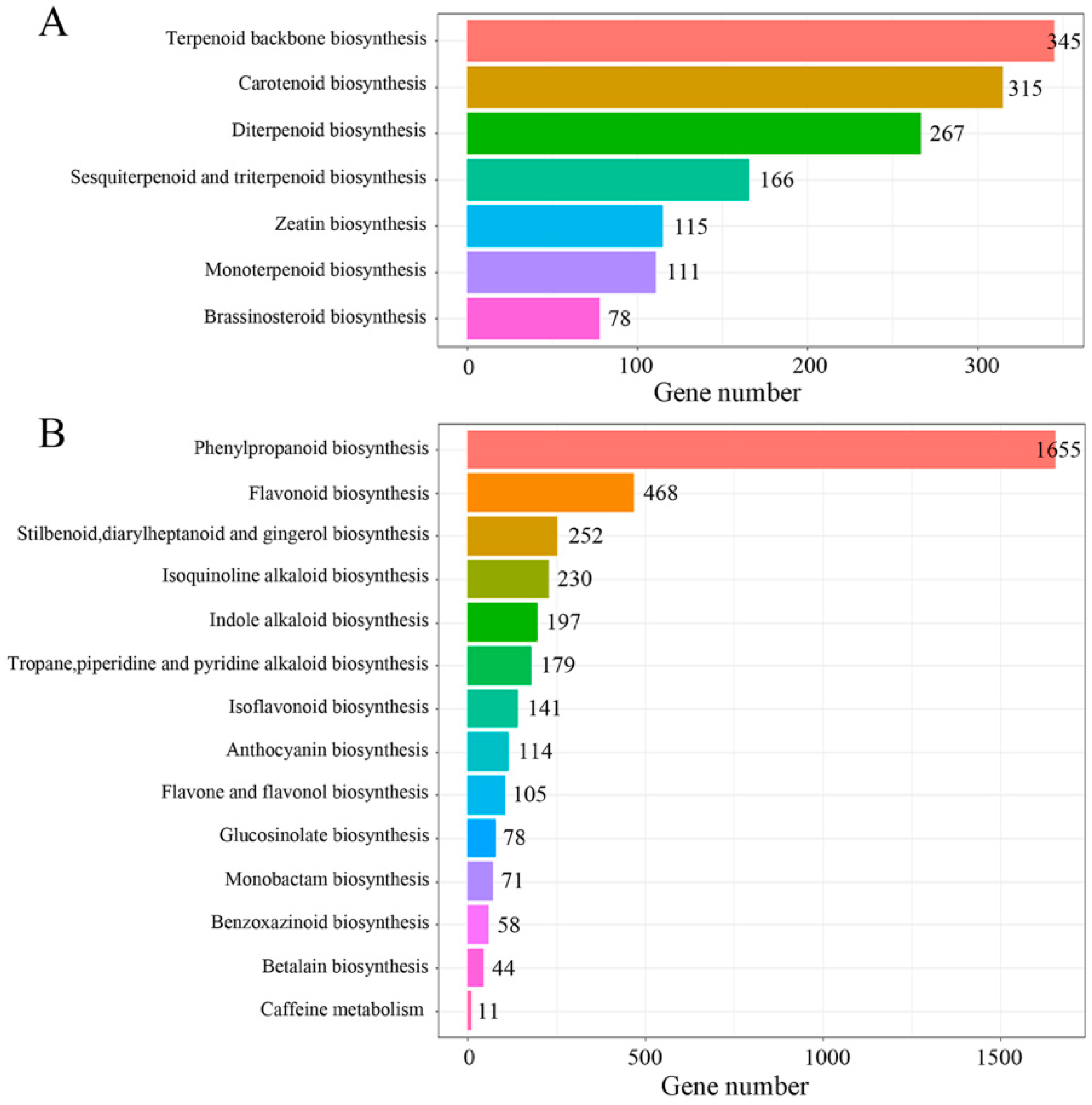
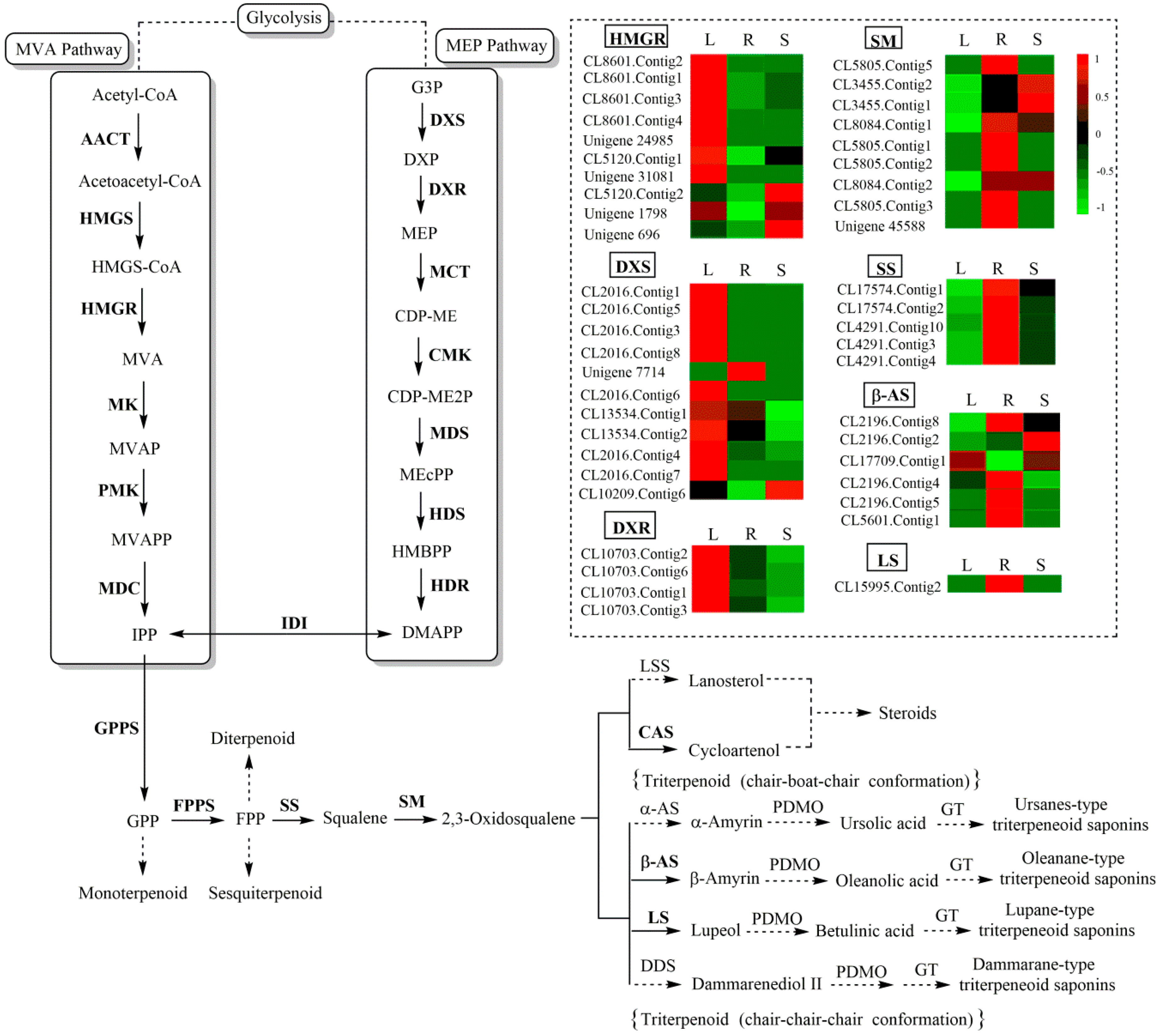
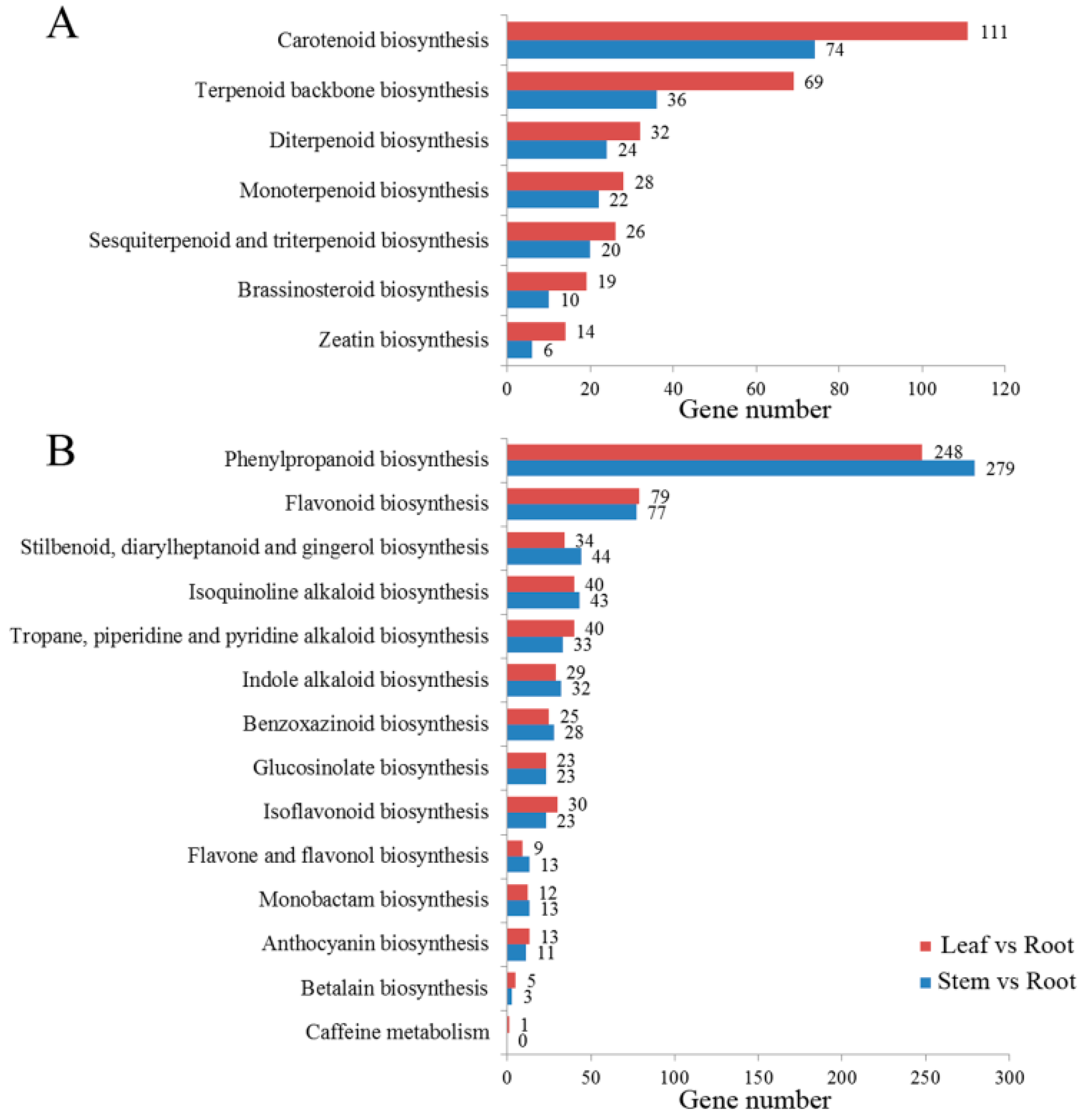
2.4. Identification of Candidate Genes Involved in Triterpenoid Saponin Biosynthesis by KEGG Pathway Analysis
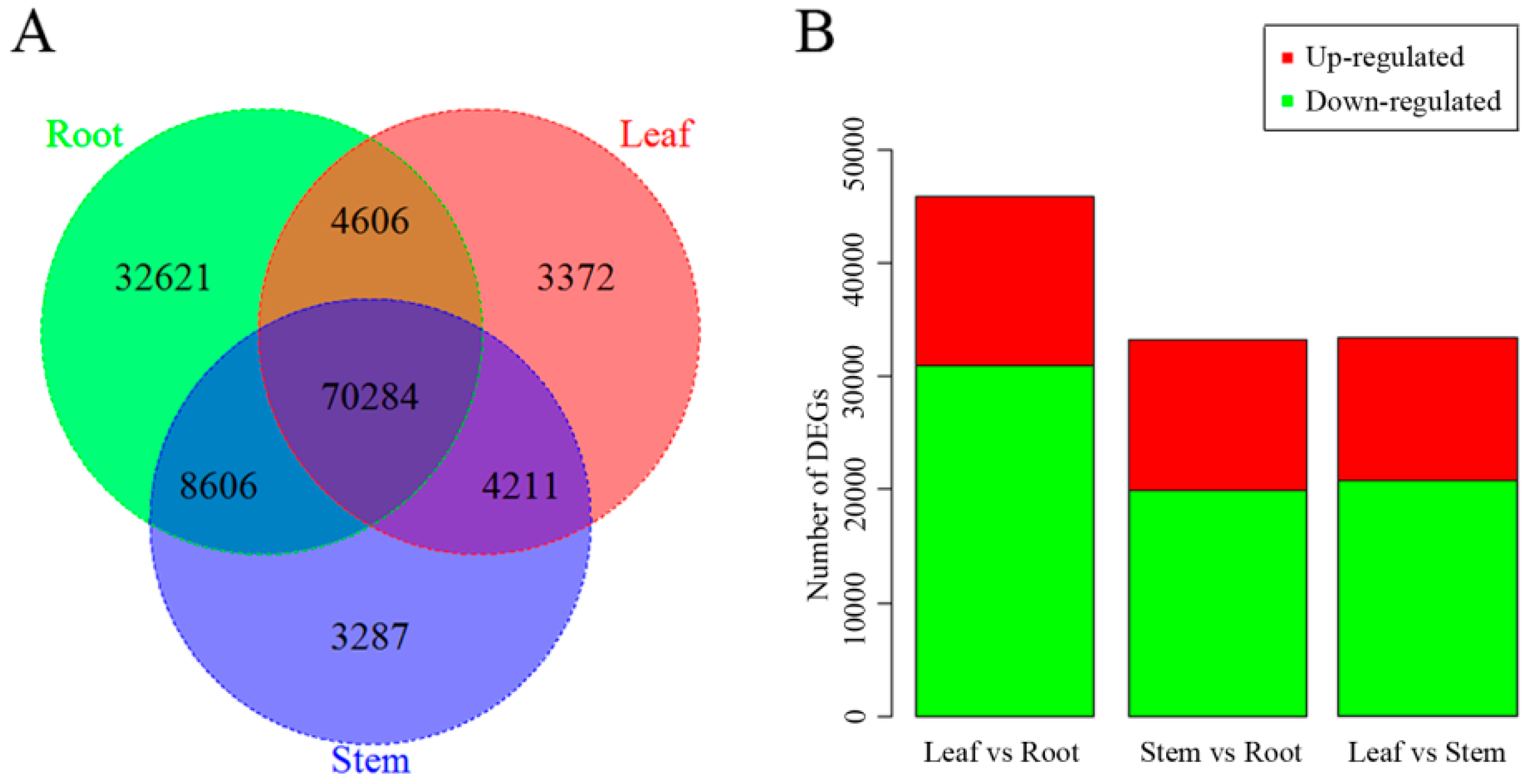
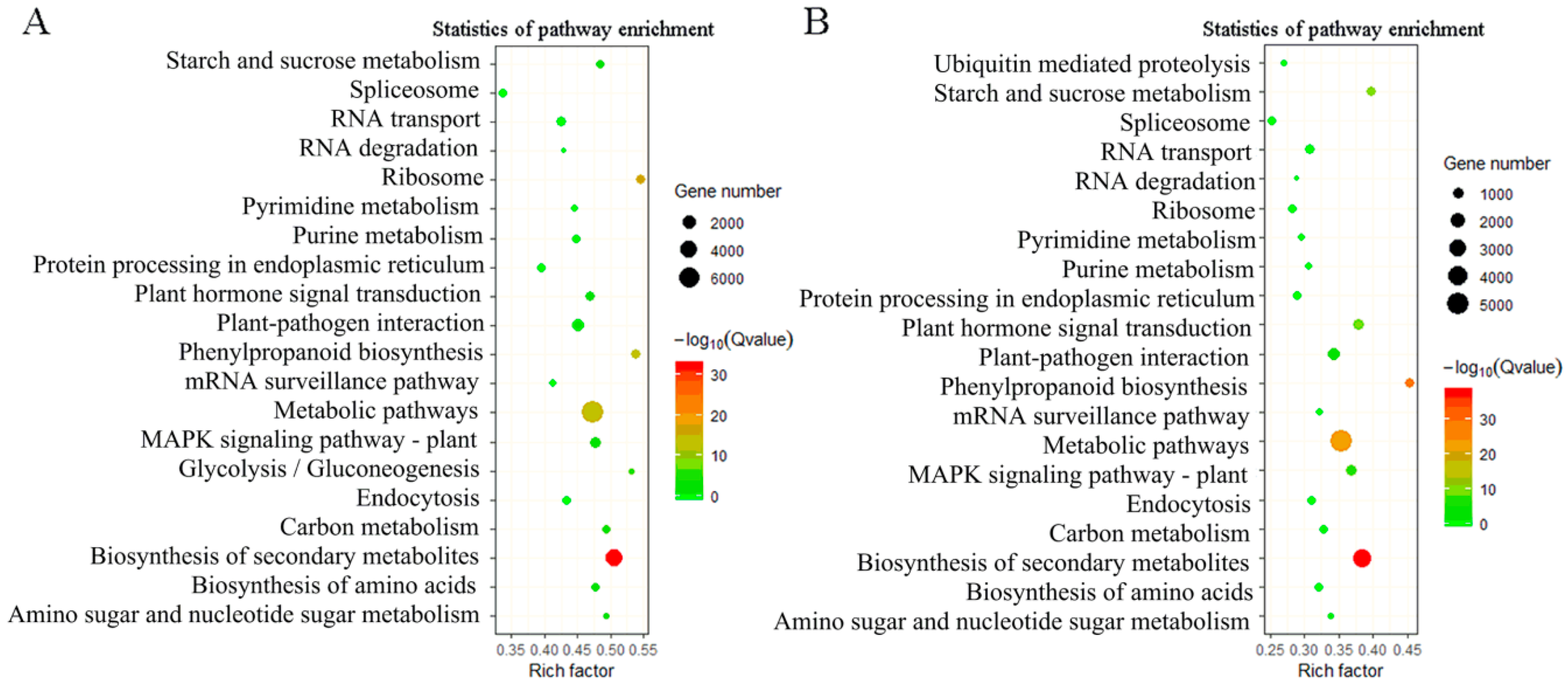
2.5. Differentially Expressed Gene Analysis in Leaf vs. Root, and Stem vs. Root Tissue
2.6. Identification of Candidate Genes Involved in Hormone Biosynthesis by MAPMAN Analysis
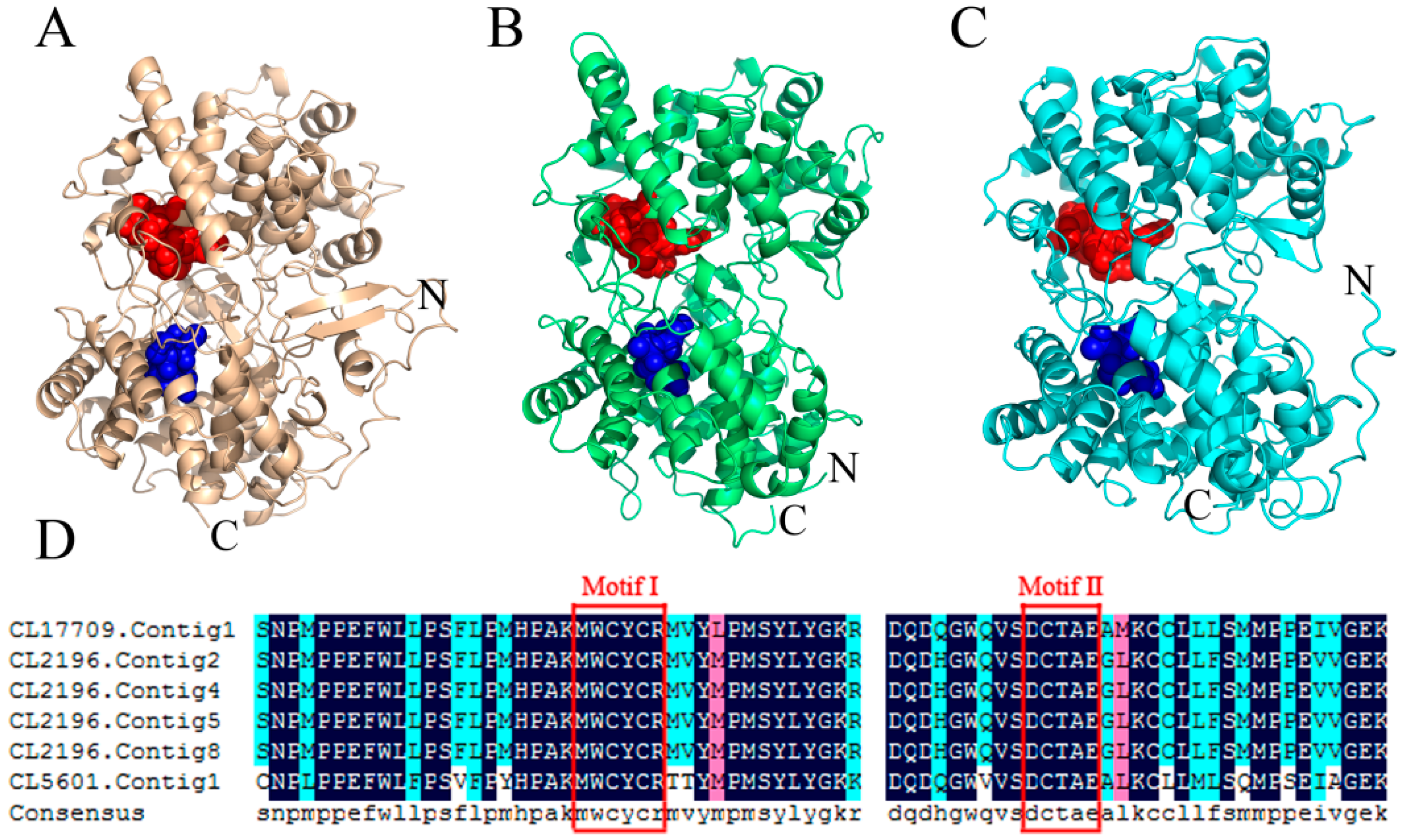
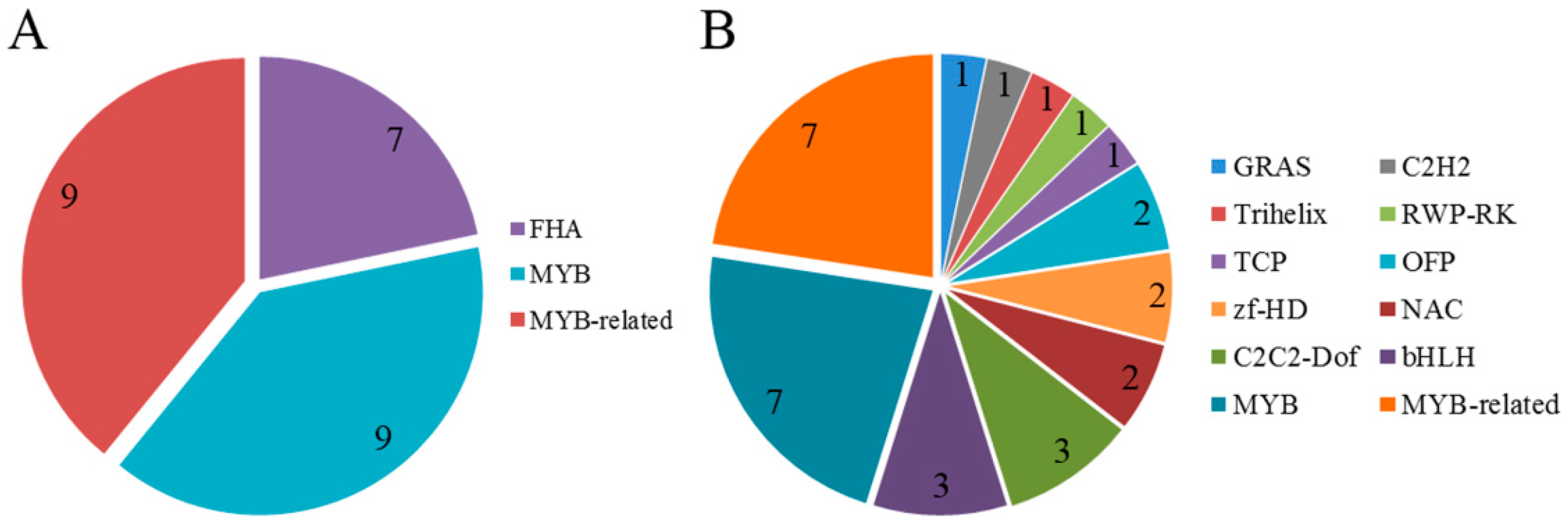
2.7. Detection of Transcription Factor Families
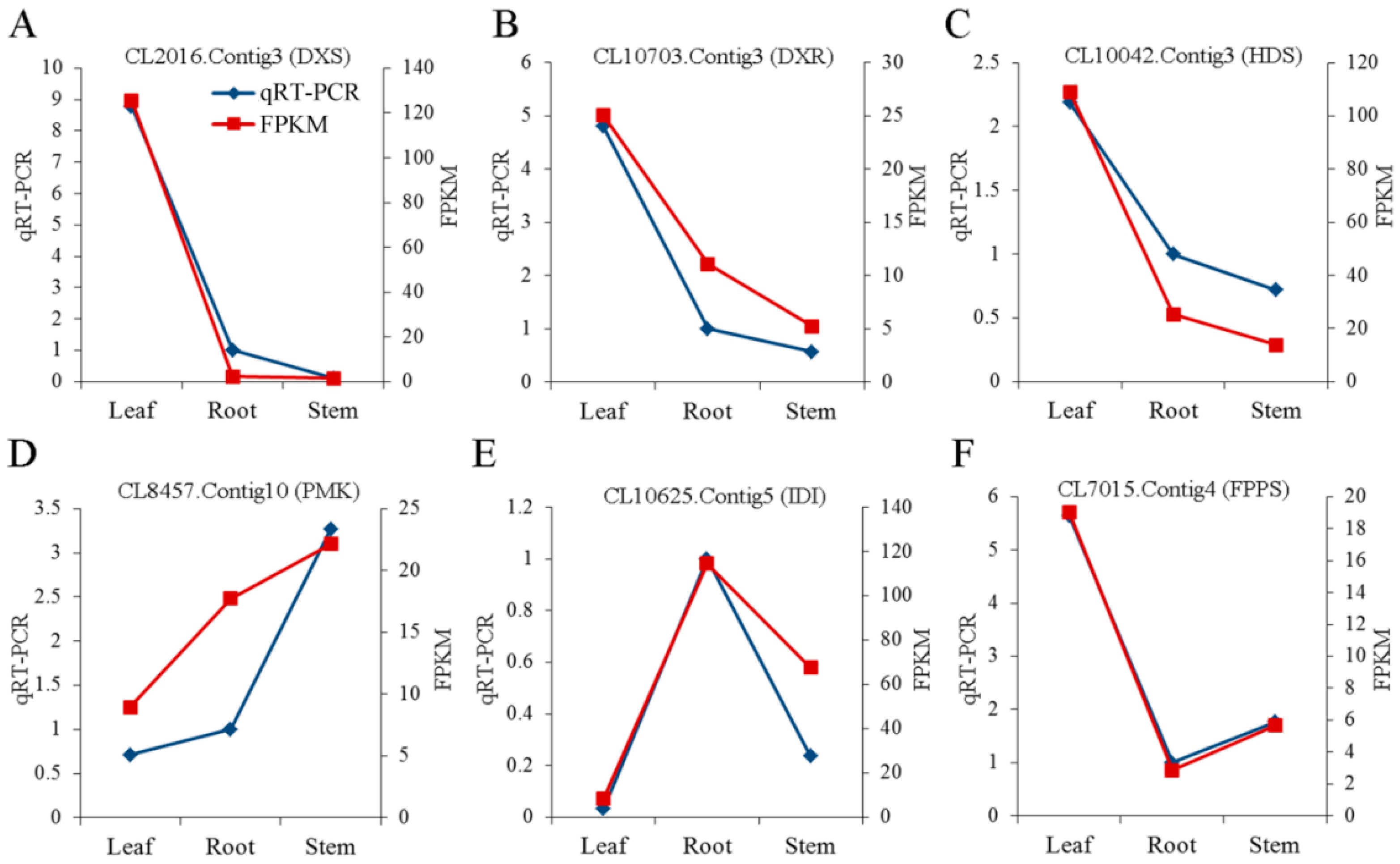
2.8. Validation of Unigenes and Gene Expression Profiling Using qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Sample Preparation for Transcriptome Sequencing and RNA Isolation
4.2. Determining Total Saponins Content
4.3. Library Construction and Sequencing
4.4. De novo Transcriptome Assembly and Unigene Functional Annotation
4.5. Analysis of Differentially Expressed Genes
4.6. Identification of Transcription Factors
4.7. qRT-PCR Analysis of Key Genes in Triterpenoid Saponin Biosynthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
Abbreviations
| AACT | Acetyl-CoA C-acetyltransferase |
| CAS | Cycloartenol synthase |
| cDNA | Complementary DNA |
| CMK | 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase |
| DDS | Dammarenediol II synthase |
| DEGs | Differentially expressed genes |
| DMAPP | Dimethylallyl diphosphate |
| DXR | 1-deoxy-D-xylulose-5-phosphate reductoisomerase |
| DXS | 1-deoxy-D-xylulose-5-phosphate synthase |
| FC | Fold change |
| FDR | False discovery rate |
| FPKM | The fragments per kilobase of transcript per Million mapped reads |
| FPPS | Farnesyl-diphosphate synthase |
| GO | Gene Ontology |
| GPPS | Geranyl-diphosphate synthase |
| GT | Glycosyltransferases |
| HDR | 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase |
| HDS | (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase |
| HMGR | Hydroxymethylglutaryl-CoA reductase |
| HMGS | Hydroxymethylglutaryl-CoA synthase |
| IDI | Isopentenyl-diphosphate Delta-isomerase |
| IPP | Isopentenyl pyrophosphate |
| LS | Lupeol synthase |
| LSS | Lanosterol synthase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOG | Clusters of euKaryotic Orthologous Groups |
| MCT | 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase |
| MDC | Diphosphomevalonate decarboxylase |
| MDS | 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase |
| MEP | 2-C-methyl-D-erythritol-4-phosphate |
| MK | Mevalonate kinase |
| MVA | Mevalonate |
| NR | NCBI non-redundant protein sequences |
| NT | NCBI nucleotide sequences |
| PDMO | Cytochrome P450-dependent monooxygenases |
| PlnTFDB | Plant transcription factor database |
| PMK | Phosphomevalonate kinase |
| qRT-PCR | Quantitative real-time PCR |
| SM | Squalene monooxygenase |
| SS | Squalene synthase |
| TF | Transcriptome factor |
| TGICL | TGI clustering tool |
| α-AS | α-amyrin synthase |
| β-AS | β-amyrin synthase |
References
- Zhong, M.L.; Xu, D.; Yu, S.C.; Sun, G.L. Advances in studies on medicinal plants in Clinopodium Linn. Chin. Tradit. Herb. Drugs 2012, 43, 820–828. [Google Scholar]
- Wang, S.; Ma, G.; Zhong, M.; Yu, S.; Xu, X.; Hu, Y.; Zhang, Y.; Wei, H.; Yang, J. Triterpene saponins from Tabellae Clinopodii. Fitoterapia 2013, 90, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Huang, Y.L.; Wen, Y.X.; Li, D.P.; Chen, W.J. Research Advance on the Saponins of Clinopodium. Nat. Prod. Res. Dev. 2010, 5, 22. [Google Scholar]
- Peng, D.Y.; Liu, Q.Y.; Min, D.; Zi, X.M.; Xu, X.X. Research on hemostatic effect of total saponins of Yinfenglun. China J. Chin. Mater. Med. 2005, 30, 909–912. [Google Scholar]
- Balik, D.; Sashka, D.; Adriana, M.; Nikola, P. In vitro screening for antitumour activity of Clinopodium vulgare L. (Lamiaceae) extracts. Biol. Pharm. Bull. 2002, 25, 499–504. [Google Scholar]
- Tian, D.N.; Wu, F.H.; Ma, S.C.; Li, D.; Dai, Y. Studies on anti-hyperglycemic effect and its mechanism of Clinopodium chinense. China J. Chin. Mater. Med. 2008, 33, 1313–1316. [Google Scholar]
- Augustin, J.M.; Kuzina, V.; Andersen, S.B.; Bak, S. Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochemistry 2011, 72, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Mondol, M.A.M.; Shin, H.J.; Rahman, M.A.; Islam, M.T. Sea Cucumber Glycosides: Chemical Structures, Producing Species and Important Biological Properties. Mar. Drugs 2017, 15, 317. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Ivanchina, N.V.; Krasokhin, V.B.; Makarieva, T.N.; Stonik, V.A. Glycosides from Marine Sponges (Porifera, Demospongiae): Structures, Taxonomical Distribution, Biological Activities and Biological Roles. Mar. Drugs 2012, 10, 1671–1710. [Google Scholar] [CrossRef]
- Zhao, C.L.; Cui, X.M.; Chen, Y.P.; Liang, Q. Key enzymes of triterpenoid saponin biosynthesis and the induction of their activities and gene expressions in plants. Nat. Prod. Commun. 2010, 5, 1147–1158. [Google Scholar]
- Haralampidis, K.; Trojanowska, M.; Osbourn, A.E. Biosynthesis of Triterpenoid Saponins in Plants. Adv. Biochem. Eng. Biotechnol. 2002, 75, 31. [Google Scholar] [PubMed]
- Vincken, J.P.; Heng, L.; de Groot, A.; Gruppen, H. Saponins, classification and occurrence in the plant kingdom. Phytochemistry 2007, 68, 275–297. [Google Scholar] [CrossRef]
- Sawai, S.; Saito, K. Triterpenoid biosynthesis and engineering in plants. Front. Plant Sci. 2011, 2, 25. [Google Scholar] [CrossRef]
- Niu, Y.; Luo, H.; Sun, C.; Yang, T.J.; Dong, L.; Huang, L.; Chen, S. Expression profiling of the triterpene saponin biosynthesis genes FPS, SS, SE, and DS in the medicinal plant. Panax notoginseng. Gene 2014, 533, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Fatih, O.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar]
- Liu, M.; Zhu, J.; Wu, J.; Zhou, M. De novo assembly and analysis of Artemisia argyi transcriptome and identification of genes involved in terpenoid biosynthesis. Sci. Rep. 2017, 8, 5824. [Google Scholar] [CrossRef]
- Zhan, C.; Li, X.; Zhao, Z.; Yang, T.; Wang, X.; Luo, B.; Zhang, Q.; Hu, Y.; Hu, X. Comprehensive Analysis of the Triterpenoid Saponins Biosynthetic Pathway in Anemone flaccida by Transcriptome and Proteome Profiling. Front. Plant Sci. 2016, 7, 1094. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Nakajima, D.; Shinpo, S.; Nakamura, M.; Kawano, N.; Kawahara, N.; Yamazaki, M.; Saito, K.; Suzuki, H.; Hirakawa, H. Identification of potential genes involved in triterpenoid saponins biosynthesis in Gleditsia sinensis by transcriptome and metabolome analyses. J. Nat. Med. 2018, 73, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ralf, T.; Tanja, S.G.; Brigitte, D.A.; Jörg, B.; Johannes, A.; Henrietta, D.; Michael, H.; Martine, S.; Armin, R. Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature 2004, 432, 118–122. [Google Scholar]
- Deshmukh, A.B.; Datir, S.S.; Bhonde, Y.; Kelkar, N.; Samdani, P.; Tamhane, V.A. De novo root transcriptome of a medicinally important rare tree Oroxylum indicum for characterization of the flavonoid biosynthesis pathway. Phytochemistry 2018, 156, 201–213. [Google Scholar] [CrossRef]
- Wang, X.; Hui, F.; Yang, Y.; Yang, S. Deep sequencing and transcriptome analysis to identify genes related to biosynthesis of aristolochic acid in Asarum heterotropoides. Sci. Rep. 2018, 8, 17850. [Google Scholar] [CrossRef]
- Nidhi, S.; Gweneal, C.; Avery, M.A.; Mccurdy, C.R. Targeting the methyl erythritol phosphate (MEP) pathway for novel antimalarial, antibacterial and herbicidal drug discovery: Inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) enzyme. Curr. Pharm. Des. 2007, 13, 1161–1177. [Google Scholar]
- Jadaun, J.S.; Sangwan, N.S.; Narnoliya, L.K.; Singh, N.; Bansal, S.; Mishra, B.; Sangwan, R.S. Over-expression of DXS gene enhances terpenoidal secondary metabolite accumulation in geranium and Withania somnifera: Active involvement of plastid isoprenogenic pathway in their biosynthesis. Physiol Plant 2017, 159, 381. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.J.; Chen, X.; Wang, M.Y.; Matich, A.J.; Perez, R.L.; Allan, A.C.; Green, S.A.; Atkinson, R.G. Natural variation in monoterpene synthesis in kiwifruit: Transcriptional regulation of terpene synthases by NAC and ETHYLENE-INSENSITIVE3-like transcription factors. Plant Physiol. 2015, 167, 1243. [Google Scholar] [CrossRef]
- Hasunuma, T.; Takeno, S.; Hayashi, S.; Sendai, M.; Bamba, T.; Yoshimura, S.; Tomizawa, K.; Fukusaki, E.; Miyake, C. Overexpression of 1-Deoxy-D-xylulose-5-phosphate reductoisomerase gene in chloroplast contributes to increment of isoprenoid production. J. Biosci. Bioeng. 2008, 105, 518–526. [Google Scholar] [CrossRef]
- Wang, Z.; Guhling, O.; Yao, R.; Li, F.; Yeats, T.H.; Rose, J.K.C.; Jetter, R. Two oxidosqualene cyclases responsible for biosynthesis of tomato fruit cuticular triterpenoids. Plant Physiol. 2011, 155, 540–552. [Google Scholar]
- Dhar, N.; Rana, S.; Razdan, S.; Bhat, W.W.; Hussain, A.; Dhar, R.S.; Vaishnavi, S.; Hamid, A.; Vishwakarma, R.; Lattoo, S.K. Cloning and functional characterization of three branch point oxidosqualene cyclases from Withania somnifera (L.) dunal. J. Biol. Chem. 2014, 289, 17249–17267. [Google Scholar] [CrossRef] [PubMed]
- Kushiro, T.; Shibuya, M.; Masuda, K.; Ebizuka, Y. Mutational Studies on Triterpene Synthases: Engineering Lupeol Synthase into β-Amyrin Synthase. J. Am. Chem. Soc. 2000, 122, 6816–6824. [Google Scholar] [CrossRef]
- Ito, R.; Masukawa, Y.; Hoshino, T. Purification, kinetics, inhibitors and CD for recombinant beta-amyrin synthase from Euphorbia tirucalli L and functional analysis of the DCTA motif, which is highly conserved among oxidosqualene cyclases. FEBS J. 2013, 280, 1267–1280. [Google Scholar] [CrossRef]
- Yanagisawa, S. Transcription factors involved in controlling the expression of nitrate reductase genes in higher plants. Plant Sci. 2014, 229, 167–171. [Google Scholar] [CrossRef]
- Mahjoub, A.; Hernould, M.; Joubès, J.; Decendit, A.; Mars, M.; Barrieu, F.; Hamdi, S.; Delrot, S. Overexpression of a grapevine R2R3-MYB factor in tomato affects vegetative development, flower morphology and flavonoid and terpenoid metabolism. Plant Physiol. Biochem. 2009, 47, 551–561. [Google Scholar]
- Sun, W.; Zhan, J.; Zheng, T. The jasmonate-responsive transcription factor regulates terpenoid biosynthetic genes to promote saponin biosynthesis in H Lév. J. Genet. 2018, 97, 1379–1388. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.j.; Gao, W. Research in the regulation mechanism of plant hormones on secondary metabolites. China J. Chin. Mater. Med. 2019. [Google Scholar]
- Ge, Q.; Zhang, Y.; Hua, W.P.; Wu, Y.C.; Jin, X.X.; Song, S.H.; Wang, Z.Z. Combination of transcriptomic and metabolomic analyses reveals a JAZ repressor in the jasmonate signaling pathway of Salvia miltiorrhiza. Sci. Rep. 2015, 5, 14048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Jiao, C.; Singer, S.D.; Gao, M.; Xu, X.; Zhou, Y.; Li, Z.; Fei, Z.; Wang, Y.; Wang, X. Gibberellin-induced changes in the transcriptome of grapevine (Vitis labrusca × V. vinifera) cv. Kyoho flowers. BMC Genom. 2015, 16, 128. [Google Scholar] [CrossRef]
- Zan, L.X. Determination of total saponins in Clinopodium herb from varieces sources by ultrasonic extraction and spectrophotometry. J. Northwest Univ. 2007, 37, 1016–1018. [Google Scholar]
- Chen, Y.-Y.; Li, D.-P.; Lu, F.-L.; Liu, J.L.; Wen, Y.-X. Determination of Buddlejasaponin Ⅳ in Clinopodium gracile by HPLC-ELSD. Agric. Res. Appl. 2011, 3. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Xian, A.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhong, H.; Lin, Y.; Chen, B.; Han, M.; Ren, H.; Lu, H.; Luber, J.M.; Xia, M.; Li, W. Assessment of the cPAS-based BGISEQ-500 platform for metagenomic sequencing. Gigascience 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Myers, E.W.; Lipman, D.J. Blast (basic local alignment search tool). Encycl. Genet. Genom. Proteom. Inform. 2008, 215, 221. [Google Scholar]
- Thimm, O.; Blaesing, O.; Gibon, Y.; Nagel, A.; Meyer, S. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Jaina, M.; Finn, R.D.; Eddy, S.R.; Alex, B.; Marco, P. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar]
- Jarvis, M.C.; Lam, H.C.; Rovira, A.; Marthaler, D.G. Complete Genome Sequence of Porcine Epidemic Diarrhea Virus Strain COL/Cundinamarca/2014 from Colomb. Genome Announc. 2016, 4, e00239-16. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2 −ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Number Annotated | Annotated Unigene Ratio (%) |
|---|---|---|
| NR | 96,353 | 70.86 |
| NT | 69,913 | 51.42 |
| SwissProt | 73,100 | 53.76 |
| KOG | 76,187 | 56.03 |
| KEGG | 74,791 | 55.01 |
| Pfam | 73,053 | 53.73 |
| GO | 51,220 | 37.67 |
| Overall | 102,154 | 75.13 |
| Abbreviation | EC number | Unigene Number | No. in Stems | No. in Roots | No. in Leaves |
|---|---|---|---|---|---|
| AACT | 2.3.1.9 | 25 | 20 | 24 | 20 |
| HMGS | 2.3.3.10 | 8 | 4 | 8 | 4 |
| HMGR | 1.1.1.34 | 15 | 11 | 14 | 8 |
| MK | 2.7.1.36 | 4 | 2 | 4 | 2 |
| PMK | 2.7.4.2 | 12 | 8 | 11 | 10 |
| MDC | 4.1.1.33 | 17 | 10 | 13 | 9 |
| DXS | 2.2.1.7 | 22 | 17 | 19 | 18 |
| DXR | 1.1.1.267 | 10 | 8 | 10 | 9 |
| MCT | 2.7.7.60 | 6 | 6 | 6 | 6 |
| CMK | 2.7.1.148 | 3 | 3 | 3 | 3 |
| MDS | 4.6.1.12 | 3 | 2 | 3 | 2 |
| HDS | 1.17.7.1, 1.17.7.3 | 6 | 5 | 6 | 5 |
| HDR | 1.17.7.4 | 18 | 13 | 15 | 11 |
| IDI | 5.3.3.2 | 13 | 7 | 13 | 7 |
| FPPS | 2.5.1.1, 2.5.1.10 | 55 | 42 | 53 | 37 |
| SS | 2.5.1.21 | 26 | 20 | 26 | 19 |
| SM | 1.14.14.17 | 18 | 15 | 17 | 15 |
| β-AS | 5.4.99.39 | 32 | 28 | 28 | 27 |
| LS | 5.4.99.41 | 6 | 4 | 5 | 4 |
| CAS | 5.4.99.8 | 20 | 14 | 16 | 13 |
| Hormone | Number of Unigenes | Number of Upregulated Genes | |
|---|---|---|---|
| Leaf vs. Root | Stem vs. Root | ||
| JA | 60 | 8 | 24 |
| GA | 10 | 0 | 1 |
| ABA | 27 | 6 | 11 |
| Total number | 97 | 14 | 36 |
| TF Family | Number of Unigenes | Number of Upregulated Genes | |
|---|---|---|---|
| Leaf vs. Root | Stem vs. Root | ||
| MYB | 572 | 100 | 75 |
| MYB-related | 433 | 66 | 54 |
| AP2-EREBP | 345 | 19 | 21 |
| bHLH | 282 | 64 | 47 |
| WRKY | 277 | 76 | 29 |
| NAC | 206 | 32 | 13 |
| GRAS | 168 | 15 | 14 |
| C3H | 160 | 16 | 17 |
| G2-like | 132 | 30 | 17 |
| C2H2 | 102 | 18 | 9 |
| MADS | 102 | 31 | 29 |
| Trihelix | 92 | 16 | 17 |
| Tify | 89 | 21 | 11 |
| HSF | 86 | 12 | 8 |
| mTERF | 78 | 21 | 17 |
| C2C2-Dof | 76 | 14 | 2 |
| FAR1 | 71 | 14 | 10 |
| RWP-RK | 68 | 16 | 7 |
| C2C2-GATA | 64 | 9 | 6 |
| ABI3VP1 | 62 | 4 | 11 |
| ARF | 61 | 0 | 14 |
| SBP | 60 | 7 | 21 |
| Alfin-like | 50 | 5 | 5 |
| TAZ | 49 | 14 | 11 |
| TUB | 47 | 0 | 2 |
| bZIP | 44 | 6 | 6 |
| LOB | 43 | 1 | 0 |
| TCP | 40 | 18 | 7 |
| LIM | 40 | 4 | 8 |
| FHA | 40 | 7 | 6 |
| other | 442 | 96 | 67 |
| Total number | 4381 | 752 | 561 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Zhang, S.; Peng, D.; Wang, C.; Zhao, D.; Ma, K.; Wu, J.; Huang, L. Transcriptome Analysis of Clinopodium chinense (Benth.) O. Kuntze and Identification of Genes Involved in Triterpenoid Saponin Biosynthesis. Int. J. Mol. Sci. 2019, 20, 2643. https://doi.org/10.3390/ijms20112643
Shi Y, Zhang S, Peng D, Wang C, Zhao D, Ma K, Wu J, Huang L. Transcriptome Analysis of Clinopodium chinense (Benth.) O. Kuntze and Identification of Genes Involved in Triterpenoid Saponin Biosynthesis. International Journal of Molecular Sciences. 2019; 20(11):2643. https://doi.org/10.3390/ijms20112643
Chicago/Turabian StyleShi, Yuanyuan, Shengxiang Zhang, Daiyin Peng, Chenkai Wang, Derui Zhao, Kelong Ma, Jiawen Wu, and Luqi Huang. 2019. "Transcriptome Analysis of Clinopodium chinense (Benth.) O. Kuntze and Identification of Genes Involved in Triterpenoid Saponin Biosynthesis" International Journal of Molecular Sciences 20, no. 11: 2643. https://doi.org/10.3390/ijms20112643