Fendiline Enhances the Cytotoxic Effects of Therapeutic Agents on PDAC Cells by Inhibiting Tumor-Promoting Signaling Events: A Potential Strategy to Combat PDAC

 and
and 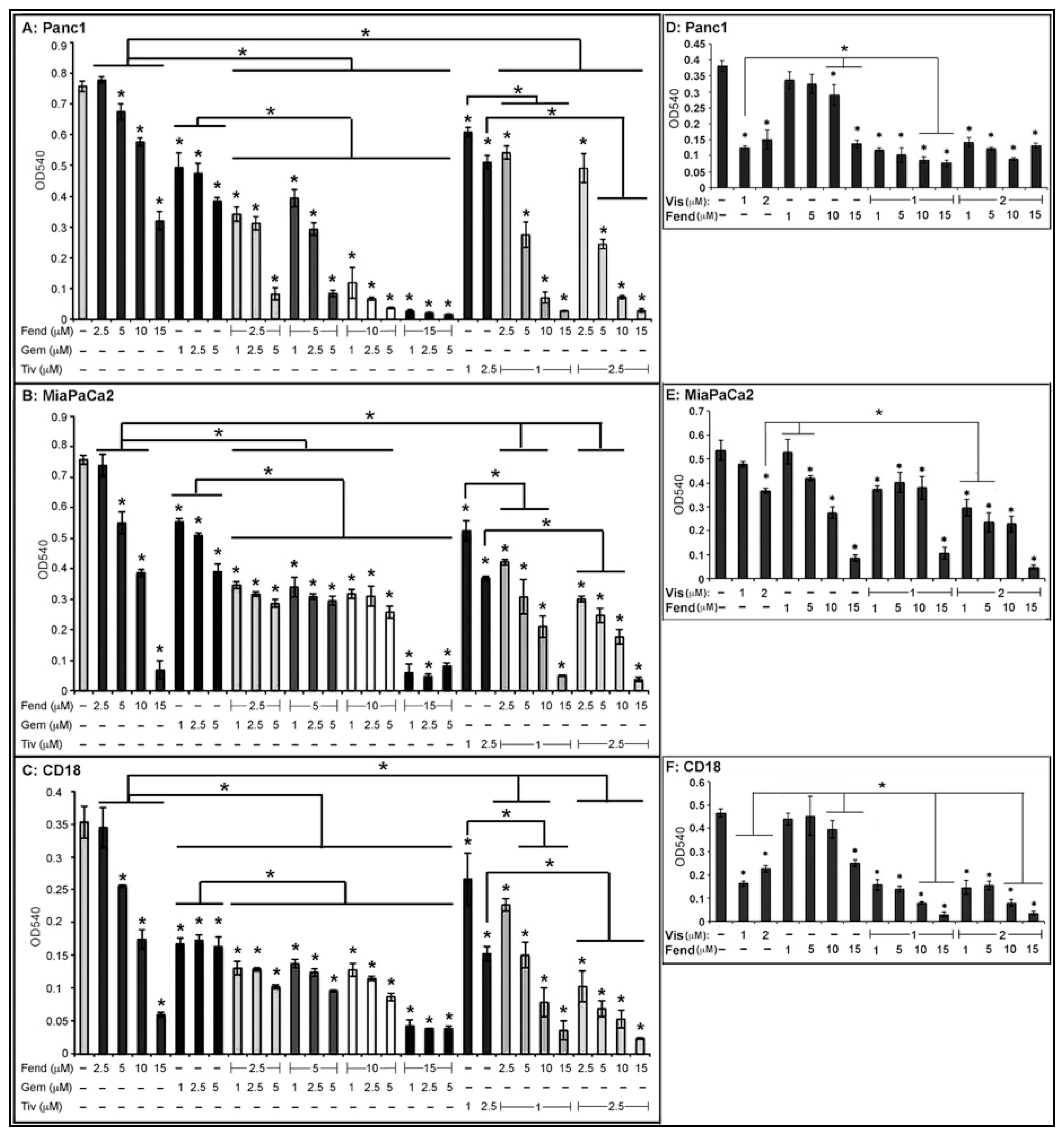{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
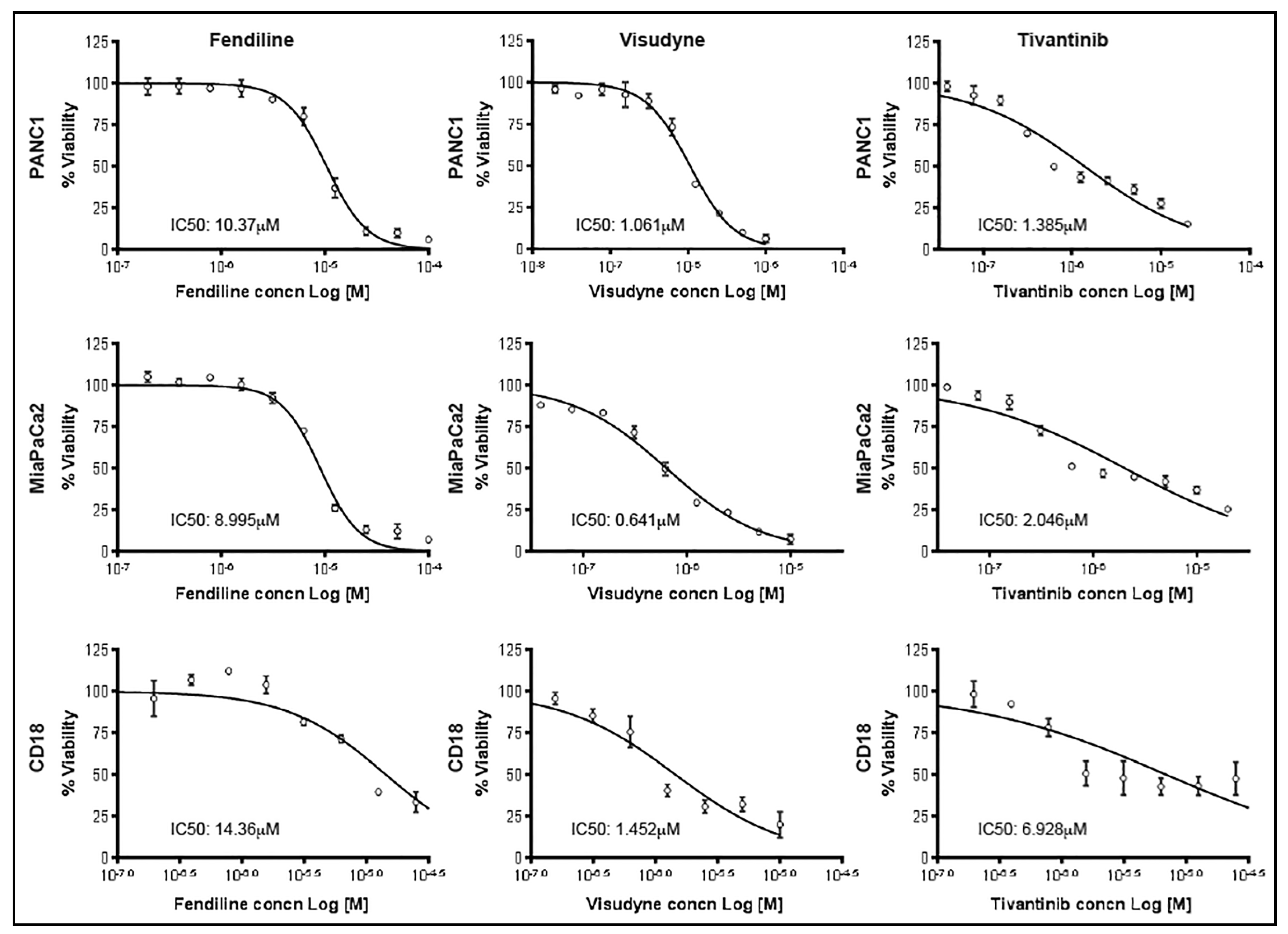
2.1. Combinatorial Treatment with Fendiline Shows Enhanced Cytotoxic Effects on PDAC Cells
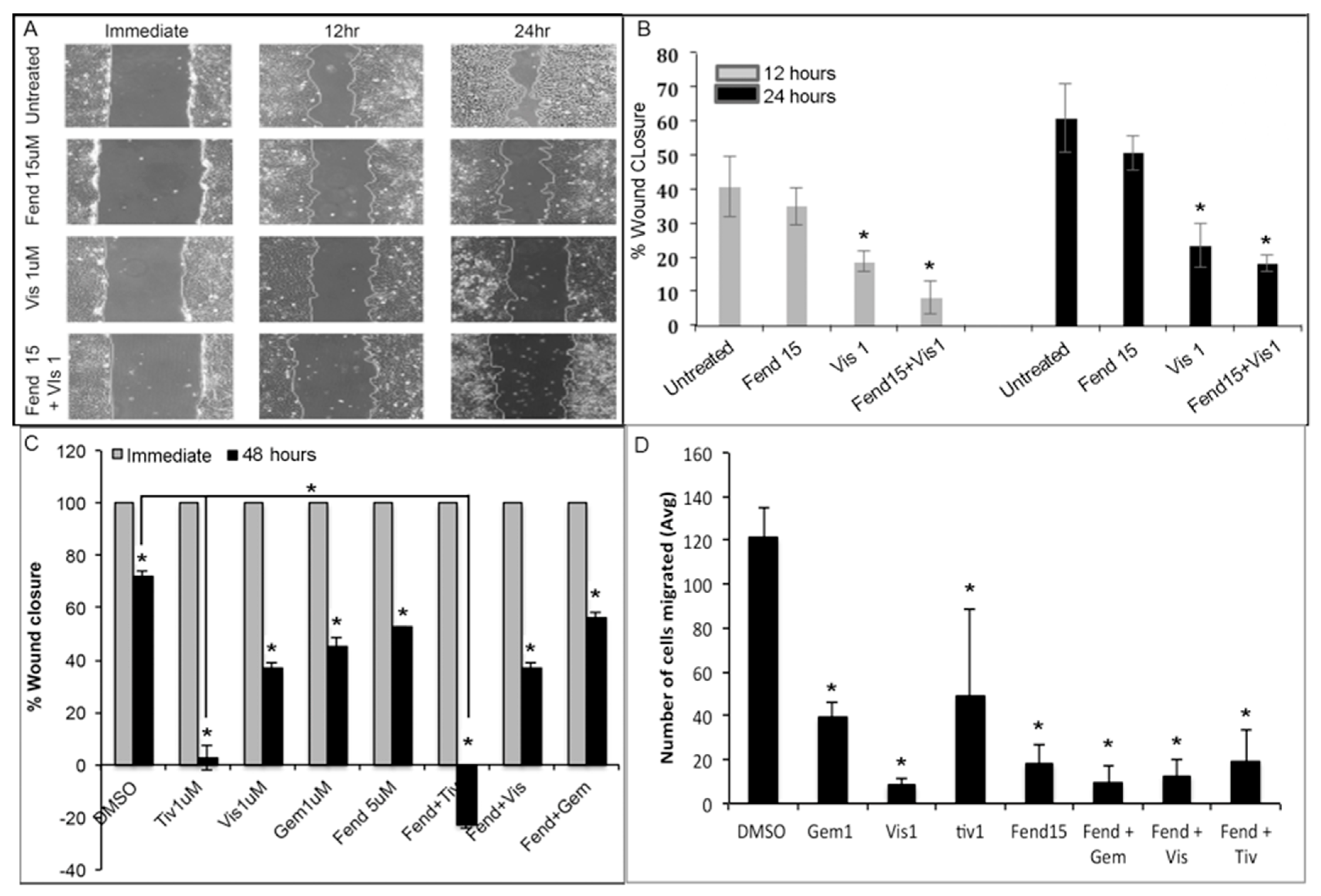
2.2. Treatment with Fendiline and the Pharmacological Agents Reduce Migration of PDAC Cells
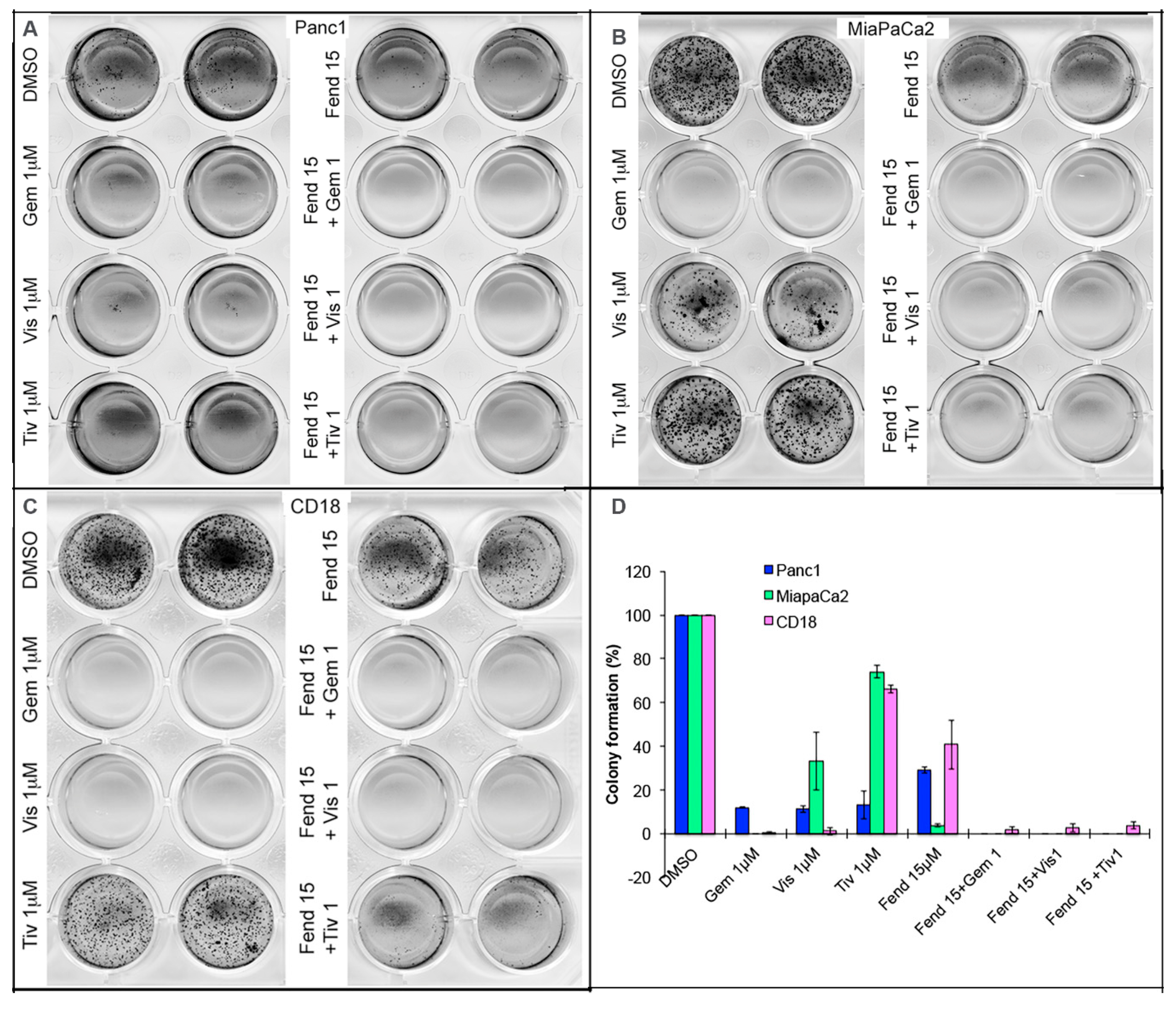
2.3. Co-Treatment with Fendiline and the Pharmacological Agents Inhibits Anchorage Independent Growth of PDAC Cells
2.4. Self-Renewal of PDAC Cells is Drastically Reduced upon Treatment with Single Agents and Completely Eliminated by Combinations of the Drugs
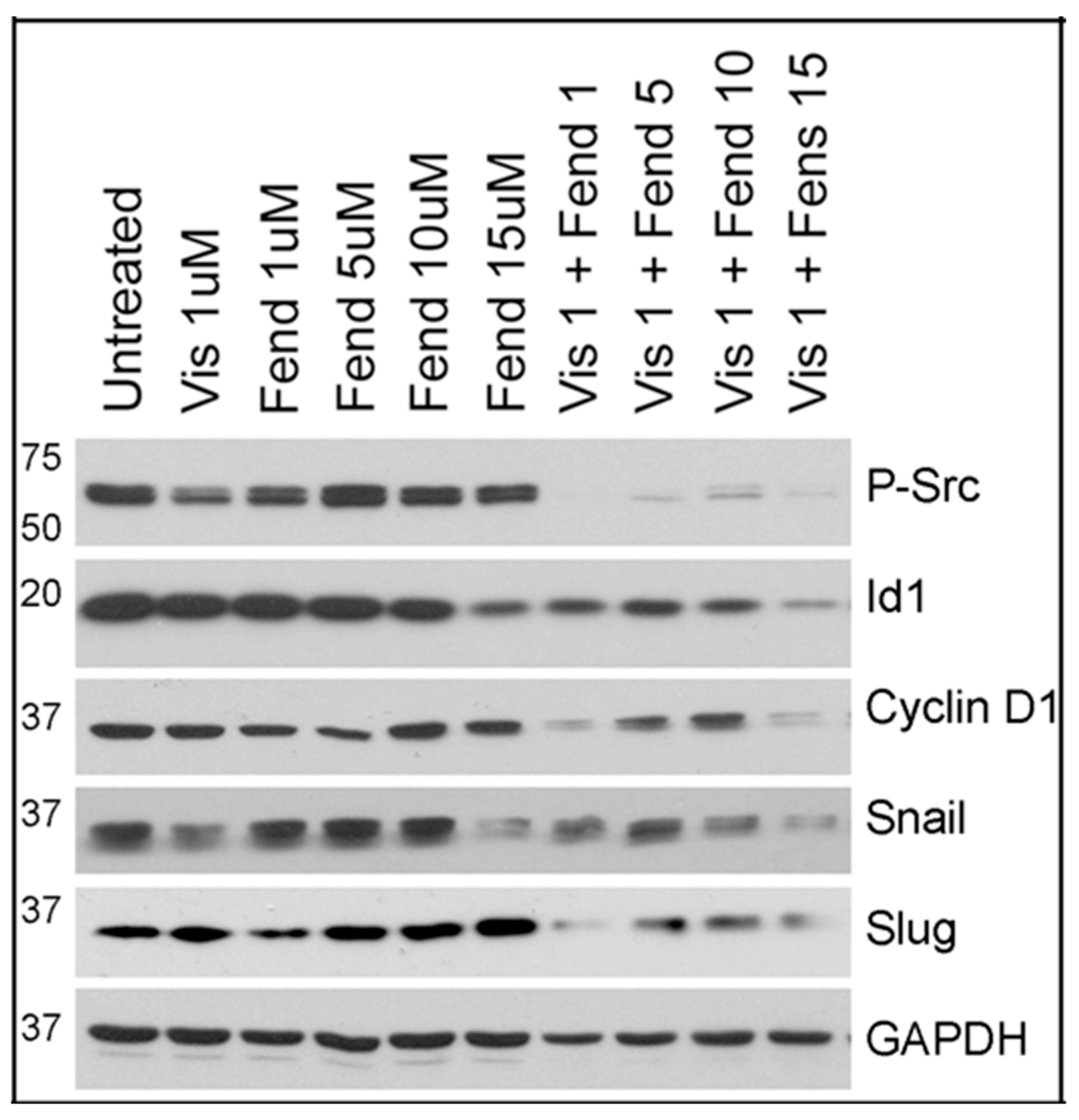
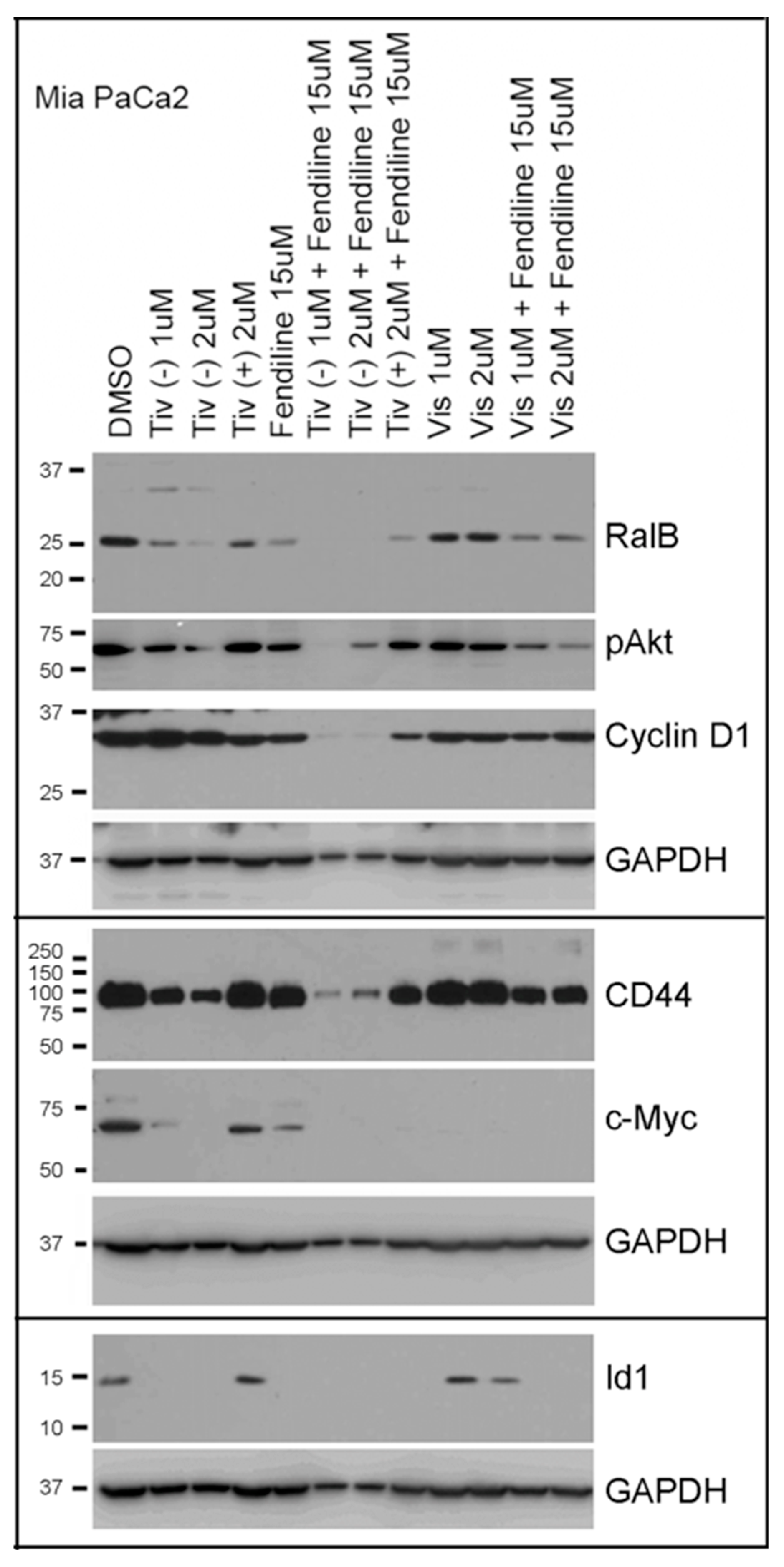
2.5. Comparative Analysis of Tivantinib, Gemcitabine and Visudyne on PDAC Cells Show Differential and Co-Operative Effects of the Drugs on Proteins Involved in Oncogenic Signaling
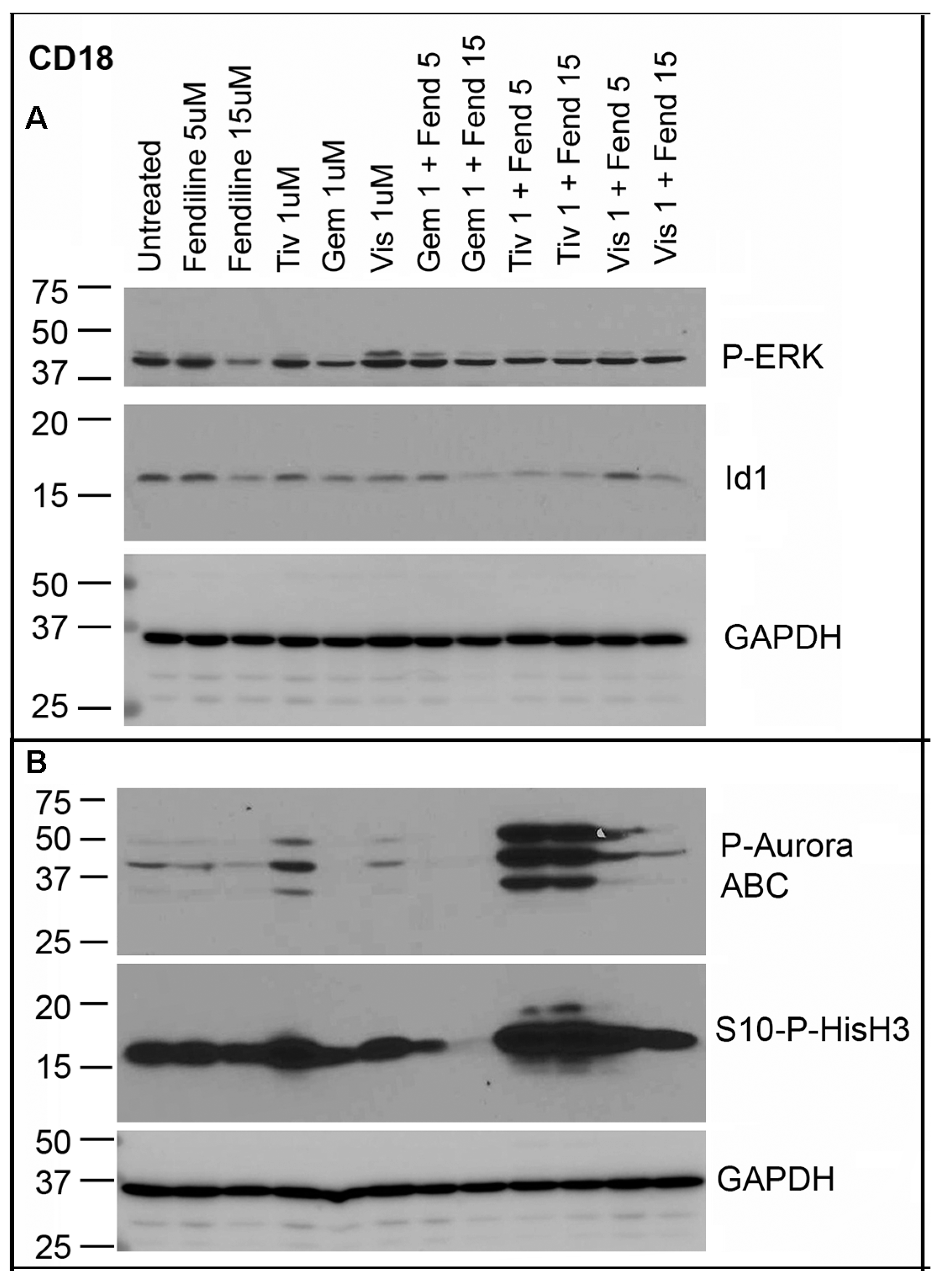
2.6. CD18 Cells Treated with Fendiline alone or along with Tivantinib, Gemcitabine or Visudyne Show Additive Effects on Inhibition of Id1
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines
4.3. MTT Assay
4.4. Soft Agar Colony Formation Assay
4.5. Sample Preparation
4.6. SDS-PAGE and Western Blotting
4.7. Sphere Formation Assay
4.8. Migration (Wound Healing) Assay
4.9. Boyden Chamber Assay
4.10. TUNEL Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| YAP1 | Yes-associated protein 1 |
| Id1 | Inhibitor of differentiation 1 |
| PARP GEM | Poly ADP ribose polymerase Genetically engineered mouse |
References
- Chiaravalli, M.; Reni, M.; O’Reilly, E.M. Pancreatic ductal adenocarcinoma: State-of-the-art 2017 and new therapeutic strategies. Cancer Treat. Rev. 2017, 60, 32–43. [Google Scholar] [CrossRef]
- Wirth, M.; Mahboobi, S.; Kramer, O.H.; Schneider, G. Concepts to Target MYC in Pancreatic Cancer. Mol. Cancer Ther. 2016, 15, 1792–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cheema, A.R.; O’Reilly, E.M. Management of Metastatic Pancreatic Adenocarcinoma. Surg. Clin. N. Am. 2016, 96, 1391–1414. [Google Scholar] [CrossRef]
- Raufi, A.G.; Manji, G.A.; Chabot, J.A.; Bates, S.E. Neoadjuvant Treatment for Pancreatic Cancer. Semin. Oncol. 2019, 46, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zeleniak, A.E.; Huang, W.; Fishel, M.L.; Hill, R. PTEN-Dependent Stabilization of MTSS1 Inhibits Metastatic Phenotype in Pancreatic Ductal Adenocarcinoma. Neoplasia 2018, 20, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.R.; Cannon, A.; Atri, P.; Wichman, C.S.; Smith, L.M.; Ganti, A.K.; Are, C.; Sasson, A.R.; Kumar, S.; Batra, S.K. Advanced pancreatic cancer: A meta-analysis of clinical trials over thirty years. Oncotarget 2018, 9, 19396. [Google Scholar] [CrossRef]
- Adesso, L.; Calabretta, S.; Barbagallo, F.; Capurso, G.; Pilozzi, E.; Geremia, R.; Delle Fave, G.; Sette, C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene 2013, 32, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef]
- Sadot, E.; Doussot, A.; O’Reilly, E.M.; Lowery, M.A.; Goodman, K.A.; Do, R.K.; Tang, L.H.; Gonen, M.; D’Angelica, M.I.; DeMatteo, R.P.; et al. FOLFIRINOX Induction Therapy for Stage 3 Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 3512–3521. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Gustafsson, A.; Andersson, R. Update on the management of pancreatic cancer: Surgery is not enough. World J. Gastroenterol. 2015, 21, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef]
- Yang, K.; Li, Y.; Lian, G.; Lin, H.; Shang, C.; Zeng, L.; Chen, S.; Li, J.; Huang, C.; Huang, K.; et al. KRAS promotes tumor metastasis and chemoresistance by repressing RKIP via the MAPK-ERK pathway in pancreatic cancer. Int. J. Cancer. 2018, 142, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Muratcioglu, S.; Tsai, C.J.; Jang, H.; Gursoy, A.; Keskin, O. The Key Role of Calmodulin in KRAS-Driven Adenocarcinomas. Mol. Cancer Res. 2015, 13, 1265–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, E.; Yachida, S. Genomic alterations in pancreatic cancer and their relevance to therapy. World J. Gastrointest Oncol. 2015, 7, 250–258. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.H.; Baines, A.T.; Fiordalisi, J.J.; Shipitsin, M.; Feig, L.A.; Cox, A.D.; Der, C.J.; Counter, C.M. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 2005, 7, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; O’Hayer, K.; Adam, S.J.; Kendall, S.D.; Campbell, P.M.; Der, C.J.; Counter, C.M. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 2006, 16, 2385–2394. [Google Scholar] [CrossRef]
- Zago, G.; Veith, I.; Singh, M.K.; Fuhrmann, L.; De Beco, S.; Remorino, A.; Takaoka, S.; Palmeri, M.; Berger, F.; Brandon, N.; et al. RalB directly triggers invasion downstream Ras by mobilizing the Wave complex. Elife 2018, 7, e40474. [Google Scholar] [CrossRef]
- Neel, N.F.; Rossman, K.L.; Martin, T.D.; Hayes, T.K.; Yeh, J.J.; Der, C.J. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol. Cell. Biol. 2012, 32, 1374–1386. [Google Scholar] [CrossRef]
- Martin, T.D.; Der, C.J. Differential involvement of RalA and RalB in colorectal cancer. Small Gtpases 2012, 3, 126–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hustinx, S.R.; Leoni, L.M.; Yeo, C.J.; Brown, P.N.; Goggins, M.; Kern, S.E.; Hruban, R.H.; Maitra, A. Concordant loss of MTAP and p16/CDKN2A expression in pancreatic intraepithelial neoplasia: Evidence of homozygous deletion in a noninvasive precursor lesion. Mod. Pathol. 2005, 18, 959–963. [Google Scholar] [CrossRef]
- Georgiadou, D.; Sergentanis, T.N.; Sakellariou, S.; Filippakis, G.M.; Zagouri, F.; Vlachodimitropoulos, D.; Psaltopoulou, T.; Lazaris, A.C.; Patsouris, E.; Zografos, G.C. VEGF and Id-1 in pancreatic adenocarcinoma: Prognostic significance and impact on angiogenesis. Eur. J. Surg. Oncol. 2014, 40, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Shuno, Y.; Tsuno, N.H.; Okaji, Y.; Tsuchiya, T.; Sakurai, D.; Nishikawa, T.; Yoshikawa, N.; Sasaki, K.; Hongo, K.; Tsurita, G.; et al. Id1/Id3 knockdown inhibits metastatic potential of pancreatic cancer. J. Surg. Res. 2010, 161, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.; Bamodu, O.A.; Tsai, W.C.; Chang, Y.M.; Lee, W.H.; Yeh, C.T.; Chao, T.Y. The therapeutic targeting of the FGFR1/Src/NF-kappaB signaling axis inhibits pancreatic ductal adenocarcinoma stemness and oncogenicity. Clin. Exp. Metastasis 2018, 35, 663–677. [Google Scholar] [CrossRef]
- Trevino, J.G.; Pillai, S.; Kunigal, S.; Singh, S.; Fulp, W.J.; Centeno, B.A.; Chellappan, S.P. Nicotine induces inhibitor of differentiation-1 in a Src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia 2012, 14, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E.; Sinnett-Smith, J.; Eibl, G. Yes-associated protein (YAP) in pancreatic cancer: At the epicenter of a targetable signaling network associated with patient survival. Signal Transduct. Target. Ther. 2018, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, L.; Purohit, V.; Shukla, S.K.; Chen, X.; Yu, F.; Fu, K.; Chen, Y.; Solheim, J.; Singh, P.K.; et al. Active YAP promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3. Oncotarget 2015, 6, 36019–36031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wei, D.; Zhang, H.; Chen, J.; Zhang, D.; Ganapathy, S.; Isakson, P.; Chen, C.; Zhu, T. PKCiota and YAP1 are crucial in promoting pancreatic tumorigenesis. Oncotarget 2018, 9, 32736–32750. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Camargo, F.D. The Hippo superhighway: Signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr. Opin. Cell. Biol. 2013, 25, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Camargo, F.D. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef]
- Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK-STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; van der Hoeven, D.; Hancock, J.F. Inhibitors of K-Ras plasma membrane localization. Enzymes 2013, 33, 249–265. [Google Scholar] [PubMed]
- Van der Hoeven, D.; Cho, K.J.; Ma, X.; Chigurupati, S.; Parton, R.G.; Hancock, J.F. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol. Cell. Biol. 2013, 33, 237–251. [Google Scholar] [CrossRef]
- Van der Hoeven, D.; Cho, K.J.; Zhou, Y.; Ma, X.; Chen, W.; Naji, A.; Montufar-Solis, D.; Zuo, Y.; Kovar, S.E.; Levental, K.R.; et al. Sphingomyelin metabolism is a regulator of KRAS function. Mol. Cell. Biol. 2017, 38, e00373-17. [Google Scholar] [CrossRef] [PubMed]
- Woods, N.; Trevino, J.; Coppola, D.; Chellappan, S.; Yang, S.; Padmanabhan, J. Fendiline inhibits proliferation and invasion of pancreatic cancer cells by interfering with ADAM10 activation and beta-catenin signaling. Oncotarget 2015, 6, 35931–35948. [Google Scholar] [CrossRef] [PubMed]
- Perra, A.; Kowalik, M.A.; Ghiso, E.; Ledda-Columbano, G.M.; Di Tommaso, L.; Angioni, M.M.; Raschioni, C.; Testore, E.; Roncalli, M.; Giordano, S.; et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J. Hepatol. 2014, 61, 1088–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keam, S.J.; Scott, L.J.; Curran, M.P. Verteporfin: A review of its use in the management of subfoveal choroidal neovascularisation. Drugs 2003, 63, 2521–2554. [Google Scholar] [CrossRef]
- Sharp, D.M.; Lai, S.; Markey, C.M. Photodynamic therapy with verteporfin for choroidal neovascularization due to age-related macular degeneration and other causes: A New Zealand outcomes study. Clin. Exp. Ophthalmol. 2007, 35, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Reeves, B.C.; Harding, S.P.; Langham, J.; Grieve, R.; Tomlin, K.; Walker, J.; Guerriero, C.; Carpenter, J.; Patton, W.P.; Muldrew, K.A.; et al. Verteporfin photodynamic therapy for neovascular age-related macular degeneration: Cohort study for the UK. Health Technol. Assess. 2012, 16, 1–200. [Google Scholar] [CrossRef]
- Zhang, Q.; Guo, Y.; Yu, H.; Tang, Y.; Yuan, Y.; Jiang, Y.; Chen, H.; Gong, P.; Xiang, L. Receptor activity-modifying protein 1 regulates the phenotypic expression of BMSCs via the Hippo/Yap pathway. J. Cell. Physiol. 2019, 234, 13969–13976. [Google Scholar] [CrossRef] [PubMed]
- Mohede, D.C.J.; de Jong, I.J.; Bank, R.A.; van Driel, M.F. Verteporfin as a Medical Treatment in Peyronie’s Disease. Sex. Med. 2018, 6, 302–308. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Tan, S.; Guo, H.; Barry, W.; Van Allen, E.; Wagle, N.; Brock, J.; Larrabee, K.; Paweletz, C.; Ivanova, E.; et al. Phase II study of tivantinib (ARQ 197) in patients with metastatic triple-negative breast cancer. Investig. New Drugs 2015, 33, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Scagliotti, G.; von Pawel, J.; Novello, S.; Ramlau, R.; Favaretto, A.; Barlesi, F.; Akerley, W.; Orlov, S.; Santoro, A.; Spigel, D.; et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2667–2674. [Google Scholar] [CrossRef]
- Azuma, K.; Hirashima, T.; Yamamoto, N.; Okamoto, I.; Takahashi, T.; Nishio, M.; Hirata, T.; Kubota, K.; Kasahara, K.; Hida, T.; et al. Phase II study of erlotinib plus tivantinib (ARQ 197) in patients with locally advanced or metastatic EGFR mutation-positive non-small-cell lung cancer just after progression on EGFR-TKI, gefitinib or erlotinib. ESMO Open. 2016, 1, e000063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanaatgar-Kasbi, S.; Khorrami, S.; Avan, A.; Aledavoud, S.A.; Ferns, G.A. Targeting the C-MET/HGF Signaling Pathway in Pancreatic Ductal Adenocarcinoma. Curr. Pharm. Des. 2018, 24, 4619–4625. [Google Scholar] [CrossRef]
- Pant, S.; Saleh, M.; Bendell, J.; Infante, J.R.; Jones, S.; Kurkjian, C.D.; Moore, K.M.; Kazakin, J.; Abbadessa, G.; Wang, Y.; et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann. Oncol. 2014, 25, 1416–1421. [Google Scholar] [CrossRef] [Green Version]
- Sierra, J.R.; Tsao, M.S. c-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3, S21–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, N.K.; Padmanabhan, J. Inhibition of amyloid precursor protein processing enhances gemcitabine-mediated cytotoxicity in pancreatic cancer cells. J. Biol. Chem. 2013, 288, 30114–30124. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Zanotto, M.; Carbone, C.; Piro, G.; Tortora, G.; Melisi, D. MEKK3 Sustains EMT and Stemness in Pancreatic Cancer by Regulating YAP and TAZ Transcriptional Activity. Anticancer Res. 2018, 38, 1937–1946. [Google Scholar] [Green Version]
- Bora-Singhal, N.; Nguyen, J.; Schaal, C.; Perumal, D.; Singh, S.; Coppola, D.; Chellappan, S. YAP1 Regulates OCT4 Activity and SOX2 Expression to Facilitate Self-Renewal and Vascular Mimicry of Stem-Like Cells. Stem Cells 2015, 33, 1705–1718. [Google Scholar] [CrossRef]
- Basilico, C.; Pennacchietti, S.; Vigna, E.; Chiriaco, C.; Arena, S.; Bardelli, A.; Valdembri, D.; Serini, G.; Michieli, P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin. Cancer Res. 2013, 19, 2381–2392. [Google Scholar] [CrossRef]
- Katayama, R.; Aoyama, A.; Yamori, T.; Qi, J.; Oh-hara, T.; Song, Y.; Engelman, J.A.; Fujita, N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013, 73, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, A.; Katayama, R.; Oh-Hara, T.; Sato, S.; Okuno, Y.; Fujita, N. Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes ABC transporter-mediated drug resistance. Mol. Cancer 2014, 13, 2978–2990. [Google Scholar] [CrossRef]
- Calles, A.; Kwiatkowski, N.; Cammarata, B.K.; Ercan, D.; Gray, N.S.; Janne, P.A. Tivantinib (ARQ 197) efficacy is independent of MET inhibition in non-small-cell lung cancer cell lines. Mol. Oncol. 2015, 9, 260–269. [Google Scholar] [CrossRef]
- Xiang, Q.; Zhen, Z.; Deng, D.Y.; Wang, J.; Chen, Y.; Li, J.; Zhang, Y.; Wang, F.; Chen, N.; Chen, H.; et al. Tivantinib induces G2/M arrest and apoptosis by disrupting tubulin polymerization in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 118. [Google Scholar] [CrossRef]
- Remsing Rix, L.L.; Kuenzi, B.M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J.M.; Haura, E.B.; Lawrence, H.R.; et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Biol. 2014, 9, 353–358. [Google Scholar] [CrossRef]
- Wei, H.; Wang, F.; Wang, Y.; Li, T.; Xiu, P.; Zhong, J.; Sun, X.; Li, J. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci. 2017, 108, 478–487. [Google Scholar] [CrossRef]
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1alpha. Exp. Cell Res. 2018, 371, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Penchev, V.R.; Chang, Y.T.; Begum, A.; Ewachiw, T.; Gocke, C.; Li, J.; McMillan, R.H.; Wang, Q.; Anders, R.; Marchionni, L.; et al. Ezrin Promotes Stem Cell Properties in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Res. 2019, 17, 929–936. [Google Scholar] [CrossRef]
- Matsuda, Y.; Kure, S.; Ishiwata, T. Nestin and other putative cancer stem cell markers in pancreatic cancer. Med. Mol. Morphol. 2012, 45, 59–65. [Google Scholar] [CrossRef]
- Li, X.P.; Zhang, X.W.; Zheng, L.Z.; Guo, W.J. Expression of CD44 in pancreatic cancer and its significance. Int. J. Clin. Exp. Pathol. 2015, 8, 6724–6731. [Google Scholar] [PubMed]
- Nagathihalli, N.S.; Merchant, N.B. Src-mediated regulation of E-cadherin and EMT in pancreatic cancer. Front. Biosci. 2012, 17, 2059–2069. [Google Scholar] [CrossRef]
- Kelber, J.A.; Reno, T.; Kaushal, S.; Metildi, C.; Wright, T.; Stoletov, K.; Weems, J.M.; Park, F.D.; Mose, E.; Wang, Y.; et al. KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res. 2012, 72, 2554–2564. [Google Scholar] [CrossRef] [PubMed]
- Shields, M.A.; Ebine, K.; Sahai, V.; Kumar, K.; Siddiqui, K.; Hwang, R.F.; Grippo, P.J.; Munshi, H.G. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol. Cancer Res. 2013, 11, 1078–1087. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Morton, J.P.; Ma, Y.; Karim, S.A.; Zhou, Y.; Faller, W.J.; Woodham, E.F.; Morris, H.T.; Stevenson, R.P.; Juin, A.; et al. Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes. Gastroenterology 2014, 146, 1386–1396. [Google Scholar] [CrossRef]
- Li, Y.; Singh, B.; Ali, N.; Sarkar, F.H. Induction of growth inhibition and apoptosis in pancreatic cancer cells by auristatin-PE and gemcitabine. Int. J. Mol. Med. 1999, 3, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Lu, Z.P.; Cai, B.B.; Zhao, L.T.; Qian, D.; Xu, Q.C.; Wu, P.F.; Zhu, Y.; Zhang, J.J.; Du, Q.; et al. Activation of pancreatic stellate cells involves an EMT-like process. Int. J. Oncol. 2016, 48, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Schneider, G.; Ellenrieder, V.; Siveke, J.T. MYC in pancreatic cancer: Novel mechanistic insights and their translation into therapeutic strategies. Oncogene 2016, 35, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; Camaj, P.; Seeliger, H.; Kleespies, A.; Guba, M.; De Toni, E.N.; Schwarz, B.; Graeb, C.; Eichhorn, M.E.; Jauch, K.W.; et al. Inhibition of Src tyrosine kinase reverts chemoresistance toward 5-fluorouracil in human pancreatic carcinoma cells: An involvement of epidermal growth factor receptor signaling. Oncogene 2008, 27, 7212–7222. [Google Scholar] [CrossRef]
- Singh, A.P.; Moniaux, N.; Chauhan, S.C.; Meza, J.L.; Batra, S.K. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res. 2004, 64, 622–630. [Google Scholar] [CrossRef]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Davis, R.; Banerjee, S.; Hug, K.; Lloyd, M.; Coppola, D.; Haura, E.; Chellappan, S.P. ARRB1-mediated regulation of E2F target genes in nicotine-induced growth of lung tumors. J. Natl. Cancer Inst. 2011, 103, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.M.; Bora-Singhal, N.; Kumar, D.M.; Chellappan, S.P. Regulation of Sox2 and stemness by nicotine and electronic-cigarettes in non-small cell lung cancer. Mol. Cancer 2018, 17, 149. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhothali, M.; Mathew, M.; Iyer, G.; Lawrence, H.R.; Yang, S.; Chellappan, S.; Padmanabhan, J. Fendiline Enhances the Cytotoxic Effects of Therapeutic Agents on PDAC Cells by Inhibiting Tumor-Promoting Signaling Events: A Potential Strategy to Combat PDAC. Int. J. Mol. Sci. 2019, 20, 2423. https://doi.org/10.3390/ijms20102423
Alhothali M, Mathew M, Iyer G, Lawrence HR, Yang S, Chellappan S, Padmanabhan J. Fendiline Enhances the Cytotoxic Effects of Therapeutic Agents on PDAC Cells by Inhibiting Tumor-Promoting Signaling Events: A Potential Strategy to Combat PDAC. International Journal of Molecular Sciences. 2019; 20(10):2423. https://doi.org/10.3390/ijms20102423
Chicago/Turabian StyleAlhothali, Marwa, Mevin Mathew, Geeta Iyer, Harshani R. Lawrence, Shengyu Yang, Srikumar Chellappan, and Jaya Padmanabhan. 2019. "Fendiline Enhances the Cytotoxic Effects of Therapeutic Agents on PDAC Cells by Inhibiting Tumor-Promoting Signaling Events: A Potential Strategy to Combat PDAC" International Journal of Molecular Sciences 20, no. 10: 2423. https://doi.org/10.3390/ijms20102423