Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance

1
Department of Biochemistry, College of Natural Sciences, Kangwon National University, Chunchon 24341, Korea
2
Institute of New Frontier Research, College of Medicine, Hallym University, Chunchon 24251, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(1), 51; https://doi.org/10.3390/ijms20010051
Submission received: 27 November 2018
/
Revised: 20 December 2018
/
Accepted: 20 December 2018
/
Published: 23 December 2018
(This article belongs to the Special Issue Histone Deacetylase Inhibitors in Health and Disease)
Abstract
:Histone modification is associated with resistance to anti-cancer drugs. Epigenetic modifications of histones can regulate resistance to anti-cancer drugs. It has been reported that histone deacetylase 3 (HDAC3) regulates responses to anti-cancer drugs, angiogenic potential, and tumorigenic potential of cancer cells in association with cancer-associated genes (CAGE), and in particular, a cancer/testis antigen gene. In this paper, we report the roles of microRNAs that regulate the expression of HDAC3 and CAGE involved in resistance to anti-cancer drugs and associated mechanisms. In this review, roles of HDAC3-miRNAs-CAGE molecular networks in resistance to anti-cancer drugs, and the relevance of HDAC3 as a target for developing anti-cancer drugs are discussed.
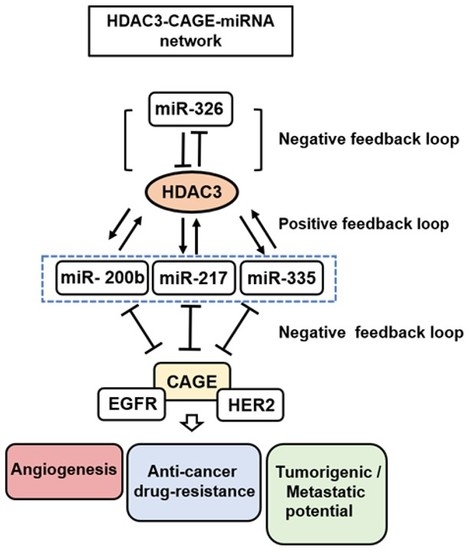
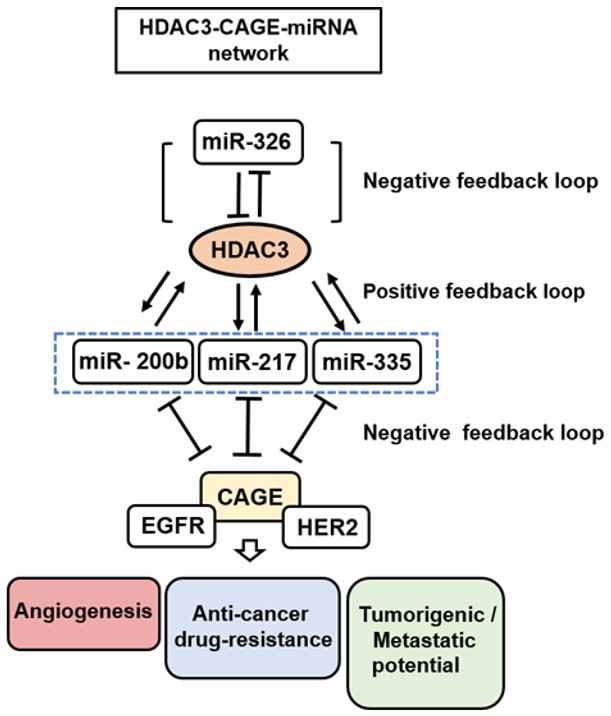{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. HDAC3 as a Target for Development of Anti-Cancer Drugs
Unlike many other histone deacetylases (HDACs), HDAC3 shows ubiquitous expression [1]. Nuclear Receptor Corepressor (N-CoR) and silencing mediator of retinoid and thyroid hormone receptors (SMRT) bind to HDAC3 and exert transcriptional repression [2]. The N-CoR-HDAC3 complex inhibits the c-jun N-terminal kinase (JNK) pathway through the GPS2 subunit [3]. HDAC3 deacetylates relA, subunit of nuclear factor-kB (NF-κB), to repress NF-κB activity [4]. HDAC3 inhibits mitogen-activated protein kinase (MAPK)-dependent activation of activating transcription factor 2 (ATF-2) to repress expression of tumor necrosis factor-α (TNF-α) [5]. These findings suggest that HDAC3 plays an important role in various cellular reactions.
HDAC3 can bind to the promoter sequences of Runt-related transcription factor 2 (Runx 2) and suppress the metastatic potential of colorectal cancers [6]. HDAC3 can also bind to cAMP response element-binding protein (CREB) and decrease the migration potential of metastatic breast cancer cells [7]. HDAC3, a downstream target of AKT serine/threonine kinase (Akt) and glycogen synthase kinase 3 beta (GSK3β), can promote neuronal death [8] and beta cell apoptosis induced by inflammatory cytokines [9]. HDAC3 can also decrease expression of Hypoxia-inducible factor-1 α (HIF-1α) by negative regulation of NF-κB in metastatic breast cancer cells [10] and regulates the signal transducer and activator of transported 3 (Stat3) which inhibits Beclin 1, a prognostic marker and autophagy in non-small cell lung cancer cells [11]. Down-regulation of HDAC3 enhances the tumorigenic potential of lung cancer cells [12]. HDAC3 can also decrease the angiogenic potential of melanoma cells [13]. The role of HDAC3 in cancer development is known to be tissue-specific. It has been reported that liver-specific deletion of HDAC3 can lead to hepatoma by causing genomic instability and DNA damage [14].
Inactivation of HDAC3 by Cre-recombinase can delay cell cycle progression and induce apoptosis in mouse embryonic fibroblast cells (MEFs) [15]. Overexpression of HDAC3 in colorectal cancer cells is known to decrease expression of p21 [16]. Down-regulation or pharmacological inhibition of HDAC3 by novel class I inhibitors, such as 4SC202, BG45, or SBHA, can inhibit cholangiocarcinoma (CCA) growth and promote apoptosis [17].
Delphinidin, a specific inhibitor of HDAC3, induces acetylation of p53 and promotes caspase-dependent apoptosis [18]. JNK phosphorylation of HDAC3 leads to increased HDAC3 activity [19]. I-7a, a novel HDAC3 inhibitor, acetylates p53 and p21 expression, which in turn leads to G1 arrest in triple-negative breast cancer (TNBC) [20]. Overexpression of HDAC3 is correlated with poor prognosis in various cancers [21,22]. HDAC3 can decrease expression of p53-upregulated modulator of apoptosis (PUMA), while down-regulation of HDAC3 leads to increased expression of PUMA by p53 [23]. The N-hydroxycinnamamide-based HDAC inhibitor with HDAC1/3 dual selectivity has shown promise as an anti-cancer drug [24]. Thus, HDAC3 can be considered as a target for the development of anti-cancer therapeutics.
2. Anti-Cancer Therapy Targeting CSCS in Relation to HDACs
Cancer stem cells (CSCs) are undifferentiated tumor cells with the capacity for self-renewal [25,26]. HDAC inhibitors can act on CSCs [27,28,29]. Pan-HDAC inhibitors can suppress CSCs via different mechanisms. AR-42 (OSU-HDAC42) is known to promote apoptosis of leukemic stem cells by inhibiting NF-κB and heat shock protein 90 (Hsp90) [30]. Suberoylanilide Hydroxamic Acid (SAHA) and 5-aza-2-deoxycytidine, an inhibitor of DNA methyl transferase I (DNMT1), can inhibit self-renewal and cellular proliferation of pancreatic cancer stem cells [31]. SAHA can reverse cisplatin resistance and decrease CSCs by down-regulating nanog, a marker of cancer stem cells [32].
Hypoxia-inducible factor-1 α (HIF-1α) links inflammation to cancer. Overexpression of HIF-1α indicates poor prognosis [33]. HIF-1α maintains CSCs by inhibiting the negative regulator of Notch1 signaling [34]. HDAC3 binds to HIF-1α and acts as a positive regulator of HIF-1α stability [35]. Arrest-defective-1(ARD1) binds to HIF-1α and mediates acetylation, leading to ubiquitination of HIF-1α [36]. However, HDAC1 [37] and the class II isoforms, HDAC4 and HDAC6 [38] prevents ARD1 from mediating ubiquitination of HIF-1α. Thus, HDAC3 can maintain CSCs by maintaining expression of HIF-1α.
Dysregulation of Stat3 signaling is known to enhance tumorigenic potential of cancer cells by increasing expression levels of angiogenesis markers, such as HIF-1α and the vascular endothelial growth factor (VEGF) [39]. HDAC3 is necessary for STAT3-dependent liver cancer [40]. The NF-κB signaling pathway contributes to the maintenance of glioblastoma stem cells [41]. Glioblastoma stem cells drive anti-cancer drug resistance [41]. NF-κB signaling is accompanied by resistance to the anti-cancer drug Temozolomide (TMZ) in glioblastoma stem cells [42]. RGFP109, a selective inhibitor of HDAC3, can enhance sensitivity to TMZ by inhibiting NF-κB signaling in a TMZ-resistant (TR) GBM cell line [42]. HDAC3 binds to the p65 subunit of NF-κB to inhibit NF-κB [43].
Abexinostat, another pan-HDAC inhibitor, exhibits anti-CSC activities by inducing differentiation [44]. Pan-HDAC inhibitors MC1742 and MC2625 can increase the level of acetylated histone H3 and induce apoptosis in sarcoma cancer stem cells [45]. Down-regulation or inhibition of HDAC3 reduces cholangiocarcinoma (CCA) cell growth and apoptosis [46]. HDACs can regulate transition of non-CSCs into anti-cancer drug-resistant CSCs by chemotherapy [47]. Thus, HDAC inhibitors can restore anti-cancer drug sensitivity by inhibiting CSC plasticity.
3. Role of HDAC3 in Anti-Cancer Drug Resistance
Trichostatin A, an inhibitor of HDACs, enhances sensitivity to anti-cancer drugs by decreasing expression of multidrug resistance protein 1(MDR1) [48]. SAHA, an inhibitor of HDAC(s), induces MDR to confer anti-apoptotic effects [49]. These results suggest the important role of HDAC3 in anti-cancer drug resistance.
Taxol-resistant ovarian tumors display altered expression of tubulins [50]. Paclitaxel resistance is accompanied by high expression levels of tubulin β3 in cancer cells such as non-small cell lung cancer cells [51,52]. It has been reported that taxol resistance is due to MAPK activation in colon cancer cells [53]. Taxol resistance is induced by the Phosphoinositide 3-kinase (PI3K)-Akt-JNK signaling pathway in osteosarcoma cells [54]. HDAC3 can inhibit JNK signaling [3] and MAPK activation [5].
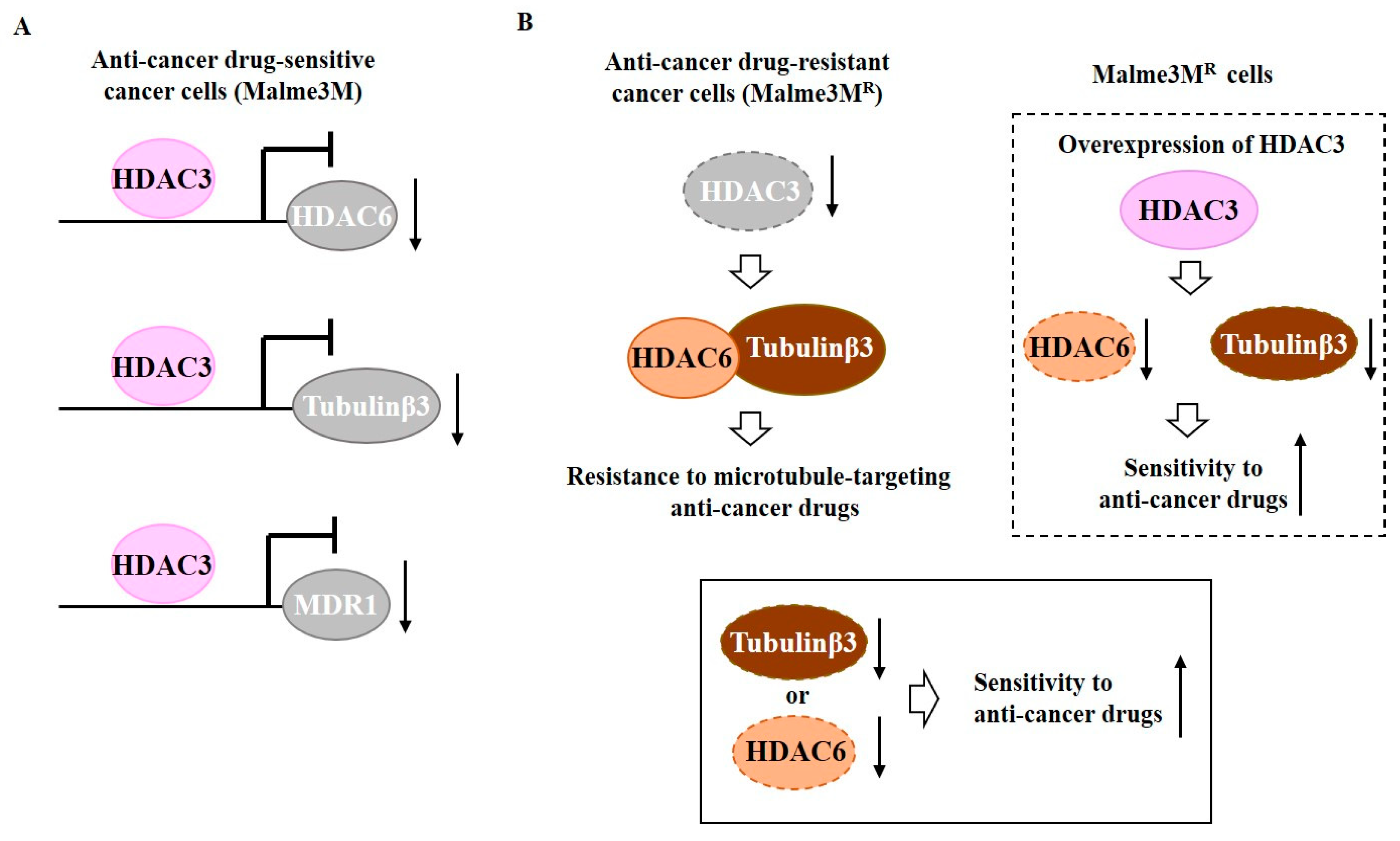
HDAC3 can bind to promoter sequences of tubulin β3, HDAC6, and MDR1, and negatively regulate expression levels of these proteins [55] (Figure 1A). Down-regulation of HDAC6 or tubulin β3 enhances sensitivity of Malme3MR cells to anti-cancer drugs [55]. HDAC6 shows an interaction with tubulin β3 that is disrupted by HDAC3 [55] (Figure 1B). HDAC6 can interact with beta tubulin in yeast two-hybrid assays and deacetylate tubulin [56]. HDAC3 functions upstream of HDAC6 and tubulin β3 (Figure 1B). Therefore, tubulin β3 is a downstream target of HDAC3 that confers resistance to anti-cancer drugs.
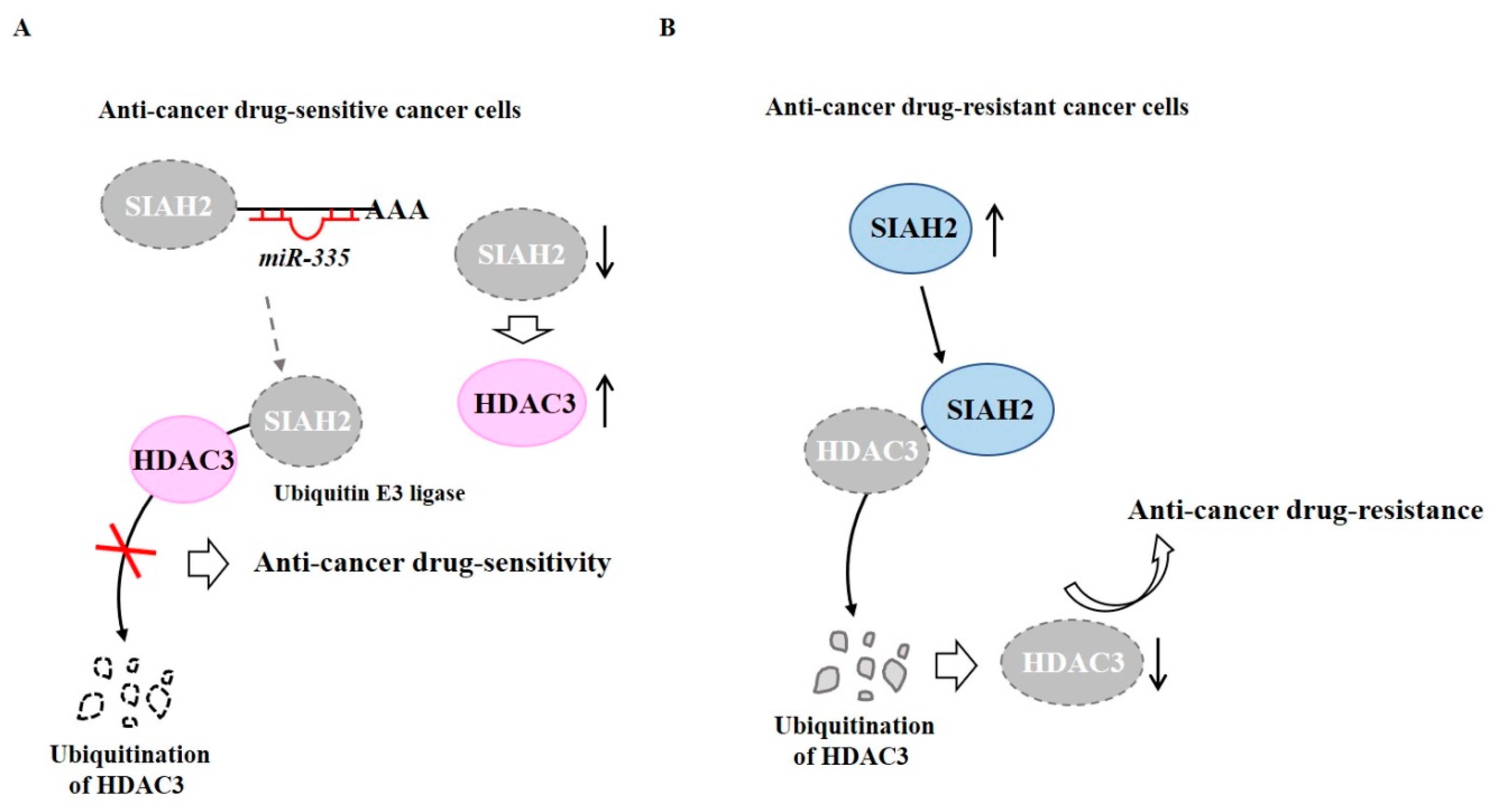
We investigated the role of ubiquitination in the expression regulation of HDAC3. The loss of Seven in Absentia Homolog 2 (SIAH2) is known to increase chemo-sensitivity [57]. E3 ubiquitin ligase activity of SIAH2 is necessary for resistance to death receptor-mediated apoptosis [58]. Inhibition of SIAH2 ubiquitin ligase blocks melanoma tumorigenesis [59]. Loss of SIAH2 suppresses tumorigenesis in a large tumor suppressor kinase 2 (LATS2)-dependent manner in a xenograft mouse model [60]. SIAH2 expression shows an inverse correlation with the tumor grade, p53, and human epidermal growth factor receptor 2 (HER2) [60]. The inhibition of SIAH2 activity reduces metastatic potential, but not tumorigenic potential, via HIF-1α [61]. SIAH2 targets p53 and regulates the acetylation status of p53 [62]. Ski protein stabilizes HDAC3 by binding to SIAH2 [63].
TargetScan analysis has predicted that miR-335 is a negative regulator of SIAH2. MiR-335 binds to 3′-UTR of SIAH2 and decreases expression of SIAH2, which in turn increases the expression of HDAC3 to confer anti-cancer drug sensitivity in anti-cancer drug-sensitive melanoma cells, such as Malme3M cells [64] (Figure 2A). HDAC3 may increase expression of miR-335. In Malme3MR cells (anti-cancer drug-resistant melanoma cells), SIAH2 can binds to HDAC3, resulting in degradation of HDAC3 by ubiquitination [64] (Figure 2B). Thus, HDAC3 may confer sensitivity to anti-cancer drugs by regulating expression of SIAH2.
4. HDAC3-miRNA Network in Angiogenesis and Anti-Cancer Drug Resistance
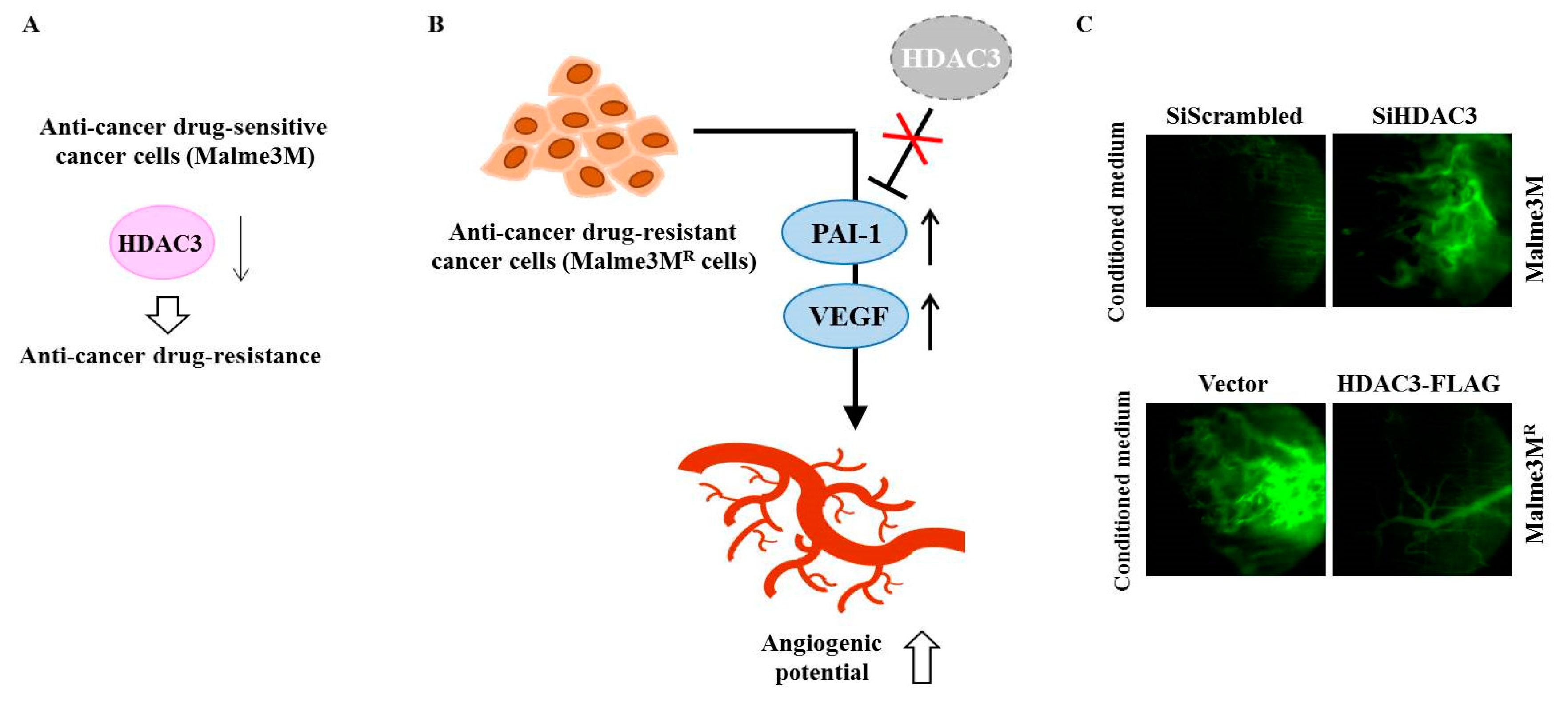
Anti-cancer drug-resistant phenotypes are under epigenetic regulation [65,66]. Among various HDACs, HDAC3 can inhibit the invasion, migration, and angiogenic potential of hepatic cancer cell lines and melanoma cell lines [67,68]. HDAC3 also confers sensitivity to anti-cancer drugs [67,69] (Figure 3A). Many reports have suggested a close relationship between angiogenic potential and anti-cancer drug resistance [68]. HDAC3 inhibits the angiogenic potential of cancer cells by decreasing expression levels of angiogenic factors, such as VEGF and plasminogen activator inhibitor-1 (PAI-1) [22] (Figure 3B). VEGF and HDAC3 can form a negative feedback loop and regulate endothelial cell tube formation [13]. Down-regulation of HDAC3 enhances angiogenic potential of Malme3M cells, while overexpression of HDAC3 decreases the angiogenic potential of Malme3MR cells [13] (Figure 3C).
Expression levels of HDAC1 and HDAC2 are higher in melanoma cells resistant to anti-cancer drugs than those in melanoma cells sensitive to anti-cancer drugs [64] (Figure 4A). It is probable that HDAC1 and HDAC-2 might be able to confer resistance to anti-cancer drugs. Malme3MR cells show lower expression of HDAC3 than parental sensitive Malme3M cells [64] (Figure 4B). Melanoma cells that are naturally resistant to anti-cancer drugs also showed lower expression of HDAC3 than anti-cancer drug-sensitive melanoma cells [64]. Overexpression of HDAC3 overcomes resistance of Malme3MR cells to anti-cancer drugs [64].
MicroRNAs (miRNAs) are small, non-coding RNAs (21–23 nucleotides) that functions in post-transcriptional regulation of gene expression. MiRNAs can regulate the expression of various oncogenes and tumor suppressor genes [70]. MicroRNA array analyses show differential expression of miRNAs, such as miR-326, miR-200b, miR-217, and miR-335 between Malme3M cells and Malme3MR cells [64]. MiR-326 negatively regulated by Notch can decrease the tumorigenic potential of glioma cells [71]. MiR-326 enhances sensitivity of etoposide (VP-16)-resistant breast cancer cell lines, MCF-7/VP to VP-16 and doxorubicin [72]. MiR-335 increases the expression of HDAC3 by preventing SIAH2 from inducing ubiquitination of HDAC3 [64]. MiR-326 directly regulated by HDAC3 (Figure 4A) can regulate responses to anti-cancer drugs [64]. Chromatin immunoprecipitation (ChIP) assays have revealed that HDAC3 can bind to promoter sequences of miR-200b, miR-217, and miR-335 in Malme3M cells (Figure 4A). Increased expressions of miR-200b, miR-217, and miR-335 confer sensitivity to anti-cancer drugs. In Malme3MRcells, miR-326 binds to 3′-UTR of the HDAC3 gene and decreases expression of HDAC3 (Figure 4B). Decreased expression of HDAC3 decreases expression levels of miR-200b, miR-217, and miR-335 to confer anti-cancer drug resistance (Figure 4B). These reports suggest the important role of the HDAC3-miRNA network in anti-cancer drug resistance.
5. Role of CAGE-miRNA Network in Anti-Cancer Drug Resistance
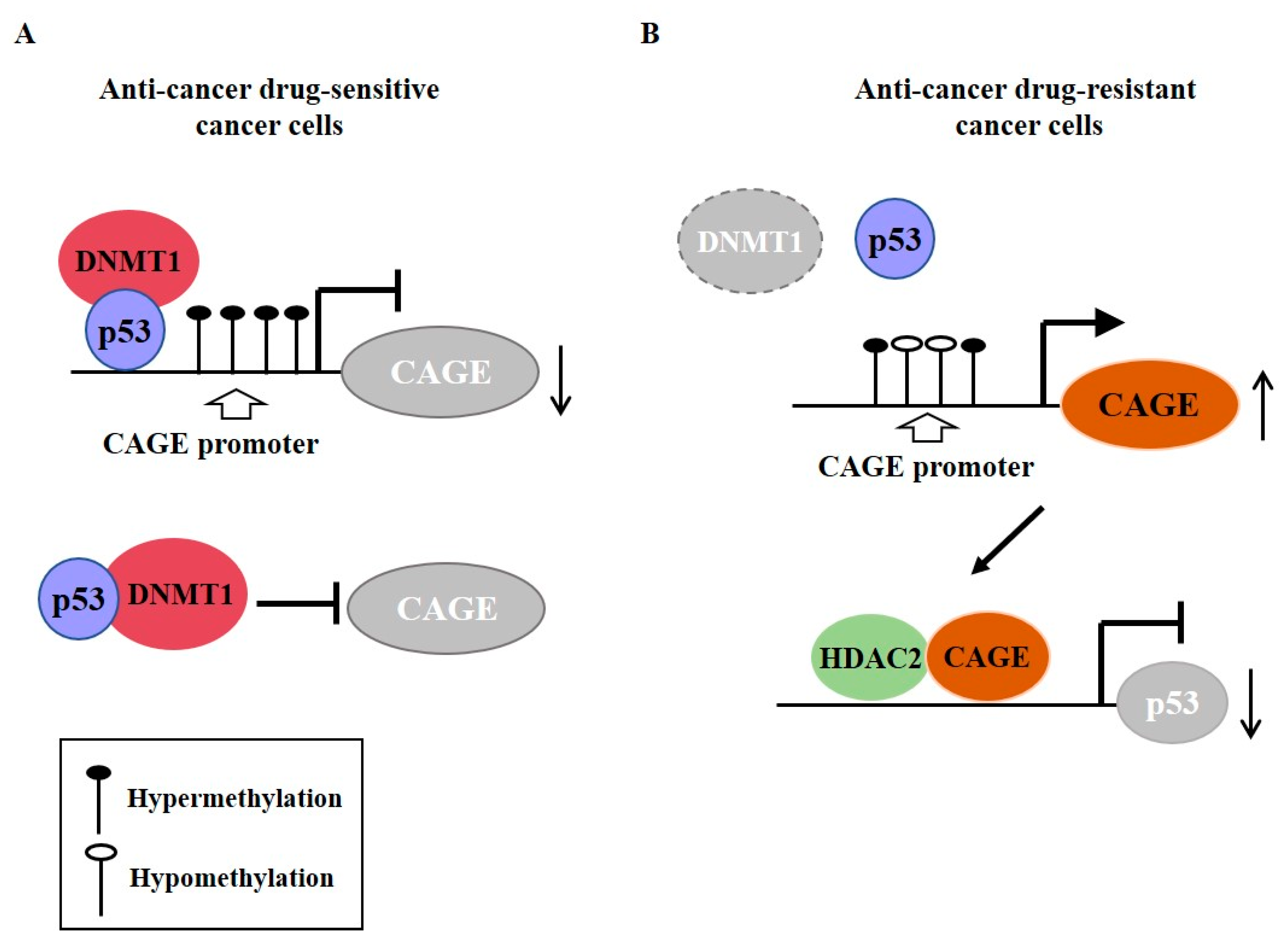
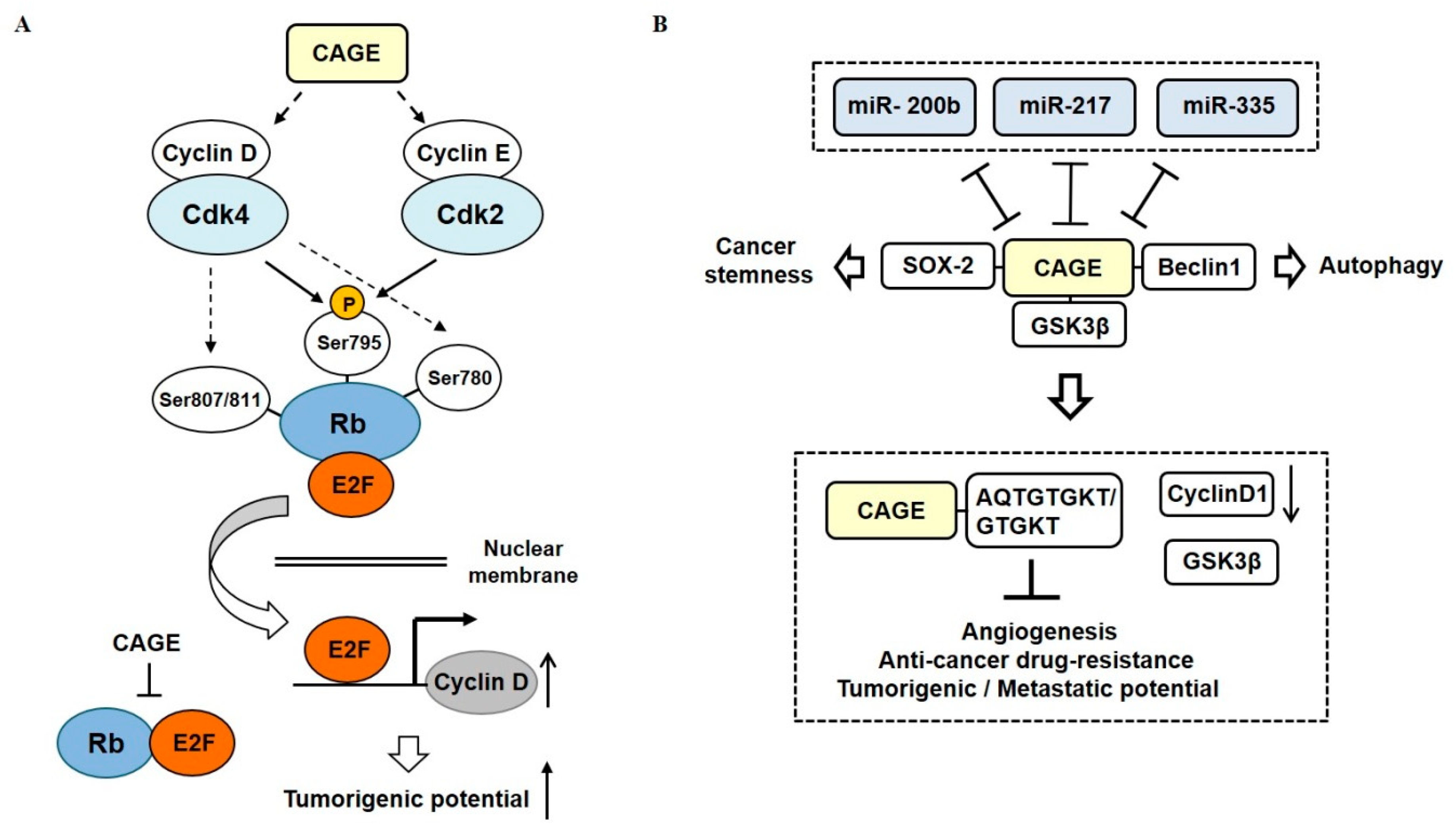
Cancer-associated genes (CAGE) have been found in the sera of patients with various cancers [73,74,75]. DNMT1 increases methylation at promoter sequences of CAGE, leading to decreased expression of CAGE [76] (Figure 5A). CAGE can bind to HDAC2 and confers resistance to various anti-cancer drugs [77] (Figure 5B). CAGE can also inactivate tumor suppressor retinoblastoma (Rb) protein by phosphorylation, increase the expression of cyclins, and act as an oncogene [78] (Figure 6A).
CAGE also binds to GSK3β and increases expression of cyclinD, thus conferring resistance to various anti-cancer drugs in melanoma cells [79] (Figure 6B). TargetScan has predicted binding of miR-200b, miR-217, and miR-335 to 3′ untranslated regions (UTR) of CAGE. MiR-200b, miR-217, and miR-335 form a negative feedback loop with CAGE (Figure 6B). CAGE-derived peptides can bind to CAGE and confer sensitivity to anti-cancer drugs in melanoma cells [79] (Figure 6B). CAGE also binds to the sex-determining region y-box 2 (SOX2) and induces cancer stem cell-like properties in melanoma cells [80] (Figure 6B). In addition, CAGE can bind to Beclin1 and induce autophagic flux (manuscript in preparation).
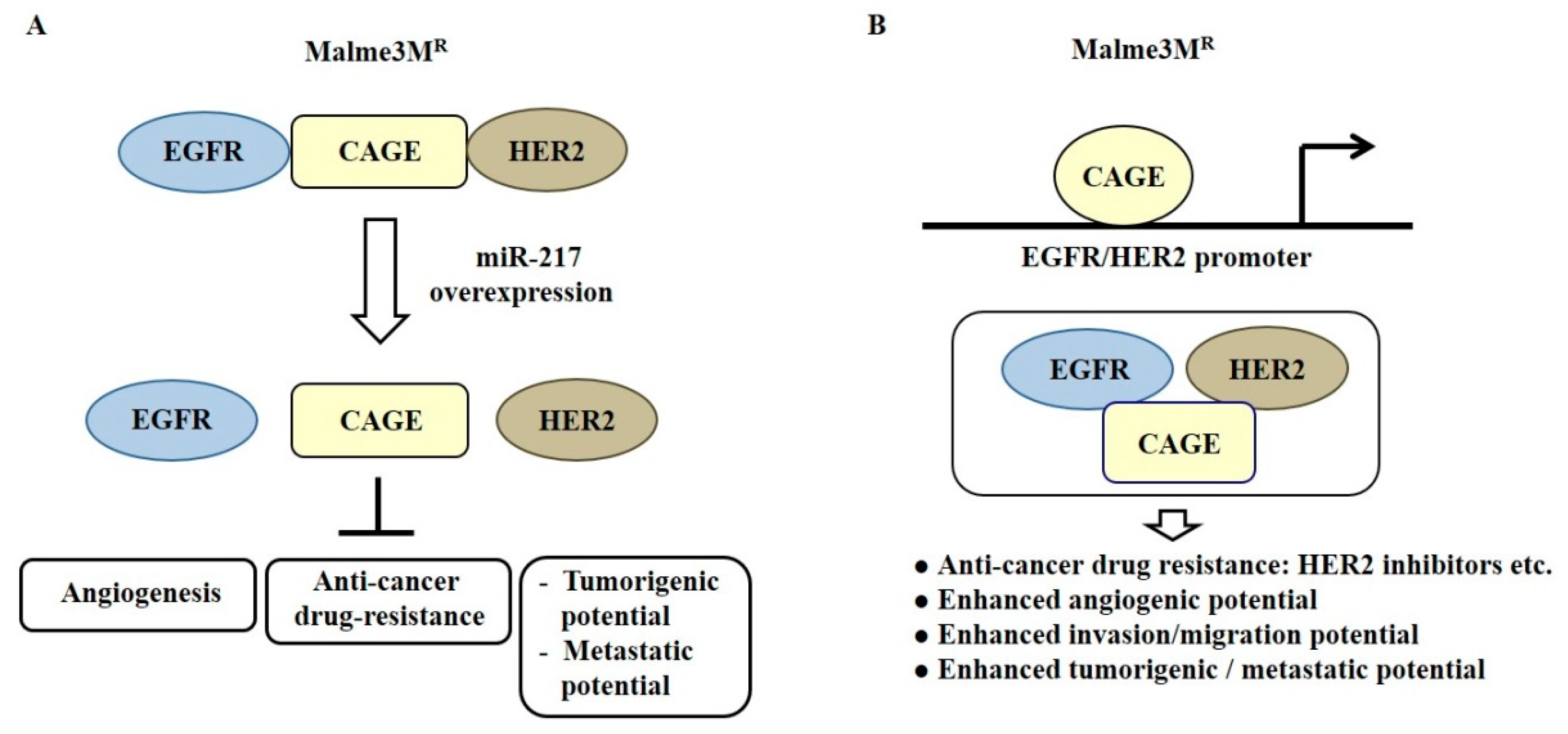
MiR-200b can form a negative feedback loop with CAGE, a cancer/testis antigen, and regulate invasion of a cancer cell line. It also regulates tumorigenic and angiogenic responses to microtubule-targeting drugs [81]. CAGE binds to miR-200b and directly regulates expression of miR-200b [81]. Recombinant CAGE proteins display angiogenic potential [81]. MiR-217 can form a negative feedback loop with CAGE and negatively regulates the tumorigenic and metastatic potential of melanoma cells [82]. CAGE binds to the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) and confers in vivo resistance to trastuzumab, an inhibitor of HER2 [82] (Figure 7A). MiR-217 overexpression prevents interactions of CAGE with EGFR and HER2 in anti-cancer drug-resistant Malme3MR cells [82] (Figure 7A). Down-regulation of EGFR or HER2 can enhance sensitivity to anti-cancer drugs, such as EGFR inhibitors [82]. CAGE directly regulates expression of HER2. It is necessary for the interaction of CAGE with EGFR and HER2 in Malme3MR cells [82] (Figure 7B).
These results indicate that HDAC3 and CAGE can exert an opposite regulation on the expression of miR-200b and miR-217. This has led us to investigate relationships between HDAC3 and CAGE in anti-cancer drug resistance.
6. HDAC3 Functions Upstream of CAGE and Targets CAGE
Anti-cancer drug-resistant cancer cell lines show an increased expression of phosphorylated epidermal growth factor receptor (pEGFRY845) and an interaction between CAGE and EGFR [82]. CAGE-derived GTGKT and AQTGTGKT peptides inhibit activation of EGFR, enhance sensitivity to gefitinib and trastuzumab, and prevent interactions of CAGE with EGFR and HER2 [82]. Vorinostat, an inhibitor of HDACs, decreases expression of EGFR and inhibits renal growth [83]. HDAC3 directly regulates the expression of EGFR by binding to promoter sequences of EGFR, while HDAC inhibitors can reverse the expression of EGFR in colorectal cancer cells [84]. HIF1β activates EGFR and extracellular regulated kinase (ERK) by HDACs [85]. EGFR signaling can increase expression levels of HDACs and inhibit osteoblast differentiation by down-regulating Runt-Related Transcription Factor 2 (Runx2) [86]. These reports imply that HDAC3 might be able to regulate EGFR signaling.
AG1478, an inhibitor of EGFR tyrosine kinase, inhibits functions of ATP-binding cassette (ABC) transporters, such as ABCB1 and ABCG2 [87]. The targeting of EGFR in cancers is quite limited due to the status of the Kirsten rat sarcoma 2 viral oncogene homolog (KRAS) mutation [88]. KRAS mutants can bypass EGFR to activate Ras/Raf/MEK/ERK signals and significantly weaken the therapeutic effect of cetuximab [89]. C-Met inhibition can enhance sensitivity to anti-cancer drugs, such as 5-Fluorouracil and taxol [90]. Combination therapy employing the inhibition of c-Met and EGFR suppresses tumorigenic potential of hepatocellular carcinoma cells [91]. These reports suggest that HDAC3 may regulate the expression of CAGE and the activation of EGFR in anti-cancer drug-resistance.
The MiR-326 inhibitor increases the expression of HDAC3, which results in the binding of HDAC3 to promoter sequences of CAGE in anti-cancer drug-resistant Malme3MR cells (Figure 8). Decreased expression of CAGE leads to enhanced sensitivity to anti-cancer drugs [69] (Figure 8). HDAC3 negatively regulates the expression of pEGFRY845 (Figure 8). Therefore, HDAC3 can target CAGE to regulate the activation of EGFR signaling, responses to various anti-cancer drugs, and the tumorigenic potential of cancer cells.
7. Discussion and Conclusions
Reports from our laboratory and others indicate that HDAC3 regulates responses to anti-cancer drugs, tumorigenic potential, and angiogenesis. MicroRNAs that regulate the expression of HDAC3 can also regulate responses to anti-cancer drugs, and the tumorigenic potential and angiogenic potential of cancer cells. Our studies provide the mechanism of anti-cancer drug resistance regulated by HDAC3. We discovered the role of CAGE, a cancer/testis antigen, in anti-cancer drug resistance and the associated mechanisms.
MicroRNAs that form negative feedback loops with CAGE can regulate responses to anti-cancer drugs and form positive feedback loops with HDAC3. HDAC3 functions upstream of CAGE and negatively regulates expression of CAGE. CAGE-promoted anti-cancer drug resistance involves direct regulation of EGFR and HER2 by CAGE and interactions of CAGE with EGFR and HER2.
To overcome anti-cancer drug-resistance, it might be necessary to employ miRNA-inhibitors or miRNA-mimics that can increase expression of HDAC3 while decreasing expression of CAGE. It would be also necessary to identify small molecules that could increase expression of HDAC3. For this, it would be necessary to develop screening tools based on the structure of HDAC3. For better understanding of anti-cancer drug resistance regulated by HDAC3, it would be necessary to employ cancer patient-derived tissues and cell lines that show low level expression levels of HDAC3 but high expression of CAGE. This approach will give better understanding of anti-cancer drug resistance and strategy to develop anti-cancer drugs. Overall, HDAC3 can serve as a target for developing anti-cancer drugs and/or drugs that can enhance sensitivity to anti-cancer drugs in clinical use.
Funding
This work was supported by National Research Foundation Grants (2017R1A2A2A05001029 and 2017M3A9G7072417), a grant from the BK21 plus Program. This work was supported by a grant from Kangwon National University (520170495). This research was also supported by (2018R1D1A1B07043498 to Y. Kim).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mahlknecht, U.; Emiliani, S.; Najfeld, V.; Young, S.; Verdin, E. Genomic organization and chromosomal localization of the human histone deacetylase 3 gene. Genomics 1999, 56, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, U.; Will, J.; Varin, A.; Hoelzer, D.; Herbein, G. Histone deacetylase 3, a class I histone deacetylase, suppresses MAPK11-mediated activating transcription factor-2 activation and represses TNF gene expression. J. Immunol. 2004, 173, 3979–3990. [Google Scholar] [CrossRef] [PubMed]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Choi, K.C.; Choi, H.K.; Kang, H.B.; Kim, M.J.; Lee, Y.H.; Lee, O.H.; Lee, J.; Kim, Y.J.; Jun, W.; et al. HDAC3 selectively represses CREB3-mediated transcription and migration of metastatic breast cancer cells. Cell. Mol. Life Sci. 2010, 67, 3499–3510. [Google Scholar] [CrossRef]
- Bardai, F.H.; D’Mello, S.R. Selective toxicity by HDAC3 in neurons: Regulation by Akt and GSK3beta. J. Neurosci. 2011, 31, 1746–1751. [Google Scholar] [CrossRef]
- Lundh, M.; Christensen, D.P.; Damgaard Nielsen, M.; Richardson, S.J.; Dahllof, M.S.; Skovgaard, T.; Berthelsen, J.; Dinarello, C.A.; Stevenazzi, A.; Mascagni, P.; et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 2012, 55, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Bendinelli, P.; Matteucci, E.; Maroni, P.; Desiderio, M.A. NF-kappaB activation, dependent on acetylation/deacetylation, contributes to HIF-1 activity and migration of bone metastatic breast carcinoma cells. Mol. Cancer Res. 2009, 7, 1328–1341. [Google Scholar] [CrossRef]
- Miao, L.J.; Huang, F.X.; Sun, Z.T.; Zhang, R.X.; Huang, S.F.; Wang, J. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in nonsmall cell lung cancer cells. Tumor Biol. 2014, 35, 7097–7103. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Alam, H.; Li, N.; Wagner, K.W.; Chung, J.; Ahn, Y.W.; Lee, M.G. Transcriptional repression of histone deacetylase 3 by the histone demethylase KDM2A is coupled to tumorigenicity of lung cancer cells. J. Biol. Chem. 2014, 289, 7483–7496. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Park, H.; Kim, Y.; Kim, H.; Jeoung, D. HDAC3 acts as a negative regulator of angiogenesis. BMB Rep. 2014, 47, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell. 2008, 30, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, L.; Wu, Y.; Zhang, R.; Zhang, M.; Liao, D.; Wang, G.; Qin, G.; Xu, R.H.; Kang, T. CBX4 Suppresses Metastasis via Recruitment of HDAC3 to the Runx2 Promoter in Colorectal Carcinoma. Cancer Res. 2016, 76, 7277–7289. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.H.; Ko, H.; Jeon, H.; Sung, G.J.; Park, S.Y.; Jun, W.J.; Lee, Y.H.; Lee, J.; Lee, S.W.; Yoon, H.G.; et al. Delphinidin induces apoptosis via cleaved HDAC3-mediated p53 acetylation and oligomerization in prostate cancer cells. Oncotarget 2016, 7, 56767–56780. [Google Scholar] [CrossRef] [Green Version]
- Hanigan, T.W.; Aboukhatwa, S.M.; Taha, T.Y.; Frasor, J.; Petukhov, P.A. Divergent JNK Phosphorylation of HDAC3 in Triple-Negative Breast Cancer Cells Determines HDAC Inhibitor Binding and Selectivity. Cell Chem. Biol. 2017, 24, 1356–1367.e8. [Google Scholar] [CrossRef]
- Yang, M.; Dang, X.; Tan, Y.; Wang, M.; Li, X.; Li, G. I-7ab inhibited the growth of TNBC cells via targeting HDAC3 and promoting the acetylation of p53. Biomed. Pharmacother. 2018, 99, 220–226. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Pan, M.; Sun, J.; Lu, H.; Shen, Q.; Zhang, S.; Jiang, T.; Liu, L.; Jin, W.; Chen, Y.; et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med. 2013, 91, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Inks, E.S.; Li, X.; Hou, J.; Chou, C.J.; Zhang, J.; Jiang, Y.; Zhang, Y.; Xu, W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J. Med. Chem. 2014, 57, 3324–3341. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Botrugno, O.A.; Santoro, F.; Minucci, S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett. 2009, 280, 134–144. [Google Scholar] [CrossRef]
- Dvorakova, M.; Vanek, T. Histone deacetylase inhibitors for the treatment of cancer stem cells. MedChemComm 2016, 7, 2217–2231. [Google Scholar] [CrossRef]
- Liu, N.; Li, S.; Wu, N.; Cho, K.S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef]
- Guzman, M.L.; Yang, N.; Sharma, K.K.; Balys, M.; Corbett, C.A.; Jordan, C.T.; Becker, M.W.; Steidl, U.; Abdel-Wahab, O.; Levine, R.L.; et al. Selective activity of the histone deacetylase inhibitor AR-42 against leukemia stem cells: A novel potential strategy in acute myelogenous leukemia. Mol. Cancer Ther. 2014, 13, 1979–1990. [Google Scholar] [CrossRef]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Yadav, A.; Lang, J.C.; Teknos, T.N.; Kumar, P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer 2015, 6, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Malek, S.N.; Zheng, P.; Liu, Y. Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011, 8, 399–411. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.W.; Park, J.A.; Lee, J.W.; Seo, J.H.; Jung, B.K.; Bae, M.K.; Kim, K.W. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1alpha protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Lu, X.F.; Cao, X.Y.; Zhu, Y.J.; Wu, Z.R.; Zhuang, X.; Shao, M.Y.; Xu, Q.; Zhou, Y.J.; Ji, H.J.; Lu, Q.R.; et al. Histone deacetylase 3 promotes liver regeneration and liver cancer cells proliferation through signal transducer and activator of transcription 3 signaling pathway. Cell Death Dis. 2018, 9, 398. [Google Scholar] [CrossRef] [Green Version]
- Rinkenbaugh, A.L.; Cogswell, P.C.; Calamini, B.; Dunn, D.E.; Persson, A.I.; Weiss, W.A.; Lo, D.C.; Baldwin, A.S. IKK/NF-kappaB signaling contributes to glioblastoma stem cell maintenance. Oncotarget 2016, 7, 69173–69187. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Li, Q.Z.; Chen, L.; Chen, B.D.; Wang, B.; Zhang, X.J.; Li, W.P. Histone Deacetylase Inhibitor RGFP109 Overcomes Temozolomide Resistance by Blocking NF-kappaB-Dependent Transcription in Glioblastoma Cell Lines. Neurochem. Res. 2016, 41, 3192–3205. [Google Scholar] [CrossRef]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; El Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.; Josselin, E.; Lelievre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low Xist expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef] [PubMed]
- Di Pompo, G.; Salerno, M.; Rotili, D.; Valente, S.; Zwergel, C.; Avnet, S.; Lattanzi, G.; Baldini, N.; Mai, A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J. Med. Chem. 2015, 58, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yin, Y.; Dorfman, R.G.; Zou, T.; Pan, Y.; Li, Y.; Wang, Y.; Zhou, Q.; Zhou, L.; Kong, B.; et al. Down-regulation of HDAC3 inhibits growth of cholangiocarcinoma by inducing apoptosis. Oncotarget 2017, 8, 99402–99413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8. [Google Scholar] [CrossRef]
- El-Khoury, V.; Breuzard, G.; Fourre, N.; Dufer, J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1) gene expression by a transcription-dependent mechanism in a drug-resistant small cell lung carcinoma cell line model. Br. J. Cancer 2007, 97, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jiang, Z.; Yin, P.; Li, Q.; Liu, J. Role for Class I histone deacetylases in multidrug resistance. Exp. Cell Res. 2012, 318, 177–186. [Google Scholar] [CrossRef]
- Kavallaris, M.; Kuo, D.Y.; Burkhart, C.A.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef]
- Levallet, G.; Bergot, E.; Antoine, M.; Creveuil, C.; Santos, A.O.; Beau-Faller, M.; de Fraipont, F.; Brambilla, E.; Levallet, J.; Morin, F.; et al. High TUBB3 expression, an independent prognostic marker in patients with early non-small cell lung cancer treated by preoperative chemotherapy, is regulated by K-Ras signaling pathway. Mol. Cancer Ther. 2012, 11, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Verdier-Pinard, P.; Wang, F.; Martello, L.; Burd, B.; Orr, G.A.; Horwitz, S.B. Analysis of tubulin isotypes and mutations from taxol-resistant cells by combined isoelectrofocusing and mass spectrometry. Biochemistry 2003, 42, 5349–5357. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Sato, N.; Yanai, K.; Akiyoshi, T.; Nagai, S.; Wada, J.; Koga, K.; Mibu, R.; Nakamura, M.; Katano, M. Enhancement of paclitaxel-induced apoptosis by inhibition of mitogen-activated protein kinase pathway in colon cancer cells. Anticancer Res. 2009, 29, 261–270. [Google Scholar] [PubMed]
- Guan, J.; Yuan, Z.; He, J.; Wu, Z.; Liu, B.; Lin, X.; Mo, L.; Mo, H. Overexpression of caveolin-1 reduces Taxol resistance in human osteosarcoma cells by attenuating PI3K-Akt-JNK dependent autophagy. Exp. Ther. Med. 2016, 12, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Jeoung, D. Tubulin Beta3 Serves as a Target of HDAC3 and Mediates Resistance to Microtubule-Targeting Drugs. Mol. Cells 2015, 38, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.S.; Sceneay, J.; House, C.M.; Halse, H.M.; Liu, M.C.; George, J.; Hunnam, T.C.; Parker, B.S.; Haviv, I.; Ronai, Z.; et al. Vascular normalization by loss of Siah2 results in increased chemotherapeutic efficacy. Cancer Res. 2012, 72, 1694–1704. [Google Scholar] [CrossRef]
- Christian, P.A.; Fiandalo, M.V.; Schwarze, S.R. Possible role of death receptor-mediated apoptosis by the E3 ubiquitin ligases Siah2 and POSH. Mol. Cancer 2011, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Stebbins, J.L.; Dewing, A.; Qi, J.; Pellecchia, M.; Ronai, Z.A. Inhibition of Siah2 ubiquitin ligase by vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and blocks melanoma tumorigenesis. Pigment Cell Melanoma Res. 2009, 22, 799–808. [Google Scholar] [CrossRef]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef]
- Chan, P.; Moller, A.; Liu, M.C.; Sceneay, J.E.; Wong, C.S.; Waddell, N.; Huang, K.T.; Dobrovic, A.; Millar, E.K.; O’Toole, S.A.; et al. The expression of the ubiquitin ligase SIAH2 (seven in absentia homolog 2) is mediated through gene copy number in breast cancer and is associated with a basal-like phenotype and p53 expression. Breast Cancer Res. 2011, 13, R19. [Google Scholar] [CrossRef] [PubMed]
- Grishina, I.; Debus, K.; Garcia-Limones, C.; Schneider, C.; Shresta, A.; Garcia, C.; Calzado, M.A.; Schmitz, M.L. SIAH-mediated ubiquitination and degradation of acetyl-transferases regulate the p53 response and protein acetylation. Biochim. Biophys. Acta 2012, 1823, 2287–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.L.; Ueki, N.; Hayman, M.J. The Ski protein negatively regulates Siah2-mediated HDAC3 degradation. Biochem. Biophys. Res. Commun. 2010, 399, 623–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Kim, H.; Park, D.; Jeoung, D. miR-335 Targets SIAH2 and Confers Sensitivity to Anti-Cancer Drugs by Increasing the Expression of HDAC3. Mol. Cells 2015, 38, 562–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Yang, X.; Xue, Y.W.; Zhang, X.F.; Wang, Y.; Liu, W.J.; Wu, X.J. Promoter methylation of glutathione S-transferase pi1 and multidrug resistance gene 1 in bronchioloalveolar carcinoma and its correlation with DNA methyltransferase 1 expression. Cancer 2009, 115, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Polgar, O.; Huff, L.M.; Morisaki, K.; Bates, S.E. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol. Cancer Res. 2008, 6, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Park, H.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. miR-326-histone deacetylase-3 feedback loop regulates the invasion and tumorigenic and angiogenic response to anti-cancer drugs. J. Biol. Chem. 2014, 289, 28019–28039. [Google Scholar] [CrossRef]
- Park, H.; Kim, Y.; Park, D.; Jeoung, D. Nuclear localization signal domain of HDAC3 is necessary and sufficient for the expression regulation of MDR1. BMB Rep. 2014, 47, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, Y.; Goh, H.; Jeoung, D. Histone Deacetylase-3/CAGE Axis Targets EGFR Signaling and Regulates the Response to Anti-Cancer Drugs. Mol. Cells 2016, 39, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; diPierro, C.G.; Li, Y.; Chiocca, E.A.; et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 2009, 29, 15161–15168. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wu, H.; Xia, J.; Li, Y.; Zhang, Y.; Huang, K.; Wagar, N.; Yoon, Y.; Cho, H.T.; Scala, S.; et al. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem. Pharmacol. 2010, 79, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.; Lim, Y.; Lee, D.Y.; Park, S.Y.; Lee, H.; Kim, W.H.; Yang, H.; Bang, Y.J.; Jeoung, D.I. Identification and characterization of a novel cancer/testis antigen gene CAGE. Biochem. Biophys. Res. Commun. 2002, 292, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Fujita, T.; Hirao, N.; Matsuzaki, Y.; Okada, T.; Mochimaru, H.; Susumu, N.; Matsumoto, E.; Sugano, K.; Yamashita, N.; et al. Frequent immune responses to a cancer/testis antigen, CAGE, in patients with microsatellite instability-positive endometrial cancer. Clin. Cancer Res. 2005, 11, 3949–3957. [Google Scholar] [CrossRef] [PubMed]
- Liggins, A.P.; Lim, S.H.; Soilleux, E.J.; Pulford, K.; Banham, A.H. A panel of cancer-testis genes exhibiting broad-spectrum expression in haematological malignancies. Cancer Immun. 2010, 10, 8. [Google Scholar] [PubMed]
- Cho, B.; Lee, H.; Jeong, S.; Bang, Y.J.; Lee, H.J.; Hwang, K.S.; Kim, H.Y.; Lee, Y.S.; Kang, G.H.; Jeoung, D.I. Promoter hypomethylation of a novel cancer/testis antigen gene CAGE is correlated with its aberrant expression and is seen in premalignant stage of gastric carcinoma. Biochem. Biophys. Res. Commun. 2003, 307, 52–63. [Google Scholar] [CrossRef]
- Kim, Y.; Park, H.; Park, D.; Lee, Y.S.; Choe, J.; Hahn, J.H.; Lee, H.; Kim, Y.M.; Jeoung, D. Cancer/testis antigen CAGE exerts negative regulation on p53 expression through HDAC2 and confers resistance to anti-cancer drugs. J. Biol. Chem. 2010, 285, 25957–25968. [Google Scholar] [CrossRef] [PubMed]
- Por, E.; Byun, H.J.; Lee, E.J.; Lim, J.H.; Jung, S.Y.; Park, I.; Kim, Y.M.; Jeoung, D.I.; Lee, H. The cancer/testis antigen CAGE with oncogenic potential stimulates cell proliferation by up-regulating cyclins D1 and E in an AP-1- and E2F-dependent manner. J. Biol. Chem. 2010, 285, 14475–14485. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeon, D.; Jeoung, D. The pentapeptide Gly-Thr-Gly-Lys-Thr confers sensitivity to anti-cancer drugs by inhibition of CAGE binding to GSK3beta and decreasing the expression of cyclinD1. Oncotarget 2017, 8, 13632–13651. [Google Scholar] [CrossRef]
- Kim, Y.; Yeon, M.; Jeoung, D. DDX53 Regulates Cancer Stem Cell-Like Properties by Binding to SOX-2. Mol. Cells 2017, 40, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, D.; Kim, H.; Choi, M.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. miR-200b and cancer/testis antigen CAGE form a feedback loop to regulate the invasion and tumorigenic and angiogenic responses of a cancer cell line to microtubule-targeting drugs. J. Biol. Chem. 2013, 288, 36502–36518. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Park, D.; Han, M.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. miR-217 and CAGE form feedback loop and regulates the response to anti-cancer drugs through EGFR and HER2. Oncotarget 2016, 7, 10297–10321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, R.E.; Huang, Q.; Thai, K.; Advani, S.L.; Lee, K.; Yuen, D.A.; Connelly, K.A.; Advani, A. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: Potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011, 79, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.W.; Wu, M.S.; Huang, W.C.; Chen, C.C. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLoS ONE 2011, 6, e18087. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.D.; Weir, L.; Romanowska, M.; Leigh, I.M.; Panteleyev, A.A. ARNT controls the expression of epidermal differentiation genes through HDAC- and EGFR-dependent pathways. J. Cell Sci 2012, 125, 3320–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Shimizu, E.; Zhang, X.; Partridge, N.C.; Qin, L. EGFR signaling suppresses osteoblast differentiation and inhibits expression of master osteoblastic transcription factors Runx2 and Osterix. J. Cell. Biochem. 2011, 112, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Tiwari, A.K.; Shukla, S.; Robey, R.W.; Kim, I.W.; Parmar, S.; Bates, S.E.; Si, Q.S.; Goldblatt, C.S.; Abraham, I.; et al. Inhibiting the function of ABCB1 and ABCG2 by the EGFR tyrosine kinase inhibitor AG1478. Biochem. Pharmacol. 2009, 77, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Gao, X.; Han, W.; Zheng, Y.; Xu, H.; Zhang, C.; He, Q.; Zhang, L.; Li, Z.; et al. c-Met targeting enhances the effect of irradiation and chemical agents against malignant colon cells harboring a KRAS mutation. PLoS ONE 2014, 9, e113186. [Google Scholar] [CrossRef]
- Steinway, S.N.; Dang, H.; You, H.; Rountree, C.B.; Ding, W. The EGFR/ErbB3 Pathway Acts as a Compensatory Survival Mechanism upon c-Met Inhibition in Human c-Met+ Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0128159. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Tubulin β3 serves as a downstream target of histone deacetylase 3 (HDAC3) and mediates resistance to microtubule-targeting drugs. (A) In anti-cancer drug-sensitive cancer cells, HDAC3 binds to the promoter sequences of histone deacetylase 6 (HDAC6), tubulin β3, and MDR1. HDAC3 decreases expression levels of HDAC6, tubulin β3, and MDR1. (B) In anti-cancer drug-resistant melanoma cells (Malme3MR cells), HDAC6 interacts with tubulin β3. Down-regulation of HDAC3 induces interaction of HDAC6 with tubulin β3. Down-regulation of tubulin β3 or HDAC6 enhances sensitivity of Malme3MR cells to anti-cancer drugs. The T-bar arrows denote inhibition of transcription. The↑rrows denotes increased expression level and ↓arrows denotes decreased expression level.
Figure 1.
Tubulin β3 serves as a downstream target of histone deacetylase 3 (HDAC3) and mediates resistance to microtubule-targeting drugs. (A) In anti-cancer drug-sensitive cancer cells, HDAC3 binds to the promoter sequences of histone deacetylase 6 (HDAC6), tubulin β3, and MDR1. HDAC3 decreases expression levels of HDAC6, tubulin β3, and MDR1. (B) In anti-cancer drug-resistant melanoma cells (Malme3MR cells), HDAC6 interacts with tubulin β3. Down-regulation of HDAC3 induces interaction of HDAC6 with tubulin β3. Down-regulation of tubulin β3 or HDAC6 enhances sensitivity of Malme3MR cells to anti-cancer drugs. The T-bar arrows denote inhibition of transcription. The↑rrows denotes increased expression level and ↓arrows denotes decreased expression level.

Figure 2.
Ubiquitination of HDAC3 leads to decreased expression of HDAC3. (A) In anti-cancer drug-sensitive cancer cells, miR-335 binds to 3′-UTR of Seven in Absentia Homolog 2 (SIAH2) (E3 ubiquitin ligase) to decrease expression of SIAH2. However, SIAH2 does not bind to HDAC3. HDAC3 confers sensitivity to anti-cancer drugs. (B) In anti-cancer drug-resistant cancer cells, SIAH2 binds to HDAC3 and causes ubiquitination of HDAC3. The lack of HDAC3 confers resistance to anti-cancer drugs. The↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.
Figure 2.
Ubiquitination of HDAC3 leads to decreased expression of HDAC3. (A) In anti-cancer drug-sensitive cancer cells, miR-335 binds to 3′-UTR of Seven in Absentia Homolog 2 (SIAH2) (E3 ubiquitin ligase) to decrease expression of SIAH2. However, SIAH2 does not bind to HDAC3. HDAC3 confers sensitivity to anti-cancer drugs. (B) In anti-cancer drug-resistant cancer cells, SIAH2 binds to HDAC3 and causes ubiquitination of HDAC3. The lack of HDAC3 confers resistance to anti-cancer drugs. The↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.

Figure 3.
HDAC3 regulates the angiogenic potential of cancer cells. (A) In anti-cancer drug-sensitive cancer cells (Malme3M cells), the down-regulation of HDAC3 confers resistance to anti-cancer drugs. (B) In anti-cancer drug-resistant cancer cells (Malme3MR cells), decreased expression of HDAC3 leads to increased expression levels of angiogenic factors, such as PAI-1 and VEGF. (C) Malme3M cells or Malme3MR cells were transfected with indicated siRNA or construct. Conditioned medium obtained after transfection was mixed with matrigel and subjected to intravital microscopy. The↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.
Figure 3.
HDAC3 regulates the angiogenic potential of cancer cells. (A) In anti-cancer drug-sensitive cancer cells (Malme3M cells), the down-regulation of HDAC3 confers resistance to anti-cancer drugs. (B) In anti-cancer drug-resistant cancer cells (Malme3MR cells), decreased expression of HDAC3 leads to increased expression levels of angiogenic factors, such as PAI-1 and VEGF. (C) Malme3M cells or Malme3MR cells were transfected with indicated siRNA or construct. Conditioned medium obtained after transfection was mixed with matrigel and subjected to intravital microscopy. The↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.

Figure 4.
HDAC3-miRNA network in anti-cancer drug resistance. (A) In anti-cancer drug-sensitive cancer cells, the HDAC3-p53 complex binds to promoter sequences of miR-326 and decreases expression of miR-326. The HDAC3-p53 complex binds to promoter sequences of miR-200b, miR-217, and miR-335 and increases expression levels of these miRNAs. (B) In anti-cancer drug-resistant cancer cells, miR-326 binds to 3‘-UTR of the HDAC3 gene to decrease expression of HDAC3. As a result, HDAC3 does not bind to promoter sequences of miR-200b, miR-217, or miR-335. Decreased expression levels of miR-200b, miR-217, and miR-335 can confer anti-cancer drug resistance, enhance tumorigenic potential and metastatic potential, and increase angiogenic potential. The T-bar arrows denote inhibition of transcription and →arrows denote activation of transcription. The ↑arrows denotes increased expression level/increased characteristics and ↓arrows denotes decreased expression level.
Figure 4.
HDAC3-miRNA network in anti-cancer drug resistance. (A) In anti-cancer drug-sensitive cancer cells, the HDAC3-p53 complex binds to promoter sequences of miR-326 and decreases expression of miR-326. The HDAC3-p53 complex binds to promoter sequences of miR-200b, miR-217, and miR-335 and increases expression levels of these miRNAs. (B) In anti-cancer drug-resistant cancer cells, miR-326 binds to 3‘-UTR of the HDAC3 gene to decrease expression of HDAC3. As a result, HDAC3 does not bind to promoter sequences of miR-200b, miR-217, or miR-335. Decreased expression levels of miR-200b, miR-217, and miR-335 can confer anti-cancer drug resistance, enhance tumorigenic potential and metastatic potential, and increase angiogenic potential. The T-bar arrows denote inhibition of transcription and →arrows denote activation of transcription. The ↑arrows denotes increased expression level/increased characteristics and ↓arrows denotes decreased expression level.

Figure 5.
Cancer-associated genes (CAGE) confers resistance to anti-cancer drug resistance. (A) In anti-cancer drug-sensitive cancer cells, DNMT1 (DNA methyl transferase I) binds to p53. DNMT causes hypermethylation of promoter sequences of CAGE. In anti-cancer drug-resistant cancer cells, expression of DNMT1 is decreased. This results in hypomethylation of CAGE and increased expression of CAGE. CAGE can bind to HDAC2. The CAGE-HDAC2 complex can bind to the promoter sequences of p53 and decrease expression of p53. The T-bar arrows denote inhibition of transcription/negative regulation and →arrows denote activation of transcription. The ↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.
Figure 5.
Cancer-associated genes (CAGE) confers resistance to anti-cancer drug resistance. (A) In anti-cancer drug-sensitive cancer cells, DNMT1 (DNA methyl transferase I) binds to p53. DNMT causes hypermethylation of promoter sequences of CAGE. In anti-cancer drug-resistant cancer cells, expression of DNMT1 is decreased. This results in hypomethylation of CAGE and increased expression of CAGE. CAGE can bind to HDAC2. The CAGE-HDAC2 complex can bind to the promoter sequences of p53 and decrease expression of p53. The T-bar arrows denote inhibition of transcription/negative regulation and →arrows denote activation of transcription. The ↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.

Figure 6.
CAGE-miRNA network in anti-cancer drug resistance. (A) CAGE increases expression levels of cyclins, which in turn increases cyclin-dependent kinase (CDK) activities. Increased CDK activities phosphorylate Rb to inactivate the Rb protein. Inactivation of Rb leads to the activation of E2 factor (E2F). Activated E2F binds to promoter sequences of cyclins. (B) MiR-200b, miR-217, and miR-335 form negative feedback loops with CAGE. CAGE binds to GSK3β, which increases expression of cyclin D1 to confer resistance to anti-cancer drugs. AQTGTGKT, as a CAGE-derived peptide can disrupt the interaction between CAGE and GSK3β and confer sensitivity to anti-cancer drugs. The T-bar arrows denote negative regulation and both side T-bar arrows denote cross inhibition. The hollow arrows denote positive regulation.
Figure 6.
CAGE-miRNA network in anti-cancer drug resistance. (A) CAGE increases expression levels of cyclins, which in turn increases cyclin-dependent kinase (CDK) activities. Increased CDK activities phosphorylate Rb to inactivate the Rb protein. Inactivation of Rb leads to the activation of E2 factor (E2F). Activated E2F binds to promoter sequences of cyclins. (B) MiR-200b, miR-217, and miR-335 form negative feedback loops with CAGE. CAGE binds to GSK3β, which increases expression of cyclin D1 to confer resistance to anti-cancer drugs. AQTGTGKT, as a CAGE-derived peptide can disrupt the interaction between CAGE and GSK3β and confer sensitivity to anti-cancer drugs. The T-bar arrows denote negative regulation and both side T-bar arrows denote cross inhibition. The hollow arrows denote positive regulation.

Figure 7.
CAGE-miRNA network in anti-cancer drug resistance. (A) In anti-cancer drug-resistant cancer cells, CAGE binds to the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2). MiR-217 decreases expression of CAGE and disrupts interactions of CAGE with EGFR and HER2. (B) In anti-cancer drug-resistant cancer cells, CAGE binds to the promoter sequences of HER2 and EGFR. This results in increased expression levels of EGFR and HER2. The CAGE-HER2-EGFR complex in Malme3MR cells confers anti-cancer drug resistance. The T-bar arrows denote negative regulation and hollow arrows denote positive regulation.
Figure 7.
CAGE-miRNA network in anti-cancer drug resistance. (A) In anti-cancer drug-resistant cancer cells, CAGE binds to the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2). MiR-217 decreases expression of CAGE and disrupts interactions of CAGE with EGFR and HER2. (B) In anti-cancer drug-resistant cancer cells, CAGE binds to the promoter sequences of HER2 and EGFR. This results in increased expression levels of EGFR and HER2. The CAGE-HER2-EGFR complex in Malme3MR cells confers anti-cancer drug resistance. The T-bar arrows denote negative regulation and hollow arrows denote positive regulation.

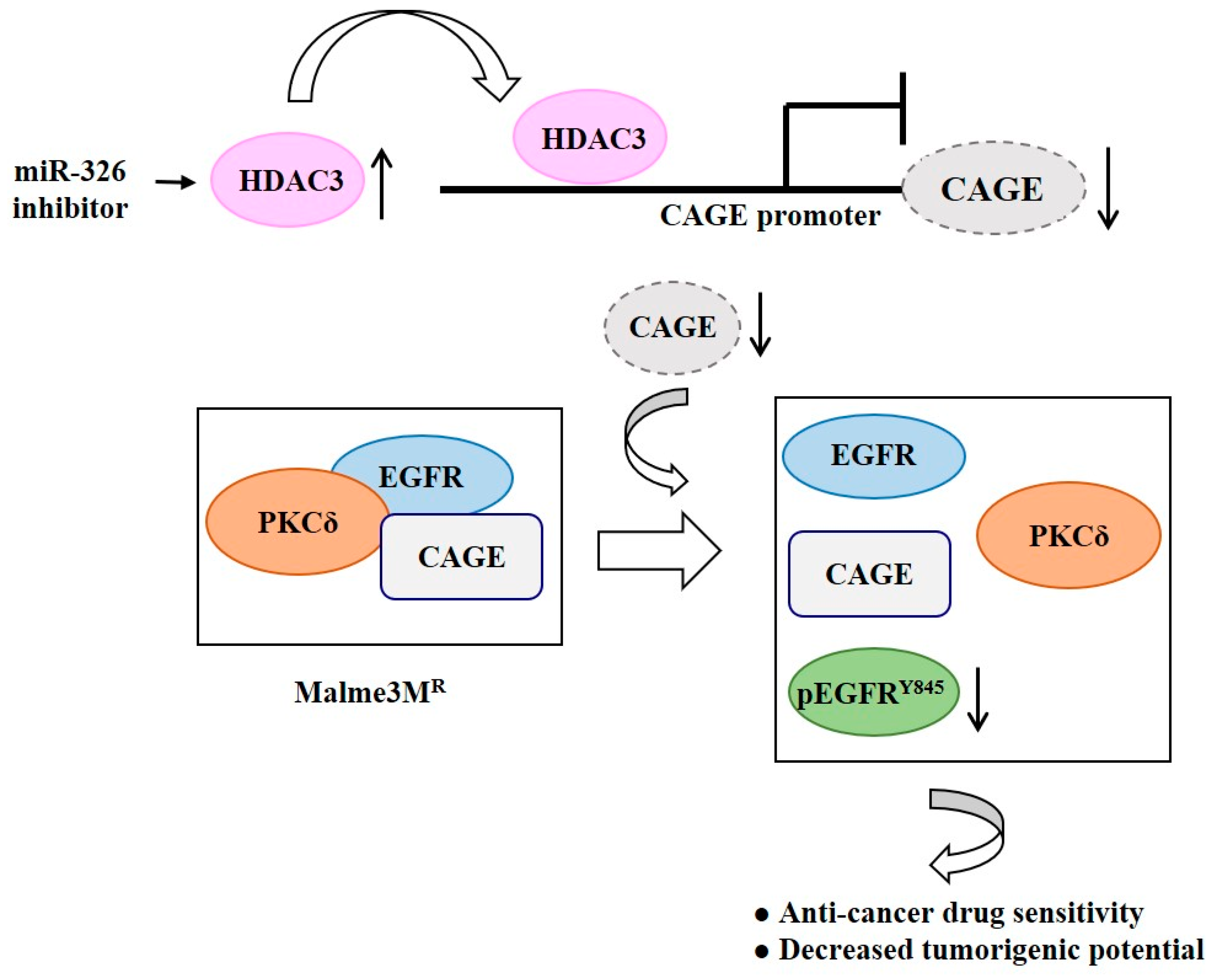
Figure 8.
HDAC3 targets CAGE to inhibit EGFR activation. The MiR-326 inhibitor increases expression of HDAC3, which in turn induces binding of HDAC3 to the promoter sequences of CAGE. Decreased expression of CAGE leads to a disruption of interactions of CAGE with EGFR and protein kinase C gamma (PKCδ) and decreased expression of pEGFR Y845 in Malme3MR cells. The ↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.
Figure 8.
HDAC3 targets CAGE to inhibit EGFR activation. The MiR-326 inhibitor increases expression of HDAC3, which in turn induces binding of HDAC3 to the promoter sequences of CAGE. Decreased expression of CAGE leads to a disruption of interactions of CAGE with EGFR and protein kinase C gamma (PKCδ) and decreased expression of pEGFR Y845 in Malme3MR cells. The ↑arrows denotes increased expression level and ↓arrows denotes decreased expression level.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kwon, Y.; Kim, Y.; Jung, H.S.; Jeoung, D. Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance. Int. J. Mol. Sci. 2019, 20, 51. https://doi.org/10.3390/ijms20010051
AMA Style
Kwon Y, Kim Y, Jung HS, Jeoung D. Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance. International Journal of Molecular Sciences. 2019; 20(1):51. https://doi.org/10.3390/ijms20010051
Chicago/Turabian StyleKwon, Yoojung, Youngmi Kim, Hyun Suk Jung, and Dooil Jeoung. 2019. "Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance" International Journal of Molecular Sciences 20, no. 1: 51. https://doi.org/10.3390/ijms20010051
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.