Functionalized Keratin as Nanotechnology-Based Drug Delivery System for the Pharmacological Treatment of Osteosarcoma

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
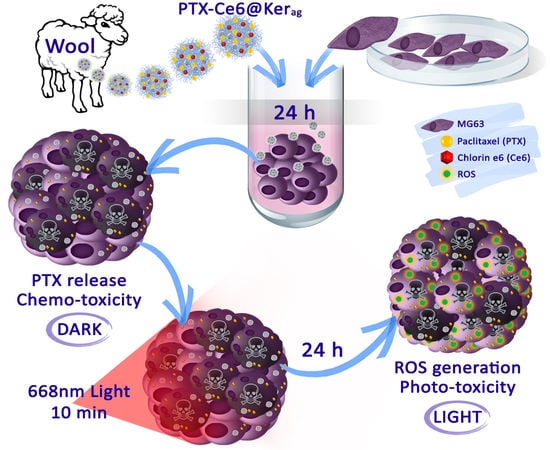
:
1. Introduction
2. Results and Discussion
2.1. Physicochemical Characterization of Keratin Nanoformulations
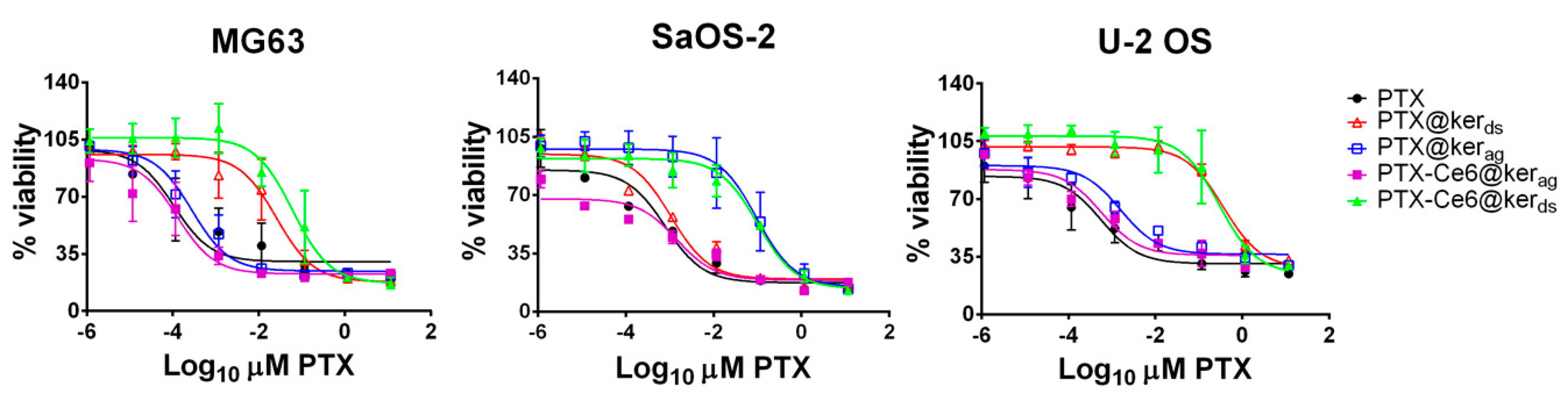
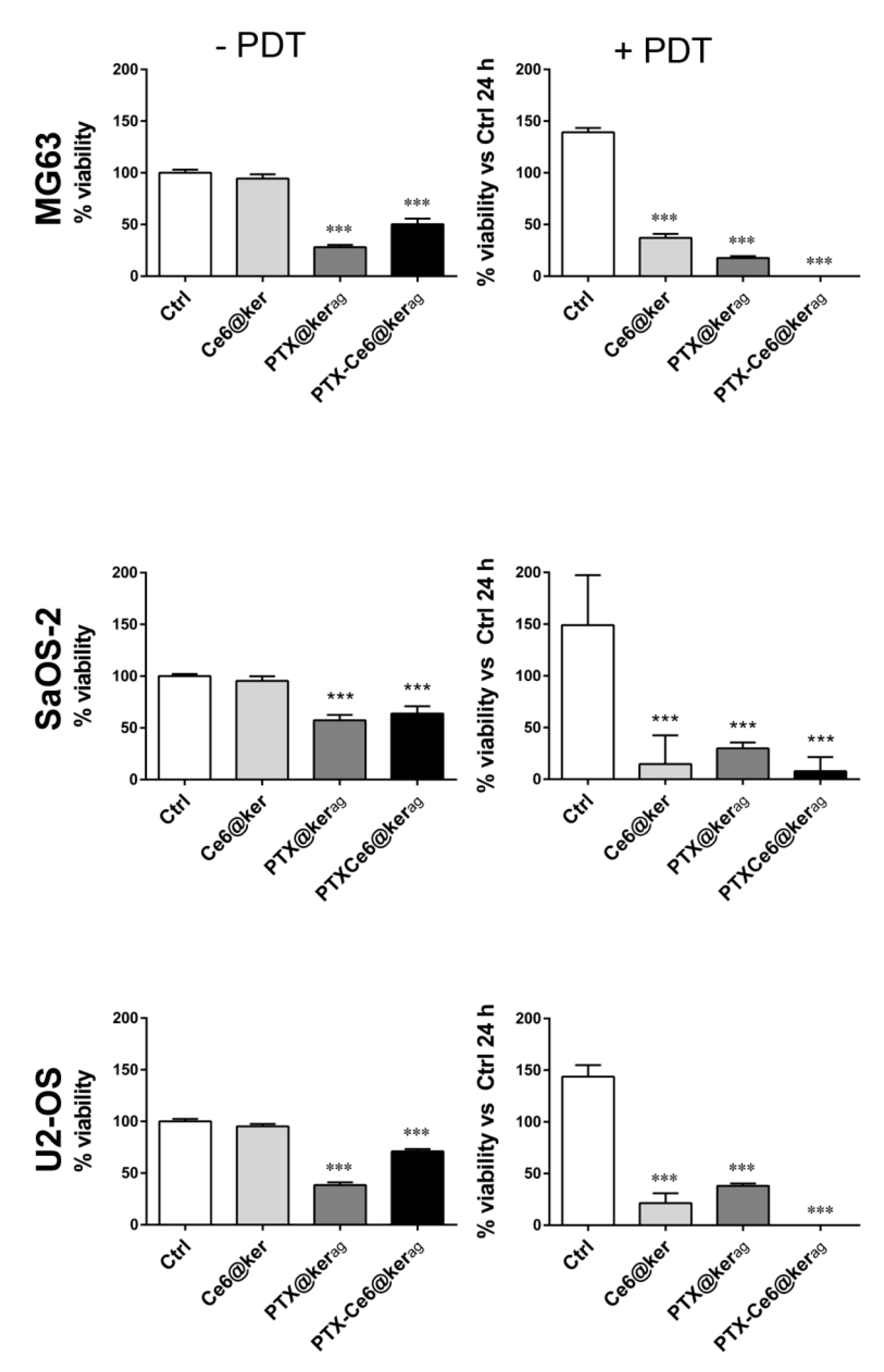
2.2. PTX Vehiculated through Keratin Nanoformulations Affect OS Cells’ Viability in 2D System
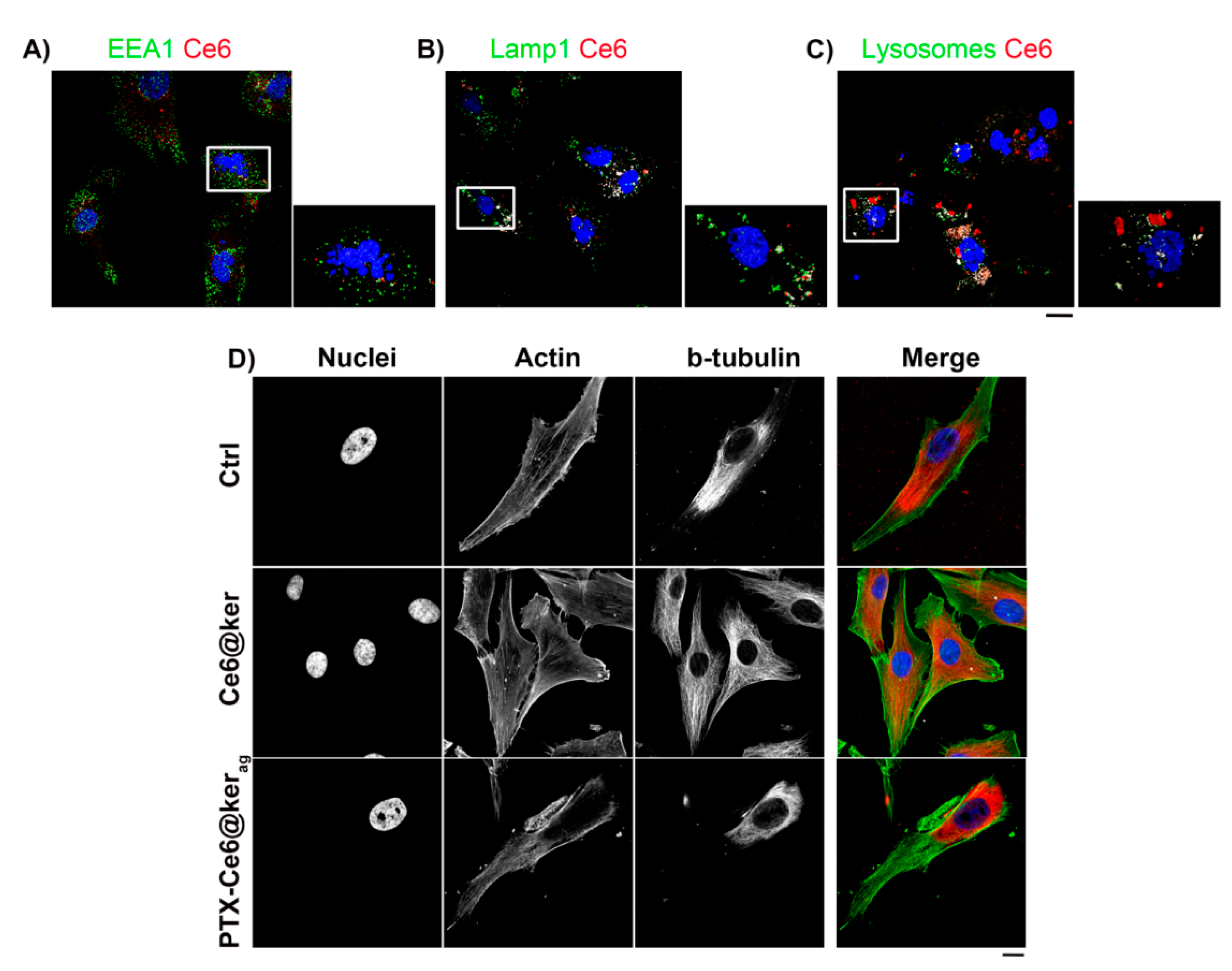
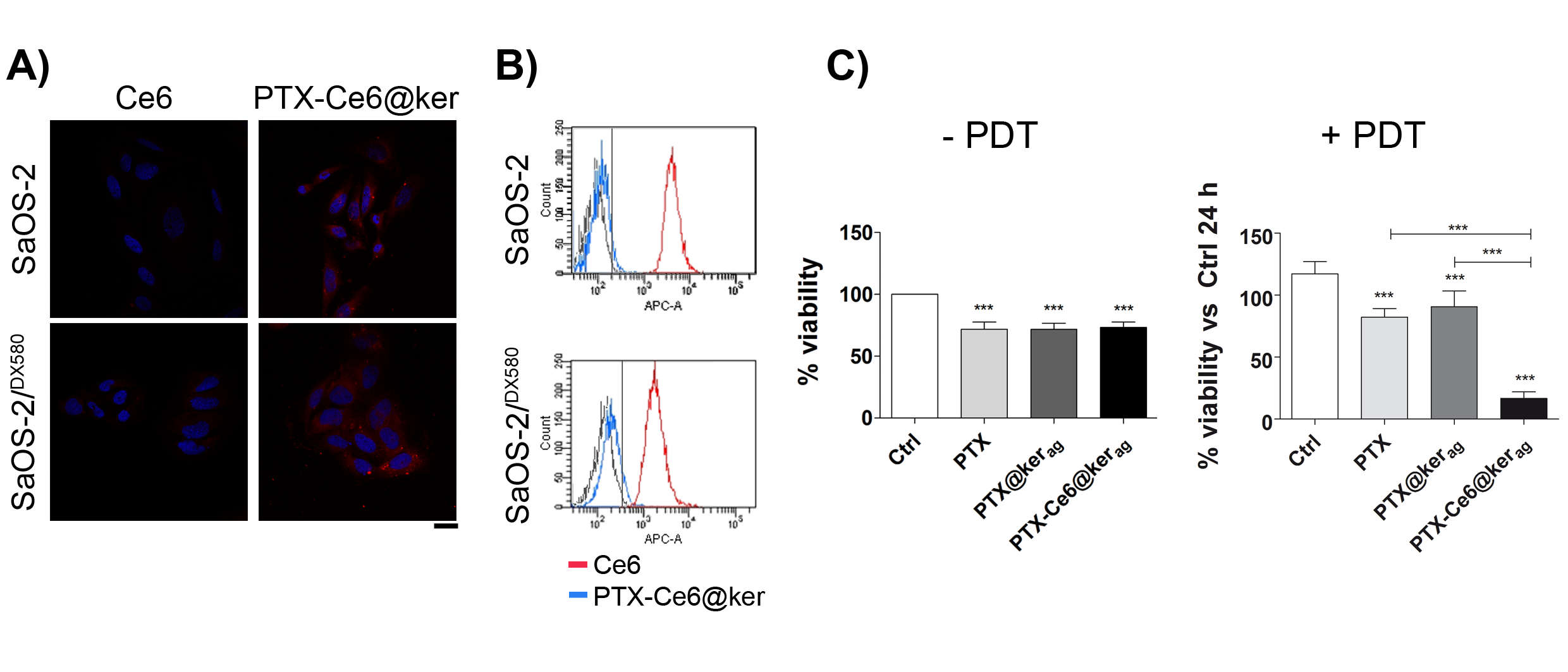
2.3. The Localization of Keratin Nanoformulations in OS Tumor Cell Lines
2.4. Impact of PTX-Ce6@Kerag on OS Cells’ Viability in 2D System
2.5. Impact of PTX-Ce6@Kerag on Chemoresistant OS Cells’ Viability in 2D System
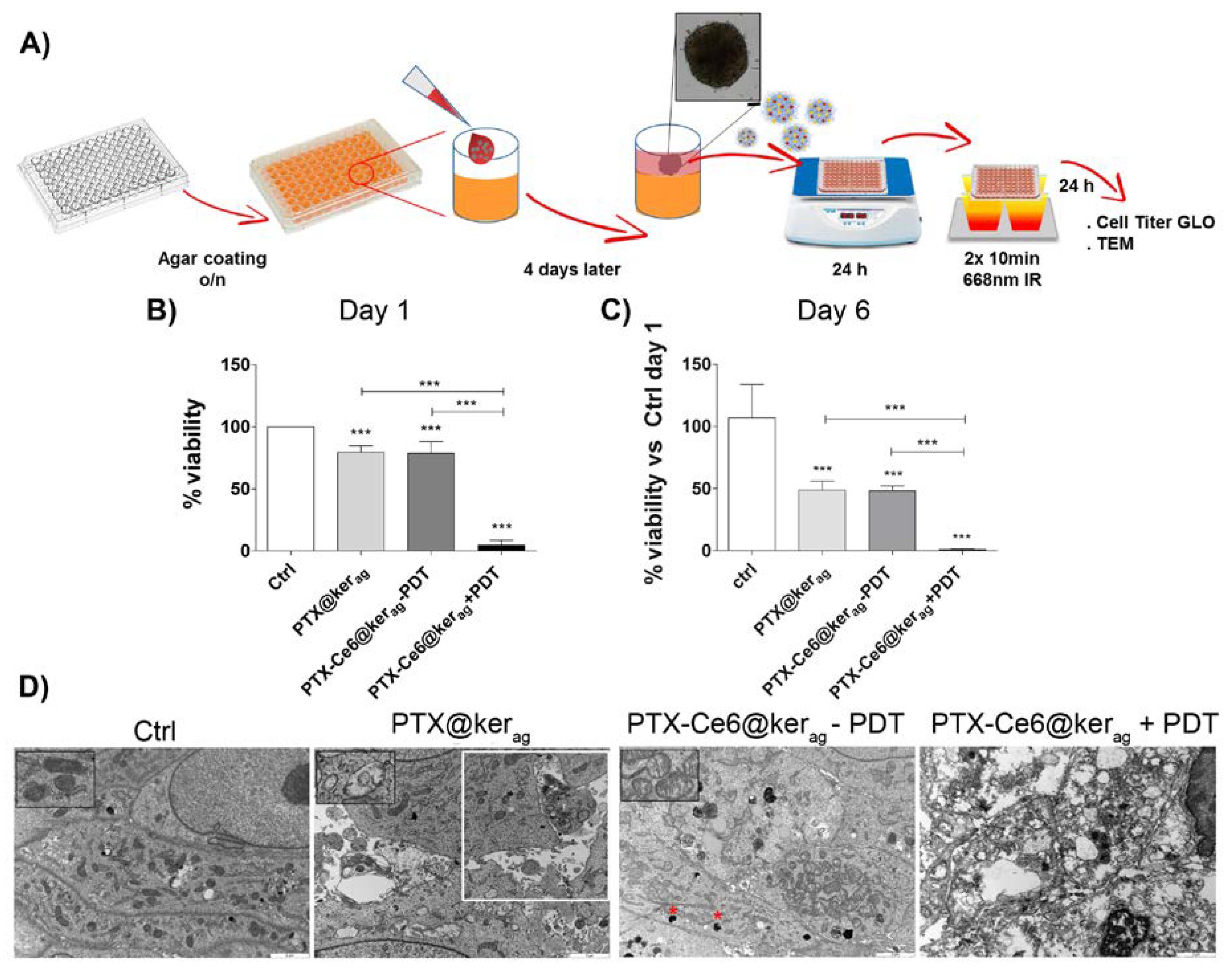
2.6. In Vitro Toxicity of Keratin Nanoformulations in 3D System
3. Experimental Section
3.1. Materials
3.2. Keratin Nanoformulations Synthesis
3.3. Keratin Nanoformulations Characterization
3.4. PTX Release
3.5. In Vitro Toxicity in 2D
3.6. Cellular localization analyses
3.7. In Vitro Toxicity in 3D
3.8. Transmission Electron Microscopy (TEM)
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindsey, B.A.; Markel, J.E.; Kleinerman, E.S. Osteosarcoma Overview. Rheumatol. Ther. 2017, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Biazzo, A.; De Paolis, M. Multidisciplinary approach to osteosarcoma. Acta Orthop. Belg. 2016, 82, 690. [Google Scholar] [PubMed]
- Wang, X.; Zheng, H.; Shou, T.; Tang, C.; Miao, K.; Wang, P.J. Effectiveness of multi-drug regimen chemotherapy treatment in osteosarcoma patients: A network meta-analysis of randomized controlled trials. Orthop. Surg. Res. 2017, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Anninga, J.K.; Gelderblom, H.; Fiocco, M.; Kroep, J.R.; Taminiau, A.H.M.; Hogendoorn, P.C.W.; Egeler, R.M. Chemotherapeutic adjuvant treatment for osteosarcoma: Where do we stand? Eur. J. Cancer 2011, 47, 2431. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.; Utset-Ward, T.J.; Reed, D.R.; Lynch, C.C. Re-calculating! Navigating through the osteosarcoma treatment roadblock. Pharmacol. Res. 2017, 117, 54. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.J.; Cram, P.; Lynch, C.F.; Buckwalter, J.A. Risk factors for metastatic disease at presentation with osteosarcoma: An analysis of the SEER database. J. Bone Joint Surg. Am. 2013, 95, e89. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Ni, J.; Huang, J. Molecular mechanisms of chemoresistance in osteosarcoma (Review). J. Oncol. Lett. 2014, 7, 1352. [Google Scholar] [CrossRef] [PubMed]
- Janeway, K.A.; Grier, H.E. Sequelae of osteosarcoma medical therapy: A review of rare acute toxicities and late effects. Lancet Oncol. 2018, 11, 670. [Google Scholar] [CrossRef]
- Xu, M.; Xu, S.F.; Yu, X.C. Clinical analysis of osteosarcoma patients treated with high-dose methotrexate-free neoadjuvant chemotherapy. Curr. Oncol. 2014, 21, 678. [Google Scholar] [CrossRef] [PubMed]
- Raj, R.; Mongia, P.; S, S.K.; Ram, A. Nanocarriers Based Anticancer Drugs: Current Scenario and Future Perceptions. Curr. Drug Targets 2016, 17, 206. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The Smart Drug Delivery System and Its Clinical Potential. Theranostics 2016, 6, 1306. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Dai, W.-M.; Guy, R.K. Chemistry and Biology of Taxol. Angew. Chem. Int. Ed. Engl. 1994, 33, 15. [Google Scholar] [CrossRef]
- Horwitz, S.B. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992, 13, 134. [Google Scholar] [CrossRef]
- Weiss, R.B.; Donehower, R.C.; Wiernik, P.H.; Ohnuma, T.; Gralla, R.J.; Trump, D.L.; Baker, J.R.; van Echo, D.A.; von Hoff, D.D.; Leyland-Jones, B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Papadopoulos, N.E.; Plager, C.; Linke, K.A.; Moseley, S.H.; Spirindonidis, C.H.; Benjamin, R. Phase II study of paclitaxel in patients with previously treated osteosarcoma and its variants. Cancer 1996, 78, 741. [Google Scholar] [CrossRef]
- Gian, V.G.; Johnson, T.J.; Marsh, R.W.; Schuhmacher, C.; Lynch, J.W. A phase II trial of paclitaxel in the treatment of recurrent or metastatic soft tissue sarcomas or bone sarcomas. J. Exp. Ther. Oncol. 1996, 1, 186. [Google Scholar] [PubMed]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M.; Yin, H.; Eaves, D.; Currier, M.; Cripe, T.P. Preclinical evaluation of nanoparticle albumin-bound paclitaxel for treatment of pediatric bone sarcoma. Pediatr. Blood Cancer 2014, 61, 2096. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Niu, X.; Zhang, Q.; Hao, L.; Ding, Y.; Xu, H. The efficacy of abraxane on osteosarcoma xenografts in nude mice and expression of secreted protein, acidic and rich in cysteine. Am. J. Med. Sci. 2012, 344, 199. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, E.; Helguera, G.; Legaspi, M.J.; Gonzalez, L.; Hocht, C.; Taira, C.; Chiappetta, D.A. Paclitaxel-loaded PCL-TPGS nanoparticles: In vitro and in vivo performance compared with Abraxane®. Colloids Surf. B Biointerfaces 2014, 113, 43. [Google Scholar] [CrossRef] [PubMed]
- Foglietta, F.; Spagnoli, G.C.; Muraro, M.G.; Ballestri, M.; Guerrini, A.; Ferroni, C.; Aluigi, A.; Sotgiu, G.; Varchi, G. Anticancer activity of paclitaxel-loaded keratin nanoparticles in two-dimensional and perfused three-dimensional breast cancer models. Int. J. Nanomed. 2018, 13, 4847. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Lan, R.L.; Tao, X.; Wu, C.Y.; Hong, H.F.; Lin, J.H. Vitronectin significantly influences prognosis in osteosarcoma. Int. J. Clin. Exp. Pathol. 2015, 8, 11364. [Google Scholar] [PubMed]
- Aluigi, A.; Ballestri, M.; Guerrini, A.; Sotgiu, G.; Ferroni, C.; Corticelli, F.; Gariboldi, M.B.; Monti, E.; Varchi, G. Organic solvent-free preparation of keratin nanoparticles as doxorubicin carriers for antitumour activity. Mater. Sci. Eng. C 2018, 90, 476. [Google Scholar] [CrossRef] [PubMed]
- Calixto, G.M.; Bernegossi, J.; de Freitas, L.M.; Fontana, C.R.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for Photodynamic Therapy of Cancer: A Review. Molecules 2016, 21, 342. [Google Scholar] [CrossRef] [PubMed]
- Chilakamarthi, U.; Giribabu, L. Photodynamic Therapy: Past, Present and Future. Chem. Rec. 2017, 17, 775. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-T.; Chen, Q.; Wang, D.-W.; Duan, Q.-Q.; Tian, S.; He, J.-W.; Ou, Y.-S.; Bai, D.-Q. Mitochondrial pathway and endoplasmic reticulum stress participate in the photosensitizing effectiveness of AE-PDT in MG63 cells. Cancer Med. 2016, 5, 3186. [Google Scholar] [CrossRef] [PubMed]
- White, B.; Rossi, V.; Baugher, P.J. Aminolevulinic Acid-Mediated Photodynamic Therapy Causes Cell Death in MG-63 Human Osteosarcoma Cells. Photomed. Laser Surg. 2016, 34, 400. [Google Scholar] [CrossRef] [PubMed]
- Coupienne, I.; Fettweis, G.; Piette, J. RIP3 expression induces a death profile change in U2OS osteosarcoma cells after 5-ALA-PDT. Lasers Surg. Med. 2011, 43, 557. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.; Huang, Q.; Ou, Y.; Du, X.; Li, K.; Tao, Y.; Yin, H. Aloe-emodin-mediated photodynamic therapy induces autophagy and apoptosis in human osteosarcoma cell line MG-63 through the ROS/JNK signaling pathway. Oncol. Rep. 2016, 35, 3209. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-R.; Yin, R.; Huang, Y.-Y.; Sheu, B.-C.; Lee, S.-C.; Hamblin, M.R. Low-level light therapy potentiates NPe6-mediated photodynamic therapy in a human osteosarcoma cell line via increased ATP. Photodiagnosis Photodyn. Ther. 2015, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Campanile, C.; Muff, R.; Born, W.; Fuchs, B. mTHPC-mediated photodynamic therapy is effective in the metastatic human 143B osteosarcoma cells. Photochem. Photobiol. 2012, 88, 721. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Aizawa, S.; Iriuchishima, T.; Goto, B.; Nagaoka, M.; Tokuhashi, Y.; Saito, A. Phototoxic effect of na-pheophorbide a toward osteosarcoma cells in vitro using a laser diode. Photomed. Laser Surg. 2014, 32, 481. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Sun, M.; Zhou, C.; Yin, F.; Wang, Z.; Hua, Y.; Cai, Z. Hematoporphyrin monomethyl ether-mediated photodynamic therapy selectively kills sarcomas by inducing apoptosis. PLoS ONE 2013, 8, e77727. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.; Botter, S.M.; Campanile, C.; Robl, B.; Gräfe, S.; Pellegrini, G.; Born, W.; Fuchs, B. Foscan and foslip based photodynamic therapy in osteosarcoma in vitro and in intratibial mouse models. Int. J. Cancer 2017, 140, 1680. [Google Scholar] [CrossRef] [PubMed]
- Nomura, J.; Yanase, S.; Matsumura, Y.; Nagai, K.; Tagawa, T. Efficacy of combined photodynamic and hyperthermic therapy with a new light source in an in vivo osteosarcoma tumor model. J. Clin. Laser Med. Surg. 2004, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Duchi, S.; Sotgiu, G.; Lucarelli, E.; Ballestri, M.; Dozza, B.; Santi, S.; Guerrini, A.; Dambruoso, P.; Giannini, S.; Donati, D.; et al. Mesenchymal stem cells as delivery vehicle of porphyrin loaded nanoparticles: Effective photoinduced in vitro killing of osteosarcoma. J. Control Release 2013, 168, 225. [Google Scholar] [CrossRef] [PubMed]
- Aluigi, A.; Sotgiu, G.; Ferroni, C.; Duchi, S.; Lucarelli, E.; Martini, C.; Posati, T.; Guerrini, A.; Ballestri, M.; Corticelli, F.; et al. Chlorin e6 keratin nanoparticles for photodynamic anticancer therapy. RSC Adv. 2016, 6, 33910. [Google Scholar] [CrossRef]
- Olivo, M.; Bhuvaneswari, R.; Lucky, S.S.; Dendukuri, N.; Thong, P.S. Targeted Therapy of Cancer Using Photodynamic Therapy in Combination with Multi-faceted Anti-Tumor Modalities. Pharmaceuticals (Basel) 2010, 3, 1507. [Google Scholar] [CrossRef] [PubMed]
- Khdair, A.; Chen, D.; Patil, Y.; Ma, L.; Dou, Q.P.; Shekhar, M.P.V.; Panyam, J. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance. J. Control. Release 2010, 141, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Zhang, P. Combinations in multimodality treatments and clinical outcomes during cancer. Oncol. Lett. 2016, 12, 4301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-J.; Hu, Q.; Feng, G.; Zhang, R.; Yuan, Y.; Lu, X.; Liu, B. Image-guided combination chemotherapy and photodynamic therapy using a mitochondria-targeted molecular probe with aggregation-induced emission characteristics. Chem. Sci. 2015, 6, 4580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, F.; Ren, C.; Yang, L.; Liu, J.; Cheng, Z.; Chu, L.; Liu, J. Targeted Chemo-Photodynamic Combination Platform Based on the DOX Prodrug Nanoparticles for Enhanced Cancer Therapy. ACS Appl. Mater. Interfaces 2017, 9, 13016. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, J.; Liang, C.; Feng, L.; Dong, Z.; Song, X.; Song, G.; Liu, Z. Drug-induced co-assembly of albumin/catalase as smart nano-theranostics for deep intra-tumoral penetration, hypoxia relieve, and synergistic combination therapy. J. Control. Release 2017, 263, 79. [Google Scholar] [CrossRef] [PubMed]
- Cunderlíková, B.; Gangeskar, L.; Moan, J. Acid-base properties of chlorin e6: Relation to cellular uptake. J. Photochem. Photobiol. B 1999, 53, 81. [Google Scholar] [CrossRef]
- André, N.; Carré, M.; Brasseur, G.; Pourroy, B.; Kovacic, H.; Briand, C.; Braguer, D. Paclitaxel targets mitochondria upstream of caspase activation in intact human neuroblastoma cells. FEBS Lett. 2002, 532, 256. [Google Scholar] [CrossRef]
- André, N.; Braguer, D.; Brasseur, G.; Gonçalves, A.; Lemesle-Meunier, D.; Guise, S.; Jordan, M.A.; Briand, C. Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells. Cancer Res. 2000, 60, 5349. [Google Scholar] [PubMed]
- Varbiro, G.; Veres, B.; Gallyas, F.; Sumegi, B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic. Biol. Med. 2001, 31, 548. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Fanelli, M.; Tavanti, E.; Vella, S.; Ferrari, S.; Picci, P.; Serra, M. Advances in emerging drugs for osteosarcoma. Expert Opin. Emerg. Drugs 2015, 20, 495. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Fanelli, M.; Tavanti, E.; Vella, S.; Riganti, C.; Picci, P.; Serra, M. Doxorubicin-resistant osteosarcoma: Novel therapeutic approaches in sight? Future Oncol. 2017, 13, 673. [Google Scholar] [CrossRef] [PubMed]
- Pautke, C.; Schieker, M.; Tischer, T.; Kolk, A.; Neth, P.; Mutschler, W.; Milz, S. Characterization of osteosarcoma cell lines MG-63, Saos-2 and U-2 OS in comparison to human osteoblasts. Anticancer Res. 2004, 24, 3743. [Google Scholar] [PubMed]
- Riccio, M.; Dembic, M.; Cinti, C.; Santi, S. Multifluorescence labeling and colocalization analyses. Methods Mol. Biol. 2004, 285, 171. [Google Scholar] [PubMed]
- Valamehr, B.; Jonas, S.J.; Polleux, J.; Qiao, R.; Guo, S.; Gschweng, E.H.; Stiles, B.; Kam, K.; Luo, T.-J.M.; Witte, O.N.; et al. Hydrophobic surfaces for enhanced differentiation of embryonic stem cell-derived embryoid bodies. Proc. Natl. Acad. Sci. USA 2008, 105, 14459. [Google Scholar] [CrossRef] [PubMed]
- Schrand, A.M.; Schlager, J.J.; Dai, L.; Hussain, S.M. Preparation of cells for assessing ultrastructural localization of nanoparticles with transmission electron microscopy. Nat. Protoc. 2010, 5, 744. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Concentration/Nanoparticles | Low | Medium | High |
|---|---|---|---|---|
| 1 | [Ce6] in PTX-Ce6@kerag | 0.84 µM | 3.35 µM | 6.7 µM |
| 2 | [PTX] in PTX-Ce6@kerag | 1.63 µM | 6.52 µM | 13.0 µM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martella, E.; Ferroni, C.; Guerrini, A.; Ballestri, M.; Columbaro, M.; Santi, S.; Sotgiu, G.; Serra, M.; Donati, D.M.; Lucarelli, E.; et al. Functionalized Keratin as Nanotechnology-Based Drug Delivery System for the Pharmacological Treatment of Osteosarcoma. Int. J. Mol. Sci. 2018, 19, 3670. https://doi.org/10.3390/ijms19113670
Martella E, Ferroni C, Guerrini A, Ballestri M, Columbaro M, Santi S, Sotgiu G, Serra M, Donati DM, Lucarelli E, et al. Functionalized Keratin as Nanotechnology-Based Drug Delivery System for the Pharmacological Treatment of Osteosarcoma. International Journal of Molecular Sciences. 2018; 19(11):3670. https://doi.org/10.3390/ijms19113670
Chicago/Turabian StyleMartella, Elisa, Claudia Ferroni, Andrea Guerrini, Marco Ballestri, Marta Columbaro, Spartaco Santi, Giovanna Sotgiu, Massimo Serra, Davide Maria Donati, Enrico Lucarelli, and et al. 2018. "Functionalized Keratin as Nanotechnology-Based Drug Delivery System for the Pharmacological Treatment of Osteosarcoma" International Journal of Molecular Sciences 19, no. 11: 3670. https://doi.org/10.3390/ijms19113670