iTRAQ-Based Comparative Proteomic Analysis Provides Insights into Molecular Mechanisms of Salt Tolerance in Sugar Beet (Beta vulgaris L.)


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Protein Identification Information by iTRAQ
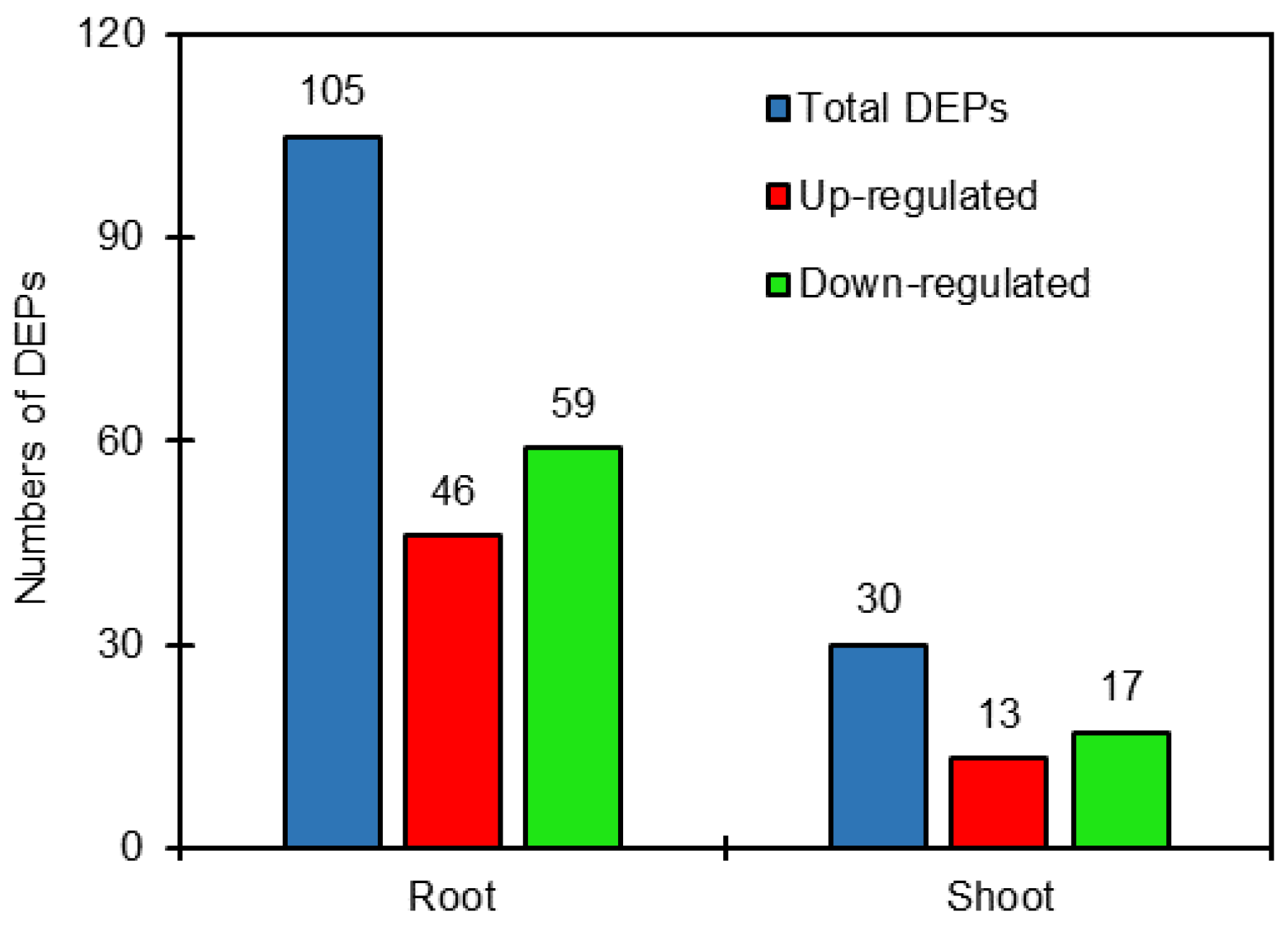
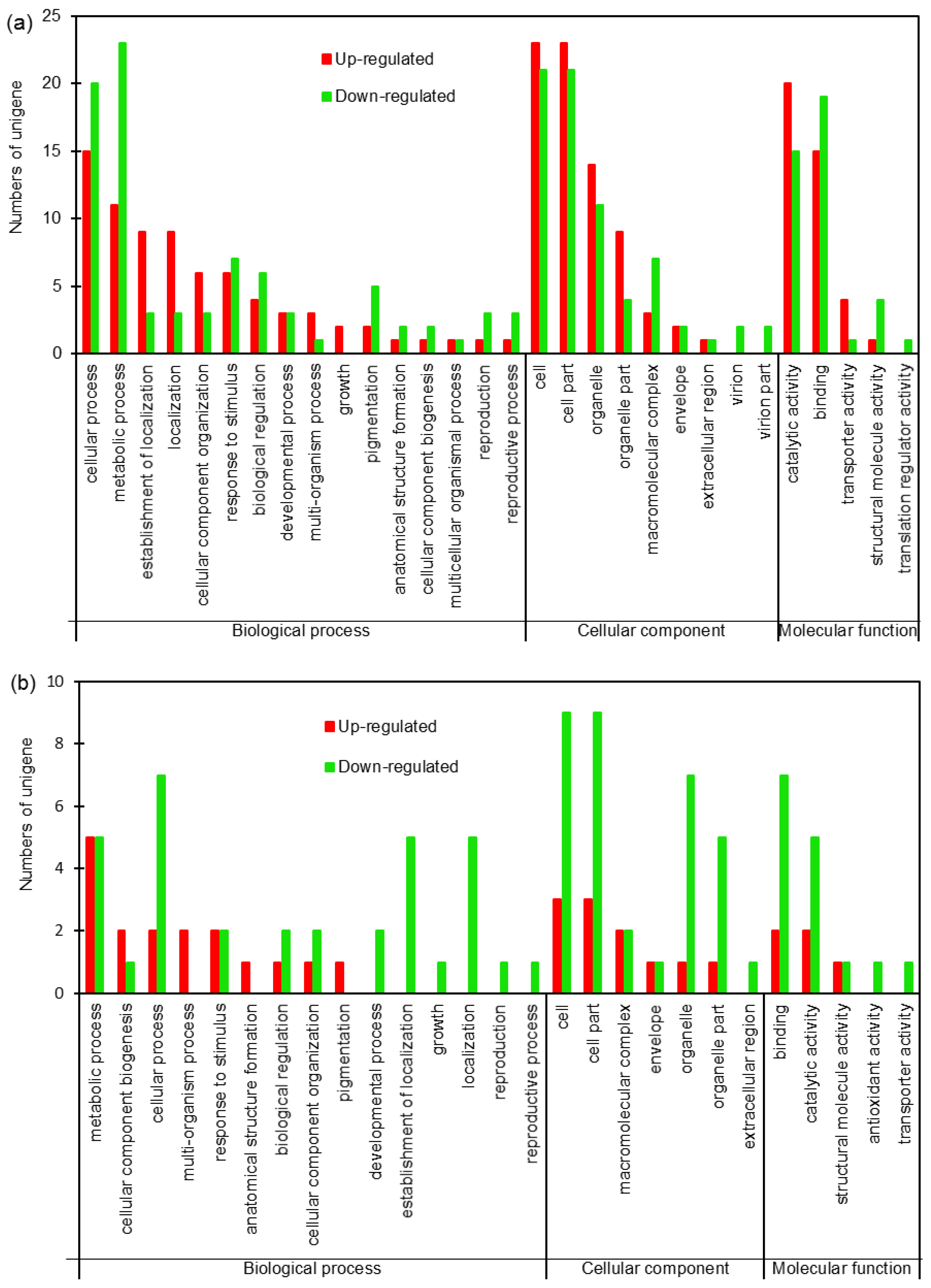
2.2. Identification of Differentially Expressed Proteins (DEPs)
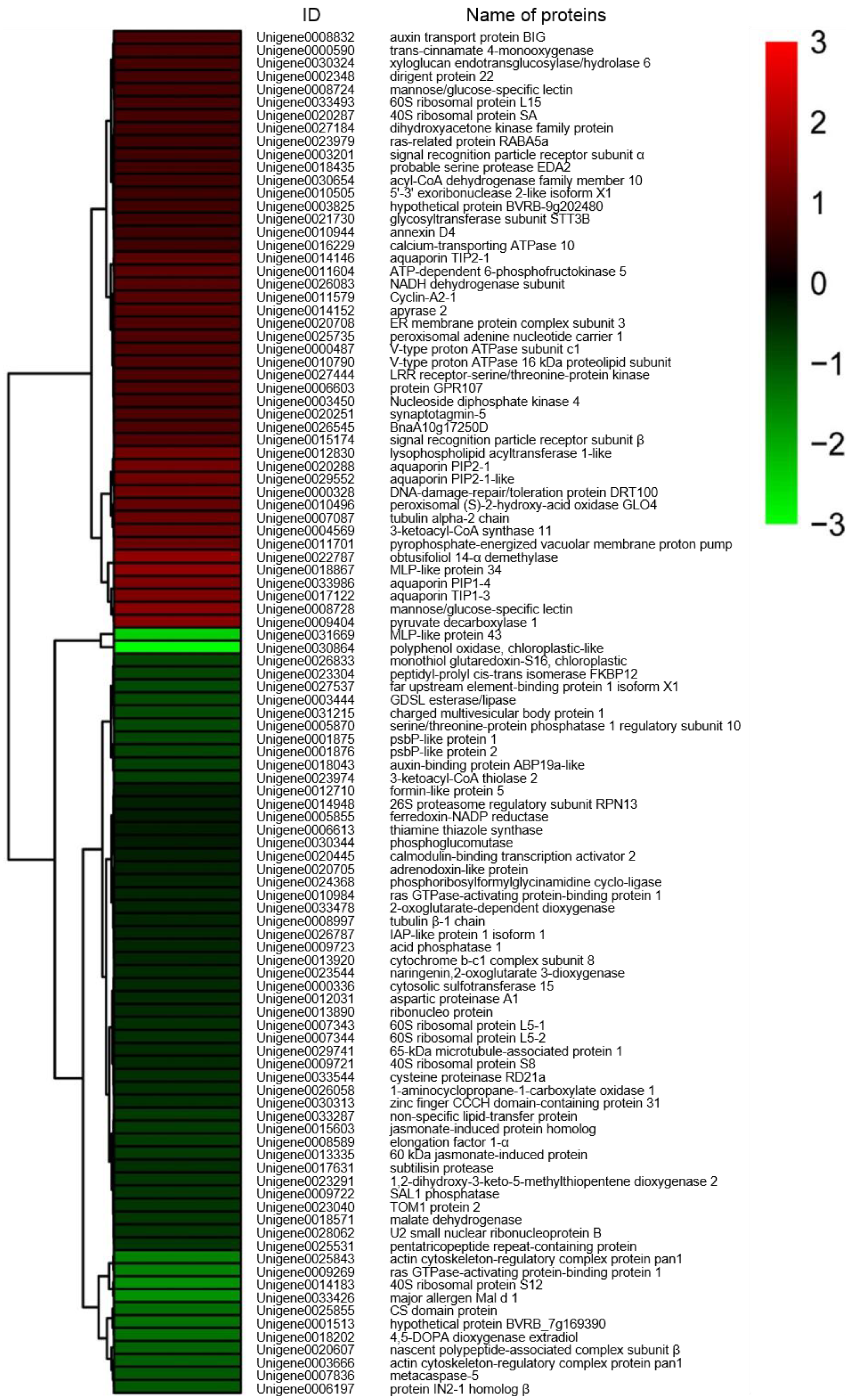
2.3. Cluster Analysis of DEPs
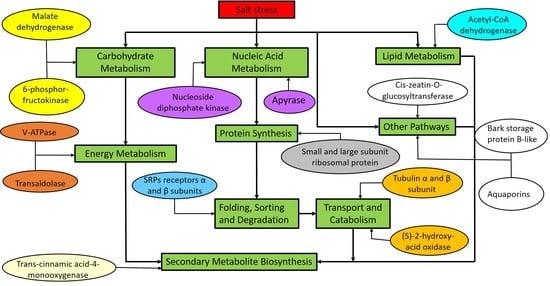
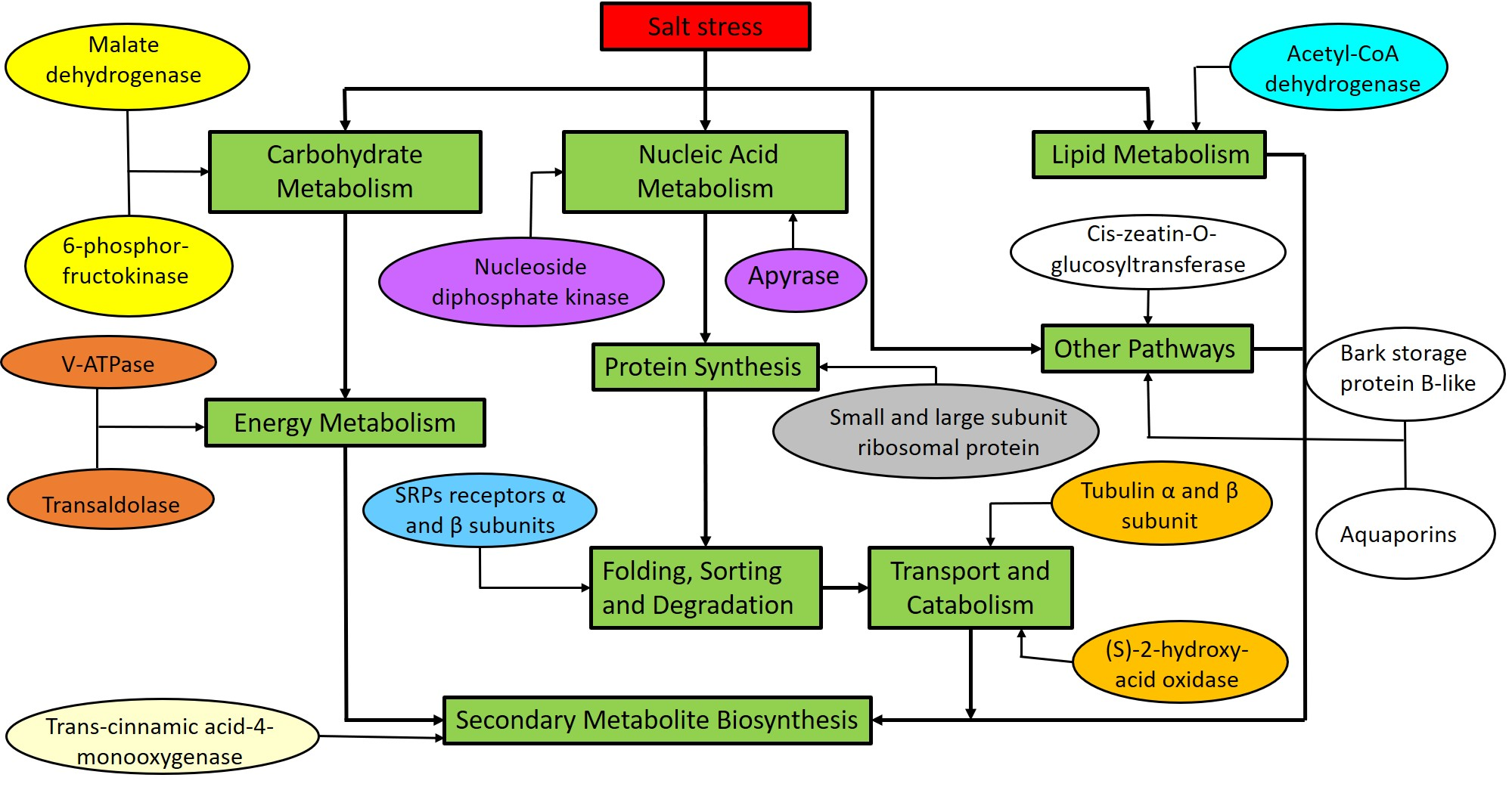
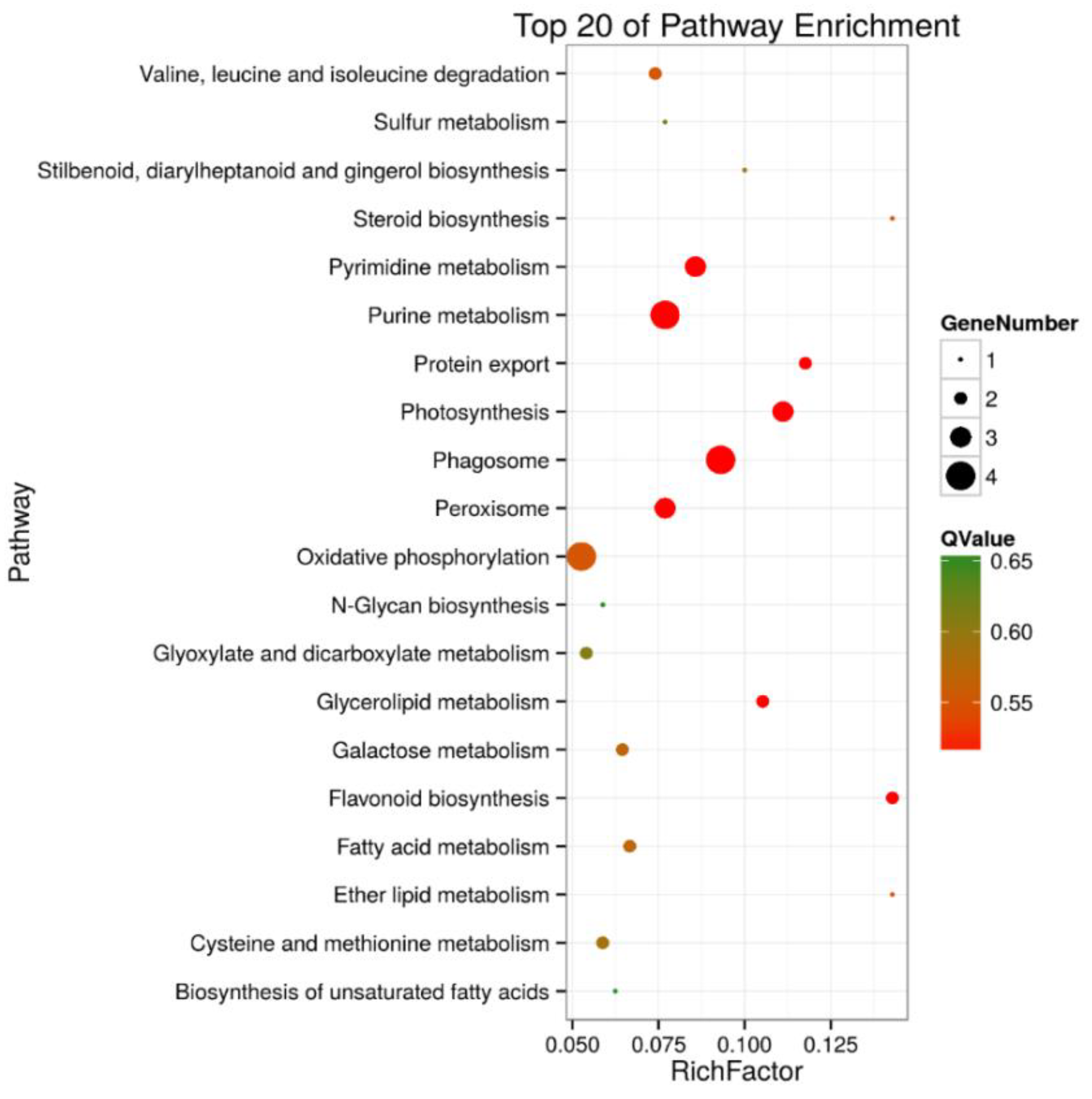
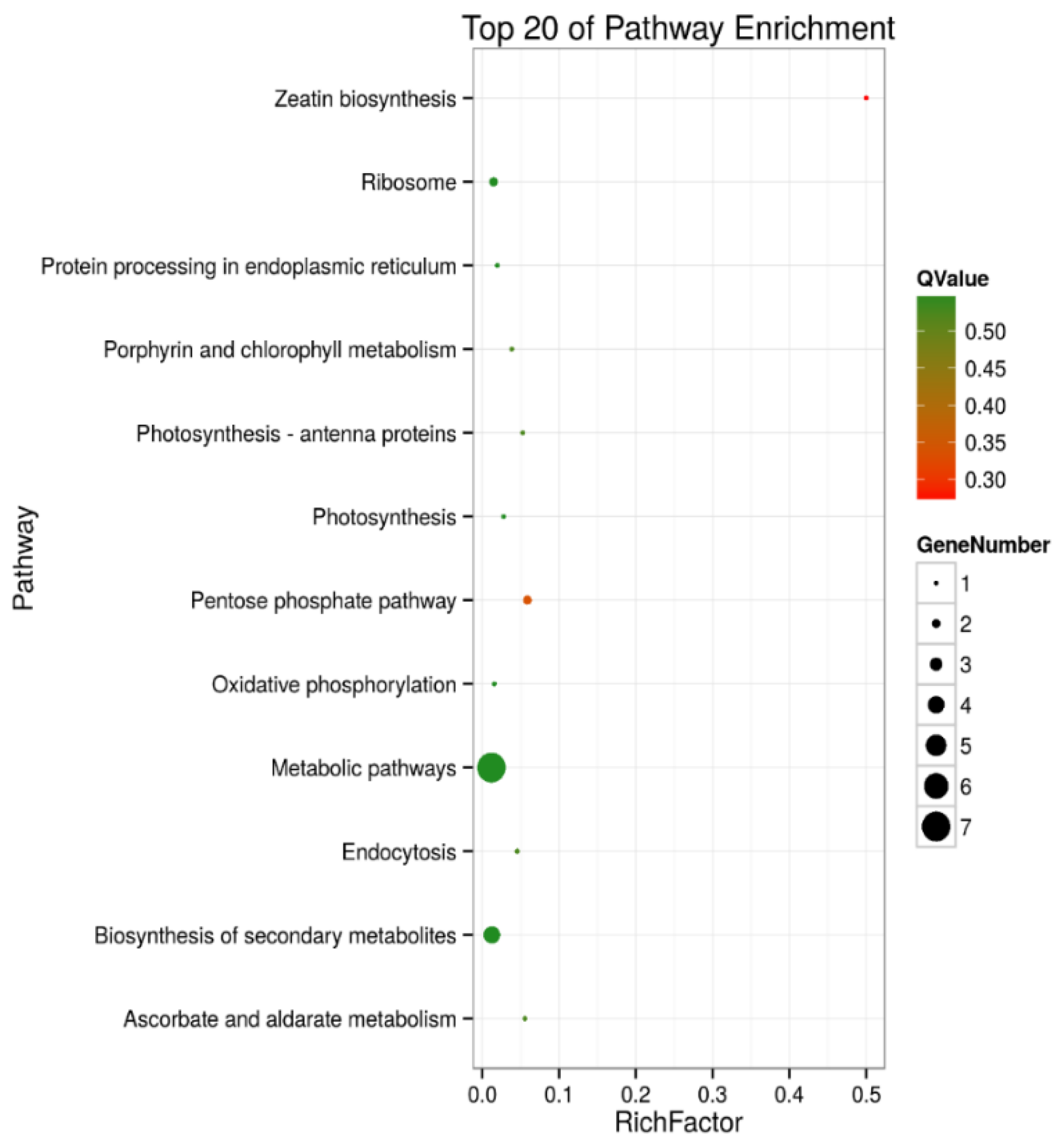
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DEPs
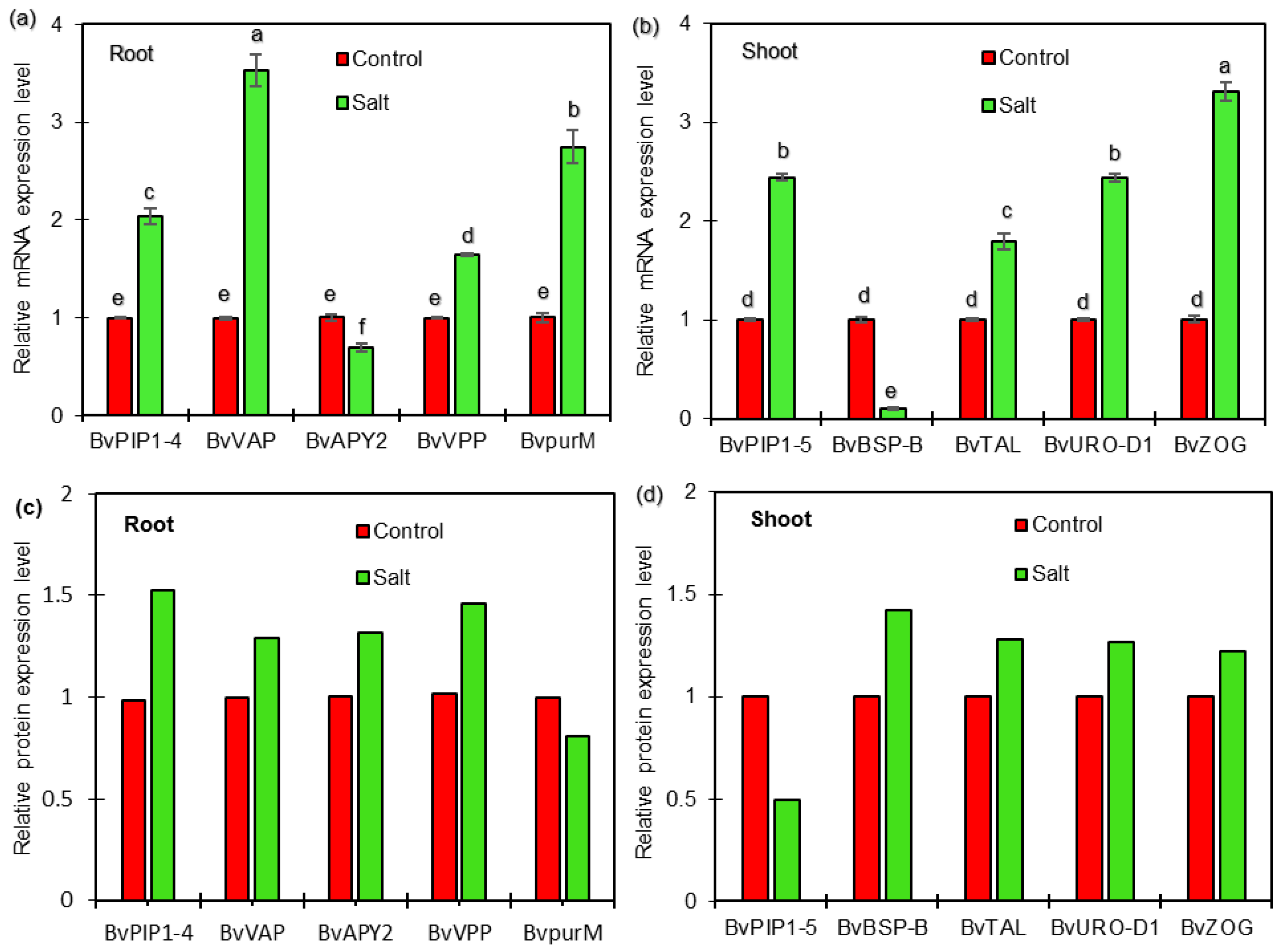
2.5. qRT-PCR Analysis of Candidate Genes Encoding DEPs
3. Discussion
3.1. Proteins Involved in Carbohydrate and Energy Metabolism
3.2. Liqid Metabolism and Secondary Metabolite Biosynthesis Retated Protein
3.3. Transcription and Protein Synthesis Related Protein
3.4. Protein Folding, Sorting and Degradation
3.5. Protein Involved in Transport
4. Materials and Methods
4.1. Plant Materials, Growth Conditions and Treatments
4.2. Protein Preparation
4.3. iTRAQ Labeling and SCX Fractionation
4.4. LC–ESI–MS/MS Analysis
4.5. Protein Identification
4.6. Bioinformatics Analysis
4.7. Quantitative Reverse Transcription PCR (qRT-PCR)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-DE | Two-dimensional gel electrophoresis |
| ACAD10 | Acyl-CoA dehydrogenase family member 10 |
| AQPs | Aquaporins |
| BSP-B | Bark storage protein B-like |
| C4H | Trans-cinnamate 4-monooxygenase |
| CK | Cytokinin |
| COG | Cluster of Orthologous Groups |
| CYP51 | Obtusifoliol 14-α demethylase |
| DEPs | Differentially expressed proteins |
| ERAD | ER-associated degradation |
| F3H | Naringenin,2-oxoglutarate 3-dioxygenase |
| GO | Gene Ontology |
| iTRAQ | isobaric Tags for Relative and Absolute Quantitation |
| KAT2 | 3-ketoacyl-CoA thiolase 2 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MDH | Malate dehydrogenase |
| MS | Mass spectrometry |
| OST | N-oligosaccharyltransferase |
| OXPHOS | Oxidative phosphorylation |
| PFK5 | ATP-dependent 6-phosphofructokinase 5 |
| PIPs | Plasma membrane intrinsic proteins |
| PPP | Pentose phosphate pathway |
| PSII | Photosystem II |
| purM | Phosphoribosylformylglycinamidine cyclo-ligase |
| qRT-PCR | quantitative Reverse Transcription-Polymerase Chain Reaction |
| RER | Rough endoplasmic reticulum |
| SCX | Strong cation exchange |
| SRP | Signal recognition particle |
| SRPR | Signal recognition particle receptor |
| TAG | Triacylglycerol |
| TAL | Transaldolases |
| TCA | Tricarboxylic acid cycle |
| TIPs | Tonoplast intrinsic proteins |
| URO-D1 | Uroporphyrinogen decarboxylase 1 |
| VAP | V-type proton ATPase subunit c1 |
| V-H+-ATPase | V-type proton ATPase |
| V-H+-PPase | Pyrophosphate-energized vacuolar membrane proton pump |
| VP | Pyrophosphate-energized vacuolar membrane proton pump |
| ZOG | Zeatin O-glucoside |
References
- Yang, Y.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt tress responses. New Phytol. 2017, 217, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotox. Environ. Safe. 2005, 60, 324–349. [Google Scholar] [CrossRef] [PubMed]
- Yazici, I.; Türkan, I.; Sekmen, A.H.; Demiral, T. Salinity tolerance of purslane (Portulaca oleracea L.) is achieved by enhanced antioxidative system, lower level of lipid peroxidation and proline accumulation. Environ. Exp. Bot. 2007, 61, 49–57. [Google Scholar] [CrossRef]
- Muchate, N.S.; Nikalje, G.C.; Rajurkar, N.S.; Suprasanna, P.; Nikam, T.D. Plant salt stress: Adaptive responses, tolerance mechanism and bioengineering for salt tolerance. Bot. Rev. 2016, 82, 371–406. [Google Scholar] [CrossRef]
- Gygi, S.P.; Corthals, G.L.; Zhang, Y.; Rochon, Y.; Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Pro. Natl. Acad. Sci. USA 2000, 97, 9390–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínezesteso, M.J.; Casadovela, J.; Sellésmarchart, S.; Pedreño, M.A.; Brumartínez, R. Differential plant proteome analysis by isobaric tags for relative and absolute quantitation (iTRAQ). Methods Mol. Biol. 2014, 1072, 155–169. [Google Scholar]
- Nogueira, F.C.; Palmisano, G.; Schwämmle, V.; Campos, F.A.; Larsen, M.R.; Domont, G.B.; Roepstorff, P. Performance of isobaric and isotopic labeling in quantitative plant proteomics. J. Proteome Res. 2012, 11, 3046–3052. [Google Scholar] [CrossRef] [PubMed]
- Casadovela, J.; Martínezesteso, M.J.; Rodriguez, E.; Borrás, E.; Elortza, F.; Brumartínez, R. Itraq-based quantitative analysis of protein mixtures with large fold change and dynamic range. Proteomics 2010, 10, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Reidegeld, K.A.; Meyer, H.E.; Warscheid, B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Bârsan, S.C.; Ivan, A.M.; Luca, L.C.; Luca, E. Sugar beet (Beta vulgaris L.) yields and potential for bioethanol production under irrigation regime. Not. Bot. Horti. Agrobo. 2015, 43, 455–461. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Saleem, A.M. Sugar beet potential to beat sugarcane as a sugar crop in Pakistan. Am-Euras. J. Agric. Environ. Sci. 2015, 15, 36–44. [Google Scholar]
- Fishman, M.L.; Chau, H.K.; Cooke, P.H.; Yadav, M.P.; Hotchkiss, A.T. Physico-chemical characterization of alkaline soluble polysaccharides from sugar beet pulp. Food Hydrocolloids 2009, 23, 1554–1562. [Google Scholar] [CrossRef]
- Rodríguez, L.A.; Toro, M.E.; Vazquez, F.; Correa-Daneri, M.L.; Gouiric, S.C.; Vallejo, M.D. Bioethanol production from grape and sugar beet pomaces by solid-state fermentation. Int. J. Hydrogen Energ. 2010, 35, 5914–5917. [Google Scholar] [CrossRef]
- Dohm, J.C.; Minoche, A.E.; Holtgräwe, D.; Salvador, C.G.; Zakrzewski, F.; Tafer, H.; Rupp, O.; Sörensen, T.R.; Stracke, R.; Reinhardt, R.; et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 2014, 505, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Ober, E.S.; Rajabi, A.; Stevanato, P. Abiotic stress in sugar beet. Sugar Tech. 2010, 12, 294–298. [Google Scholar] [CrossRef]
- Jamil, M.; Bae, L.D.; Yong, J.K.; Ashraf, M. Effect of salt (NaCl) stress on germination and early seedling growth of four vegetables species. J. Cent. Eur. Agric. 2006, 7, 273–282. [Google Scholar]
- Yang, L.; Ma, C.; Chen, S.; Li, H. Salt stress induced proteome and transcriptome changes in sugar beet monosomic addition line M14. J. Plant Physiol. 2012, 19, 839–850. [Google Scholar] [CrossRef]
- Yu, B.; Li, J.; Koh, J.; Dufresne, C.; Yang, N.; Qi, S.S.; Zhang, Y.X.; Ma, C.Q.; Duong, B.V.; Chen, S.X.; et al. Quantitative proteomics and phosphoproteomics of sugar beet monosomic addition line M14 in response to salt stress. J. Proteomics 2016, 143, 286–297. [Google Scholar] [CrossRef]
- Wakeel, A.; Asif, A.R.; Pitann, B.; Schubert, S. Proteome analysis of sugar beet (Beta vulgaris L.) elucidates constitutive adaptation during the first phase of salt stress. J. Plant Physiol. 2011, 168, 519–526. [Google Scholar] [CrossRef]
- Pi, Z.; Stevanato, P.; Sun, F.; Yang, Y.; Sun, X.; Zhao, H.; Geng, G.; Yu, L. Proteomic changes induced by potassium deficiency and potassium substitution by sodium in sugar beet. J. Plant Res. 2016, 129, 527–538. [Google Scholar] [CrossRef]
- Li, H.Y.; Pan, Y.; Zhang, Y.X.; Wu, C.; Ma, C.; Yu, B.; Zhu, N.; Koh, J.; Chen, S. Salt stress response of membrane proteome of sugar beet monosomic addition line M14. J. Proteomics 2015, 127, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Q.; Feng, R.J.; Zhang, J.J. Evaluation of salinity tolerance in seedlings of sugar beet (Beta vulgaris L.) cultivars using proline, soluble sugars and cation accumulation criteria. Acta Physiol. Plant. 2013, 35, 2665–2674. [Google Scholar] [CrossRef]
- Wu, G.Q.; Wang, C.M.; Su, Y.Y.; Zhang, J.J.; Feng, R.J.; Liang, N. Assessment of drought tolerance in seedlings of sugar beet (Beta vulgaris L.) cultivars using inorganic and organic solutes accumulation criteria. Soil Sci. Plant Nutr. 2014, 60, 565–576. [Google Scholar] [CrossRef]
- Wu, G.Q.; Feng, R.J.; Liang, N.; Yuan, H.J.; Sun, W.B. Sodium chloride stimulates growth and alleviates sorbitol-induced osmotic stress in sugar beet seedlings. Plant Growth Regul. 2015, 75, 307–316. [Google Scholar] [CrossRef]
- Mustroph, A.; Stock, J.; Hess, N.; Aldous, S.; Dreilich, A.; Grimm, B. Characterization of the phosphofructokinase gene family in rice and its expression under oxygen deficiency stress. Front. Plant Sci. 2013, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, A.; Sonnewald, U.; Biemelt, S. Characterisation of the ATP-dependent phosphofructokinase gene family from Arabidopsis thaliana. FEBS Lett. 2007, 581, 2401–2410. [Google Scholar] [CrossRef]
- Liu, C.; Chang, T.; Hsu, Y.; Wang, A.Z.; Yen, H.; Wu, Y.; Wang, C.; Lai, C. Comparative proteomic analysis of early salt stress responsive proteins in roots and leaves of rice. Proteomics 2014, 14, 1759. [Google Scholar] [CrossRef]
- Tesfaye, M.; Samac, D.A. Overexpression of malate dehydrogenase in transgenic alfalfa enhances organic acid synthesis and confers tolerance to aluminum. Plant Physiol. 2001, 127, 1836–1844. [Google Scholar] [CrossRef]
- Wu, L.; Zhao, F.; Fang, W.; Xie, D.; Hou, J.; Yang, X. Identification of early salt stress responsive proteins in seedling roots of upland cotton (Gossypium hirsutum L.) employing iTRAQ-based proteomic technique. Front. Plant Sci. 2015, 6, 732. [Google Scholar]
- Du, C.X.; Fan, H.F.; Guo, S.R.; Tezuka, T.; Li, J. Proteomic analysis of cucumber seedling roots subjected to salt stress. Phytochemistry 2010, 71, 1450–1459. [Google Scholar] [CrossRef]
- Dooki, A.D.; Mayerposner, F.J.; Askari, H.; Zaiee, A.A.; Salekdeh, G.H. Proteomic responses of rice young panicles to salinity. Proteomics 2006, 6, 6498–6507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Chen, J.; Zhang, J.; Shi, S.; Zhou, Y.; Lu, L.; Wang, P.; Jiang, Z.; Yang, J.; Zhang, S.; et al. Physiological and proteomic analyses of leaves from the halophyte Tangut nitraria reveals diverse response pathways critical for high salinity tolerance. Front. Plant Sci. 2015, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Tester, M.; Davenport, R. Na+ tolerance and Na+ transport in higher plants. Ann. Bot. 2003, 91, 503–527. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, S.; Maslowski, P. Effect of inorganic pyrophosphate on respiration and oxidative phosphorylation in higher plants. Phytochemistry 1981, 20, 2611–2615. [Google Scholar] [CrossRef]
- Li, M.; Zhang, K.; Long, R.; Sun, Y.; Kang, J.; Zhang, T.; Cao, S.H. iTRAQ-based comparative proteomic analysis reveals tissue-specific and novel early-stage molecular mechanisms of salt stress response in Carex Rigescens. Environ. Exp. Bot. 2017, 143, 99–114. [Google Scholar] [CrossRef]
- Wang, J.; Yao, L.; Li, B.; Meng, Y.; Ma, X.; Lai, Y.; Si, E.; Ren, P.; Yang, K.; Shang, X.; et al. Comparative proteomic analysis of cultured suspension cells of the halophyte Halogeton glomeratus by iTRAQ provides insights into response mechanisms to salt stress. Front. Plant Sci. 2016, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tang, S.; Peng, X.; Yan, X.; Zeng, X.; Li, J.; Li, X.; Wu, G. Elucidation of cross-talk and specificity of early response mechanisms to salt and PEG-simulated drought stresses in Brassica napus using comparative proteomic analysis. PLoS ONE 2015, 10, e0138974. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Kerkeb, L.; Donaire, J.P.; Rodríguez-Rosales, M.P. Plasma membrane H-ATPase activity is involved in adaptation of tomato calli to NaCl. Physiol. Plant. 2001, 111, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, B.; Harris, N.S.; Deyholos, M.K. Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. J. Exp. Bot. 2007, 58, 3591–3607. [Google Scholar] [CrossRef] [PubMed]
- Belghith, I.; Senkler, J.; Hildebrandt, T.; Abdelly, C.; Braun, H.P.; Debez, A. Comparative analysis of salt-induced changes in the root proteome of two accessions of the halophyte Cakile maritima. Plant Physiol. Biochem. 2018, 130, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, C.; Gu, J.; Duan, W.; Zhao, M.; Ma, C. Overexpression of VP, a vacuolar H+-pyrophosphatase gene in wheat (Triticum aestivum L.), improves tobacco plant growth under Pi and N deprivation, high salinity, and drought. J. Exp. Bot. 2014, 65, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fang, Y.; Liang, Z.; Huang, L. Enhanced expression of vacuolar H+-ATPase subunit E in the roots is associated with the adaptation of Broussonetia papyriferato salt stress. PLoS ONE 2012, 7, e48183. [Google Scholar]
- Xiong, J.; Sun, Y.; Yang, Q.; Tian, H.; Zhang, H.; Liu, Y. Proteomic analysis of early salt stress responsive proteins in alfalfa roots and shoots. Proteome Sci. 2017, 15, 19. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Weisz, D.A.; Vidavsky, I.; Gross, M.L.; Pakrasi, H.B. MS-based cross-linking analysis reveals the location of the PsbQ protein in cyanobacterial photosystem II. Pro. Natl. Acad. Sci. USA 2014, 111, 4638–4643. [Google Scholar] [CrossRef]
- Germain, V.; Rylott, E.L.; Larson, T.R.; Sherson, S.M.; Bechtold, N.; Carde, J.P.; Bryce, J.H.; Graham, I.A.; Smith, S.M. Requirement for 3-ketoacyl-coa thiolase-2 in peroxisome development, fatty acid β-oxidation and breakdown of triacylglycerol in lipid bodies of Arabidopsis seedlings. Plant J. 2001, 28, 1–12. [Google Scholar] [CrossRef]
- Carrie, C.; Murcha, M.W.; Millar, A.H.; Millar, H.; Smith, S.M.; Whelan, J. Nine 3-ketoacyl-CoA thiolases (KATs) and acetoacetyl-CoA thiolases (ACATs) encoded by five genes in Arabidopsis thaliana are targeted either to peroxisomes or cytosol but not to mitochondria. Plant Mol. Biol. 2007, 63, 97. [Google Scholar] [CrossRef]
- Ghisla, S.; Thorpe, C. Acyl-coa dehydrogenases. a mechanistic overview. Eur. J. Biochem. 2004, 271, 494–508. [Google Scholar] [CrossRef]
- ƠBrien, M.; Chantha, S.C.; Rahier, A.; Matton, D.P. Lipid signaling in plants. cloning and expression analysis of the obtusifoliol 14 alpha-demethylase from Solanum chacoense bitt. a pollination- and fertilization-induced gene with both obtusifoliol and lanosterol demethylase activity. Plant Physiol. 2005, 139, 734–749. [Google Scholar] [CrossRef]
- Zhu, S.; Gong, Y.; Hang, Y.; Liu, H.; Wang, H. Transcriptome analysis of Dunaliella viridis. Hereditas 2015, 37, 828–836. [Google Scholar] [PubMed]
- Kahn, R.A.; Bak, S.; Olsen, C.E.; Svendsen, I.; Moller, B.L. Isolation and reconstitution of the heme-thiolate protein obtusifoliol 14 alpha-demethylase from Sorghum bicolor (L.). Moench. J. Biol. Chem. 1996, 271, 32944–32950. [Google Scholar] [CrossRef] [PubMed]
- Cabane, M.; Afif, D.; Hawkins, S. Lignins and abiotic stresses. In Lignins: Biosynthesis Biodegradation Bioengineering; Jouann, L., Lapierre, C., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 61, pp. 219–262. [Google Scholar]
- Wu, Y.; Guo, J.; Zhou, Q.; Xin, Y.; Wang, G.; Xu, L. De novo transcriptome analysis revealed genes involved in flavonoid biosynthesis, transport and regulation in Ginkgo biloba. Ind. Crop. Prod. 2018, 124, 226–235. [Google Scholar] [CrossRef]
- Shi, L.; Chen, X.; Chen, W.; Zheng, Y.; Yang, Z. Comparative transcriptomic analysis of white and red Chinese bayberry (Myrica rubra) fruits reveals flavonoid biosynthesis regulation. Sci. Hortic. 2018, 235, 9–20. [Google Scholar] [CrossRef]
- Martin, R.C.; Smets, R.; Mok, M.C. Development of transgenic tobacco tarboring a Zeatin O-Glucosyltransferase gene from Phaseolus. Vitro Cell. Dev-Pl. 2001, 37, 354–360. [Google Scholar] [CrossRef]
- Martin, R.C.; Mok, M.C. Isolation of a cytokinin gene, ZOG1, encoding zeatin O-glucosyltransferase from Phaseolus lunatus. Pro. Natl. Acad. Sci. USA 1999, 96, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Shang, X.; Xie, R.; Tian, H.; Wang, Q.; Guo, F. Putative zeatin O-glucosyltransferase osczogl regulates roots and shoots development and formation of agronomic traits in rice. Chin. Bull. Bot. 2016, 58, 627–641. [Google Scholar]
- Havlova, M.; Dobrev, P.I.; Motyka, V.; Storchova, H.; Libus, J.; Dobra, J.; Malbeck, J.; Gaudinova, A.; Vankova, R. The role of cytokinins in responses to water-deficit in tobacco plants over-expressing trans-zeatin O-glucosyltransferase under 35S or SAG12 promoters. Plant Cell Environ. 2008, 31, 341–353. [Google Scholar] [CrossRef]
- Gong, W.; Xu, F.; Sun, J.; Peng, Z.; He, S.; Pan, Z.; Du, X. iTRAQ-based comparative proteomic analysis of seedling leaves of two upland cotton genotypes differing in salt tolerance. Front. Plant Sci. 2017, 8, 2113. [Google Scholar] [CrossRef]
- Lv, D.W.; Zhu, G.R.; Zhu, D.; Bian, Y.W.; Liang, X.N.; Cheng, Z.W.; Deng, X.; Yan, Y.M. Proteomic and phosphoproteomic analysis reveals the response and defense mechanism in leaves of diploid wheat T. monococcum under salt stress and recovery. J. Proteomics 2016, 143, 93–105. [Google Scholar] [CrossRef]
- Delille, J.; Peterson, E.C.; Johnson, T.; Moore, M.; Kight, A.; Henry, R. A novel precursor recognition element facilitates posttranslational binding to the signal recognition particle in chloroplasts. Pro. Natl. Acad. Sci. USA 2000, 97, 1926. [Google Scholar] [CrossRef] [PubMed]
- Dumax-Vorzet, A.; Roboti, P.; High, S. Ost4 is a subunit of the mammalian oligosaccharyltransferase required for efficient n-glycosylation. J. Cell Sci. 2013, 126, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, C.; Sun, Y.; Wang, N.; Li, X.; Dong, Y.; Yao, N.; Liu, X.; Chen, X.; Wang, Z.; et al. Overexpression of vacuolar proton pump ATPase (V-H+-ATPase) subunits B, C and H confers tolerance to salt and saline-alkali stresses in transgenic alfalfa (Medicago sativa L.). J. Integr. Agr. 2016, 15, 2279–2289. [Google Scholar] [CrossRef]
- Dabbous, A.; Saad, R.B.; Brini, F.; Farhat-Khemekhem, A.; Zorring, W.; Abdely, C.; Hamed, K.B. Over-expression of a subunit E1 of a vacuolar H+-ATPase gene (Lm VHA-E1) cloned from the halophyte Lobularia maritima improves the tolerance of Arabidopsis thaliana to salt and osmotic stresses. Environ. Exp. Bot. 2017, 137, 128–141. [Google Scholar] [CrossRef]
- Damaris, R.N.; Li, M.; Liu, Y.; Chen, X.; Murage, H.; Yang, P. A proteomic analysis of salt stress response in seedlings of two African rice cultivars. Biochimica et Biophysica Acta 2016, 1864, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Westermann, S.; Weber, K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Kjellbom, P.; Larsson, C.; Johansson, I.; Karlsson, M.; Johanson, U. Aquaporins and water homeostasis in plants. Trend Plant Sci. 1999, 4, 308–314. [Google Scholar] [CrossRef]
- Gaspar, M.; Bousser, A.; Sissoëff, I.; Roche, O.; Hoarau, J.; Mahé, A. Cloning and characterization of ZmPIP1-5b, an aquaporin transporting water and urea. Plant Sci. 2003, 165, 21–31. [Google Scholar] [CrossRef]
- Danielson, J.A.; Johanson, U. Unexpected complexity of the aquaporin gene family in the moss Physcomitrella patens. BMC Plant Biol. 2008, 8, 45. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, C.; Wang, Y.; Wang, Y.; Yang, C.; Lu, M.; Wang, C. Tamarix hispida aquaporin ThPIP2;5 confers salt and osmotic stress tolerance to transgenic Tmarix and Arabidopsis. Environ. Exp. Bot. 2018, 152, 158–166. [Google Scholar] [CrossRef]
- Katsuhara, M.; Koshio, K.; Shibasaka, M.; Hayashi, Y.; Hayakawa, T.; Kasamo, K. Over-expression of a barley aquaporin increased the shoot/root ratio and raised salt sensitivity in transgenic rice plants. Plant Cell Physiol. 2003, 44, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ballesta Mdel, C.; Bastias, E.; Zhu, C.; Schaffner, A.R. Boric acid and salinity effects on maize roots. Response of aquaporins ZmPIP1 and ZmPIP2, and plasma membrane H+-ATPase, in relation to water and nutrient uptake. Physiol. Plant 2008, 132, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of proteindye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.-Q.; Wang, J.-L.; Feng, R.-J.; Li, S.-J.; Wang, C.-M. iTRAQ-Based Comparative Proteomic Analysis Provides Insights into Molecular Mechanisms of Salt Tolerance in Sugar Beet (Beta vulgaris L.). Int. J. Mol. Sci. 2018, 19, 3866. https://doi.org/10.3390/ijms19123866
Wu G-Q, Wang J-L, Feng R-J, Li S-J, Wang C-M. iTRAQ-Based Comparative Proteomic Analysis Provides Insights into Molecular Mechanisms of Salt Tolerance in Sugar Beet (Beta vulgaris L.). International Journal of Molecular Sciences. 2018; 19(12):3866. https://doi.org/10.3390/ijms19123866
Chicago/Turabian StyleWu, Guo-Qiang, Jin-Long Wang, Rui-Jun Feng, Shan-Jia Li, and Chun-Mei Wang. 2018. "iTRAQ-Based Comparative Proteomic Analysis Provides Insights into Molecular Mechanisms of Salt Tolerance in Sugar Beet (Beta vulgaris L.)" International Journal of Molecular Sciences 19, no. 12: 3866. https://doi.org/10.3390/ijms19123866