Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases


1
EuroEspes Biomedical Research Center, Institute of Medical Science and Genomic Medicine, 15165 La Coruña, Spain
2
Chair of Genomic Medicine, Continental University Medical School, Huancayo 12000, Peru
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(10), 3199; https://doi.org/10.3390/ijms19103199
Submission received: 19 September 2018
/
Revised: 5 October 2018
/
Accepted: 8 October 2018
/
Published: 16 October 2018
(This article belongs to the Special Issue Genomics of Brain Disorders)
Abstract
:Cerebrovascular and neurodegenerative disorders affect one billion people around the world and result from a combination of genomic, epigenomic, metabolic, and environmental factors. Diagnosis at late stages of disease progression, limited knowledge of gene biomarkers and molecular mechanisms of the pathology, and conventional compounds based on symptomatic rather than mechanistic features, determine the lack of success of current treatments, including current FDA-approved conventional drugs. The epigenetic approach opens new avenues for the detection of early presymptomatic pathological events that would allow the implementation of novel strategies in order to stop or delay the pathological process. The reversibility and potential restoring of epigenetic aberrations along with their potential use as targets for pharmacological and dietary interventions sited the use of epidrugs as potential novel candidates for successful treatments of multifactorial disorders involving neurodegeneration. This manuscript includes a description of the most relevant epigenetic mechanisms involved in the most prevalent neurodegenerative disorders worldwide, as well as the main potential epigenetic-based compounds under investigation for treatment of those disorders and their limitations.
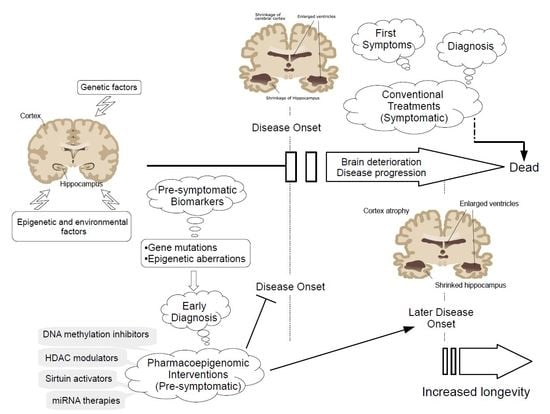
1. Introduction
Brain disorders with a vascular and/or neurodegenerative component affect one billion people worldwide, according to the World Health Organization. Most of these pathologies share an onset of dementia. Disability caused by dementia increases dramatically with aging, by affecting 9 per 1000 of the population aged 65 to 74 years to 83 per 1000 in the population over 85 years old [1]. Alzheimer’s disease (AD) is the major cause of dementia in Western countries, affecting 45 to 60% of the population, followed by vascular dementia and mixed dementia with prevalences of 30 to 40% and 10 to 20%, respectively [2,3]. Alzheimer’s disease (AD) is a complex polygenic disorder defined clinically by a progressive neurodegenerative disorder, resulting in a gradual, irreversible loss of memory and cognitive function and neuropathologically by gross atrophy of the brain and the accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles. Early stages of AD are characterized by mild cognitive impairment and several histopathological hallmarks including neuritic plaques, neurofibrillary tangles, and loss of basal forebrain cholinergic neurons. AD progression results in senile plates and neurofibrillary tangles corresponding to β-amyloid (Aβ) aggregation and hyperphosphorylation of tau protein, respectively, which results in loss of synapses, neuronal degeneration, and subsequent memory impairment, dementia, and functional decline [2,3,4]. Parkinson’s disease (PD) is the second in the ranking of the most common neurodegenerative disorders, after AD, affecting 2% of the population over 60 years of age in the world [5], and involves genetic, environmental, cerebrovasular, and epigenetic factors [6,7,8,9,10]. PD is a complex neurodegenerative disease characterized by progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta and the formation of intracytoplasmatic inclusions made of accumulations of α-synuclein known as Lewy bodies [11,12]. Clinical features of PD include rigidity, resting tremor, bradykinesia, and postural imbalance.
Neurodegeneration process begins with a series of earlier events, affecting cell development, metabolism, and axonal transport, which progressively lead to a massive cell death rate. Brain often compensates those premature features that remain rather asymptomatic. The lack of success of the current treatment strategies relies on our still limited knowledge of the pathogenic genomic variants. Importantly, currently detected polymorphic variants in pathogenic genes associate directly with a very low rate of Alzheimer’s or Parkinson’s disease patients. Personalization of treatments according to individual pharmacogenomic profiles would also increase the rate of success and reduce unnecessary costs. In addition, detection of the first symptomatic features normally occurs after a high rate of cell death and damaged tissue, which significantly affect brain function and hinders potential treatments. In this regard, novel epigenetically-based treatments are gaining a great interest as a potential novel treatments for complex multigenic neurodegenerative diseases [3,13,14,15,16,17].
Complex diseases often result from the interplay of genetic variants (genomics) and the environmental influence on gene function (epigenomics). The epigenetic machinery controls metabolic pathways by regulating gene expression through chemical and structural modifications on the genome, such as DNA/RNA methylation, chromatin structure, and non-coding RNA binding. The study of the epigenetic gene regulation might allow the identification of early biomarkers corresponding to previously undetectable features in the progression of neurodegeneration. In addition, these epigenetic aberrations may be restored with the use of appropriate epigenetic-based therapies. In this regard, during the last decade, scientists have been highly motivated in the search of epigenetic aberrations that occur during presymptomatic disease stages in order to establish novel treatment approaches using compounds (epidrugs) which target the epigenetic aberrations occurring during the progression of neurodegenerative processes [2,13,18,19,20,21]. These novel approaches will potentially reduce or delay the onset of these diseases, which would improve or elongate life quality of the patients, and would reduce disease management costs. Despite of the promising results on cell and animal models, epidrugs must fulfill certain requirements for the proper evaluation of their efficacy and safety in clinical trials, including (i) more physiological IC50 ranges and efficient drug delivery strategies; (ii) more specific targets; and (iii) personalized treatments according to the individual pharmacogenomic and pharmacoepigenomic profiles.
2. Current Gene Targets and Pharmacological Treatments for Alzheimer’s and Parkinson’s Diseases
2.1. Alzheimer’s Disease (AD)
The complexity of AD pathogenesis relies on the combination of genetic, epigenetic, and environmental factors. Current research accumulates data from over 600 single-nucleotide polymorphisms (SNPs), as well as Mendelian and mitochondrial mutations, in genes potentially associated with AD progression [13,22,23,24]. Mendelian mutations affect AD pathogenic genes, including presenilins (PSEN1 and PSEN2), Aβ-precursor protein (APP), apolipoprotein E (APOE), and the alpha-2-macroglobulin (A2M).
APP cleavage, Aβ clearance, and microtubule stability regulated by phosphorylation of microtubule-associated tau protein, are key targets of AD progression. Presenilins are important determinants for the β-secretase-mediated APP cleavage. Polymorphic variants on PSEN1 and PSEN2 genes, detected in some AD patients, correlate with an impaired APP cleavage and Aβ aggregation into senile plates. Polymorphisms in the gene encoding the microtubule-associated protein tau (MAPT) promote tau protein hyperphosphorylation which results in microtubule destabilization leading to neurofibrillary degeneration [2,3,23,25]. Polymorphic variants in the gene encoding apolipoprotein E (APOE), which associate with hypercholesterolemia and vascular disorders, constitute one of the most relevant genetic hallmark s of AD. Presence of the APOE-ε4/ε4 haplotype represents a 60 to 80% probability of an early AD onset [25,26,27]. Although the molecular mechanisms are not clear, several studies associate APOE-ε4 with an impaired APP metabolism leading to Aβ aggregation promoting tau hyperphosphorylation resulting in the formation of fibrillary tangles, as well as lipid metabolism and transport impairment and oxidative and neuroinflammatory processes leading to a massive cell death rate [25,26,28,29,30]. Importantly, the presence of APOE-ε2, tightly linked to pathologies with a vascular component, is nevertheless protective against dementia [25,26]. The A2M gene, encoding for the alpha-2-macroglobulin (a protease inhibitor), is also localized in amyloid plaques and interacts with Aβ and APOE. The polymorphism 2998 G > A (rs669) in homozygosis increases the risk for the onset of AD by 4-fold compared with the general population [23,25,26].
Most current pharmacological approaches for AD treatment rely on promoting cholinergic synapses, reducing neuronal cytotoxicity, or preventing the formation of senile plates [2,13,31,32,33,34,35]. Despite numerous attempts during the last thirteen years, the only five drugs approved by FDA tacrine, donepezil, rivastigmine, galantamine, and memantine, demonstrated limited success. AD-related impaired memory and learning tasks as well as lack of attention, associate with a loss of cholinergic neurons [31,32]. Therefore, the first pharmacological strategies relied on the generation of cholinesterase inhibitors in order to promote acetylcholine levels at cholinergic synapses. Unfortunately, the positive effects of these compounds were rather controversial [2,13,33]. The high affinity antagonist of glutamatergic N-methyl-d-aspartate (NMDA) receptors, memantine, is a current alternative strategy for patients with moderate or severe stages of the disease. Memantine reduces neuronal excitotoxicity by inhibiting the prolonged influx of Ca2+ ions from extrasynaptic receptors. Nevertheless, efficacy of this drug is under debate [2,13,34,35]. Lack of success of alternative approaches based on preventing the formation of senile plates by β-secretase inhibitors or immunotherapy relied on undesirable side effects on detriment of the scarce beneficial effects [13].
2.2. Parkinson’s Disease (PD)
Recent studies provide explanations about the implications of α-synuclein in PD at the molecular level. It has been recently established the interaction of α-synuclein with mitochondrial membranes [36,37,38,39,40] and its implication in mitochondrial impairment leading to cell death [41,42]. α-synuclein affects Complex I [38] and Complex IV [43] of the mitochondrial respiratory chain, leading to a bioenergetic dysregulation, resulting in ROS production and cell death. Experiments in vitro and in yeast mitochondria corroborate these results finding that α-synuclein was able to translocate from cytosol to the mitochondrial inner membrane through the voltage-dependent anion channel (VDAC) and target the mitochondrial respiratory chain [44].
Besides the SCNA gene, which encodes α-synuclein, over 100 other pathogeneic genes may be involved in PD, from which 15 PD loci (PARK1-15) as well as other loci might be a direct cause of the disease [45]. Mutations in synuclein-alpha (SNCA), parkin 2 (PARK2), PTEN-induced putative kinase 1 (PINK1), parkin 7 (PARK7, DJ1), and leucine-rich repeat kinase 2 (LRRK2) genes are associated with the genetic etiology of PD, whereas other loci, such as, microtubule-associated protein tau (MAPT), spatacsin, polymerase (DNA) gamma, catalytic subunit (POLG1), glucosylceramidase beta (GBA), and ataxin (SCA1, SCA2), might be susceptibility genes associated with sporadic PD, normally associated with toxic or environmental exposure [8,46].
The loss of dopaminergic neurons during development of PD results in concomitant loss of dopamine in the affected areas (especially the nigrostriatal system) which is manifested with classic motor symptoms (resting tremor, rigidity, bradykinesia, postural instability, and slowness of movements which ends up in muscle atrophy), and other non-motor symptoms (depression, obsessive compulsive behavior, sleep disturbance, and cognitive impairment, among others). Current pharmacological treatments for PD are based on restoring the dopamine levels using different strategies: (i) increase dopamine availability by treatments with dopamine precursors, such as L-DOPA (levodopa), or dopaminergic agonists (amantadine, apomorphine, bromocriptine, lisuride, cabergoline, pergolide, pramipexole, ropinirole, and rotigotine) and (ii) inhibition of dopamine catabolism or degradation, by using monoamine-oxidase B (MOB) inhibitors, such as rasagiline and selegiline, or catechol-O-methylatransferase (COMPT) inhibitors, such as entacapone and tolcapone. Unfortunately, all these pharmacological treatments only provide a symptomatic relief rather than stopping or delaying the progression of the disease. Furthermore, the chronic administration of antiparkinsonian drugs currently induces the “wearing-off phenomenon”, with additional psychomotor and autonomic complications [7,17].
Combination of current drugs with novel compounds, especially bioproducts seem to reduce these clinical complications and provide dopaminergic neuroprotection in order to enhance dopaminergic neurotransmission and reduce premature neurodegeneration [17].
3. Main Epigenetic Hallmarks of Neurodegeneration
The analysis of the whole genome sequencing unveils a wide range of gene variants which facilitate the diagnosis of a number of complex, multigenic disorders. These polymorphic variants and gene mutations also serve as targets for developing more accurate and less expensive treatments. Nevertheless, genetic factors define only partially the onset of neurodegenerative diseases, which normally arouse due a complex interplay of genetic and epigenetic mechanisms. The strict epigenetic regulatory processes result in the control of gene expression in response to the metabolic demands of the organism. Epigenetic mechanisms regulate gene expression at both, transcriptionally and post-transcriptionally levels. While DNA methylation status and modulation of chromatin structure, mediated by ATP-dependent chromatin regulator complexes (ATP-CRCs) and post-translational histone modifications, exert a transcriptional control, non-coding RNAs suppress gene expression post-transcriptionally [47].
Neurophysiologic mechanisms integrated in upper level complex processes, such as memory acquisition, learning, or motor coordination are, to a large extent, epigenetically regulated [48,49,50]. Therefore, alterations in this meticulously controlled epigenetic machinery increase the risk for onset of disorders that involve mental decline, memory and motor impairments, brain deterioration, and neurodegeneration. These epigenetic aberrations target genes directly related to the pathogenesis, such as those modulating synaptic plasticity, immune response, cell development, and apoptosis, and also genes indirectly involved in the disease [2,18,19,21].
3.1. DNA Methylation
DNA methylation consists in the incorporation of methyl groups into cytosine molecule, normally located at CpG islands where CG content is greater than 60%. The level and location of DNA methylation affect gene function in different manners. Levels of methylation at the promoter regions affect gene expression. Generally, the higher is the level of methylation at the promoter region, the lower is the expression of this gene, and vice versa. Promoter hypermethylation promotes the binding of transcription repressors or inhibits transcription factors leading to a reduced gene expression [18,51,52]. Three main DNA methyltransferases (DNMTs) are responsible for DNA methylation process in mammals. DNMT3a and DNMT3b add methyl groups to new unmethylated cytosines, whereas DNMT1 maintains the methylated status [53,54]. As a counterpart, DNA demethylases reduce DNA methylation levels and promote transcription. Three enzyme families mediate DNA demethylation process: (i) the ten-eleven translocation (TET) family, which converts of 5-methyl-cytosine (5mC) into 5-hydroxymethyl-cytosine (5hmC); (ii) the AID/APOBEC family, acting as mediators of 5mC or 5hmC deamination; and (iii) the BER (base excision repair) glycosylase family involved in DNA repair [19].
3.1.1. Global DNA Methylation and Neurodegeneration
The two most prevalent neurodegenerative disorders worldwide, Alzheimer’s (AD) and Parkinson’s diseases (PD), share a reduced DNA methylation in brains and blood from animal models and human subjects [2,18,46,55,56,57,58,59,60,61,62,63,64]. Reduced expression of DNMTs and impaired Vitamin B activity are the main players of this global hypomethylation. Vitamin B complex (vitamins B2, B6, B12, and folate) displays important brain protective benefits and restores the proper DNA methylation levels by promoting homocysteine (Hcy) methylation by the S-adenosyl-l-methionine-dependent methyltransferase (SAMe) [18,65]. Indeed, both AD [66,67,68,69] and PD [62,63,64] associate with reduced levels of SAM, which result in a defective methylation of Hcy, which promotes promoter demethylation. Vitamin B deficiency also induces hypomethylation leading to overexpression of specific genes involved in AD pathogenesis. In this regard, deprivation of folate and vitamins B6 and B12 led to hypomethylation and overexpression of the β-secretase 1 (BACE1) and presenilin 1 (PSEN1) genes in cell cultures, transgenic AD animal models, and post-mortem brains of AD patients [70,71,72,73,74]. This excessive β-secretase activity resulted in impaired APP cleavage and promoted Aβ aggregation into senile plates. Some studies also suggest a link between the formation of alpha synuclein protein (α-Syn) aggregates, a PD hallmark, with reduced SAMe levels [75,76]. According to these studies, defective SAMe activity may reduce methylation levels at the promoter region of the α-Syn-encoding gene (SCNA), which would promote the expression and aggregation of α-Syn.
Contrary to AD and PD, the epigenetic pattern of the Amyotrophic Lateral Sclerosis (ALS) involves a general DNA hypermethylation [77] enforced by increased DNMT expression [78,79] and impaired demethylation machinery [80]. According to Al-Chalabi and colleges [81], approximately 10% of ALS forms are familial and caused by gene mutations whereas 90% are sporadic, i.e., influenced by surrounding environment [81].
3.1.2. Gene Specific Methylation and Neurodegeneration
Most aberrant DNA methylation patterns leading to neurodegeneration target pathogenic genes directly involved in the disease, genes involved in neuroinflammatory pathways, and genes related to neurodevelopment and synaptic processes.
Despite of the importance of β-secretase activity and APP cleavage in the progression of AD [2,3,23,25], except for some exceptions [82,83,84,85], most of the studies find no direct correlation between APP methylation and AD progression [46,55,56,57,58,59,86]. Hyperphosphorylation of protein tau is one of the molecular hallmarks of AD-related neurodegeneration. Excessive protein tau phosphorylation reduces the binding affinity of this protein to cytoskeleton which detaches and accumulates into free aggregates forming neurofibrillary tangles (NFTs). Protein tau detachment also leads to a concomitant destabilization of cytoskeleton and cell structure. Vitamin B deficit in AD patients reduces methylation of the glycogen synthase kinase 3β gene (GSK3β) at the promoter region, which promotes the expression of this protein kinase that induces tau phosphorylation, NFT aggregation, loss of cytoskeleton integrity, and cell death [87]. Promoter hypomethylation also induces the expression of genes involved in cell death and neuroinflammation, such as bridging factor 1 (BIN1), complement receptor 1 (CR1), the CD33 molecule (CD33), and the tumor necrosis factor (TNF-α) [2,21,56,88]. However, promoter hypermethylation reduces the expression of the sortilin-related receptor (SORL1) and neprylisin (NEP) genes involved in the Aβ degradation and clearance. Sanchez-Mut et al. [89] found that hypermethylation in promoter regions of the thromboxane A2 receptor (TBXA2R), sorbin and SH3 domain containing 3 (SORBS3), and spectrin beta 4 (SPTBN4) genes in AD animal models and human subjects. Authors suggest that the activation of the cyclic AMP response element-binding protein (CREB) pathway and the axon initial segment might contribute to the pathogenesis of AD [89].
Although APOE gene haplotypes are among the most reliable biomarkers for AD diagnosis, information available about the epigenetic modulation of this gene is scarce. Some studies suggest that the C > T transition in the 3’-CpG island, which is specific of APOE-ε4, might prevent methylation of this site and promote APOE-ε4 expression in AD patients [18,71,90].
Genome wide association studies found a direct implication of methylation status of α-synuclein and development of PD. The putative gene promoter, located in the intron 1 of SCNA gene, was significantly hypomethylated in blood and brain samples from PD patients as compared to controls [91]. This hypomethylation was associated with the overexpression of α-synuclein and protein aggregation leading to PD [7]. This hypomethylation/overexpression is observed in substantia nigra, putamen, and cortex of sporadic PD cases [62,92].
Other genes were also found epigenetically regulated in PD. Increased TNF-α levels are associated with neuroinflammation and dopaminergic cell death in PD. Therefore, the higher vulnerability to TNF-α regulation found in dopaminergic neurons suggests the gene promoter is hypomethylated [93]. Importantly, TNF-α overexpression is usually detected in the cerebrospinal fluid of PD patients, as TNF-α induces apoptosis in neuronal cells [93]. It was recently reported the aberrant expression of clock genes in animal models of PD [94,95]. Methylation level of seven clock gene promoters was analyzed finding a reduced methylation in PD compared to controls [96]. In addition, DNA methylation, among other epigenetic mechanisms, plays an important role in mesodiencephalic dopaminergic neurons, which are severely affected in PD patients [97].
Other studies revealed that methylation aberrations may associate with imprinting mechanisms, such as those responsible for huntingtin overexpression in Huntington’s disease patients [98], or the risk of triggering intergenerational extension or instability of CAG repeat expansions by changes in DNA methylation during epigenetic reprogramming [99,100].
3.2. Histone Post-Translational Modifications Affecting Chromatin Remodeling
Chromatin stability and conformation regulates gene expression and silencing of transposable elements, as well as maintains genome integrity. ATP-dependent chromatin regulator complexes (ATP-CRCs) and post-translational histone modifications control chromatin structure.
Histone post-translational modifications alter the chromatin package into a tight (hetrochromatin) or loose (euchromatin) conformation, which affects gene accessibility to the transcription machinery. Histone acetylation, mediated by histone lysine-acetyltransferases (HATs or KATs) reduces the electrostatic interaction between DNA and histones which results in a looser chromatin conformation that allows gene accessibility and thus activates transcription [101,102]. Gcn5-related N-acetyltransferases (GNATs), which include GCN5, p300/cAmp-response element binding protein (CBP)-associated factor (PCAF), KAT6-8, CREB-binding protein/CBP (CREBBP/CBP), and EP300 promote histone acetylation [55,101,102,103,104,105,106]. On the other hand, histone deacetylases (HDAC), reduce the level of acetyl groups and thus the negative charge of histones which enhances the electrostatic binding to DNA and promotes a compact chromatin conformation with the subsequent repressed gene transcription [2,18,48,101,102,103,104,105,107]. This conformation limits the access for transcription factors but also for DNA repair machinery, which negatively affects to the number of synapses leading to impaired memory and learning abilities. Disruption of HAT/HDAC equilibrium associates with histone acetylation decay, which increases with aging and drastically declines during AD progression, especially in the temporal lobe of AD patients [108,109] and in AD animal models [48,110]. Overexpression of nuclear EP300 interacting inhibitor of differentiation 1 (EID1) in cortical neurons of AD patients and animal models promotes histone hypoacetylation mediated by inhibition of EP300 and CREBBP [111,112]. HDAC2-mediated acetylation decay located in prefrontal cortex and hippocampus reduced neuroplasticity, as well as downregulated genes involved in learning, memory, and synaptic plasticity in AD mice [48,113]. Some studies also suggest that one of the neurotoxic effects of α-synuclein in PD involves its direct binding to histones, preventing H3 acetylation [114,115]. Indeed, treatment with HDAC inhibitors reduced α-synuclein neurotoxicity in neuroblastoma cells and transgenic Drosophila [114,115,116]. Alternatively, the class III NAD+-dependent HDACs, sirtuins (SIRT), promote lifespan and healthy aging by delaying the onset of neurodegenerative processes [21,46,117,118]. Importantly, several studies using animal models indicate that sirtuin expression drifts with aging [119,120,121] as well as with age-related neurodegenerative disorders [122,123].
Histone methylation and demethylation, mediated by histone methylases (HMTs) and demethylases (HDMTs), respectively, also contribute to neurodegenerative progression. Those enzymes have a high specificity as they usually modify one single lysine per histone which may be translated into activation or repression of transcription [19,55,101,102,124,125]. Histone methylations H3K4, H3K36, and H3K79 are associated with the activation of gene expression, whereas methylations at H3K9, H3K27, and H4K20, correspond to gene silencing. Histone methylation has also been associated with DNA repair [1,55,101,126].
Histone methylation levels (H3K14, HeK9me2, among others) increased significantly in young preplaque AD transgenic mice as compared with wild-type mice [110,127,128]. Some studies suggest the role of histone methylation promotes polarization of microglial activation pathways involving dopaminergic cell loss during PD progression. Frequency of classical (M1 phenotype) and alternative (M2 phenotype) activation pathways determines the detrimental or beneficial effects for CNS. Histone demethylase H3K27me3 Jumonji domain containing 3 (Jmjd3) is essential for M2-type activation. Suppression of Jmjd3 magnifies M1-mediated microglial overactivation leading to extensive cell death in substantia nigra in MPTP-intoxicated PD transgenic mice [129]. MPTP-mediated toxicity also reduces H3K4me3 levels in the striatum of mice and non-human primates, which can be reverted through chronic treatment with L-DOPA [130].
3.3. Non-Coding RNAs
Differential expression of non-coding RNAs (ncRNAs) modulates gene expression post-transcriptionally. Aberrant expression of a number of ncRNAs alters expression of genes involved in metabolic pathways leading to neurodegeneration.
These regulatory RNAs include long ncRNAs (lncRNAs), which target pathogenic genes directly involved in the disease or epigenetically regulated genes, small interference RNAs (siRNAs), piwi RNAs (piRNAs), and microRNAs (miRNAs). These last ones induce mRNA degradation by binding to the 3′ untranslated region and are the most popular biomarkers for disease diagnosis and progression. Altered expression of miRNAs affects modulation of direct pathogenic genes or those involved in other neurophysiological roles indirectly associated with the disease. A number of cell free blood and cerebrospinal fluid-circulating miRNAs are informative biomarkers of the stage and progression of the disease in vivo, with a rapid and noninvasive liquid biopsy. Circulating miRNAs are thus good candidates as presymptomatic biomarkers and for early diagnosis of the disease.
3.4. Epigenetic Regulation of Telomeres
Telomeres are protective structures located at the end of the chromosomes, which contain a number of TTAGGG repeats. Shortening of telomere length increases with aging and age-related and neurodegenerative disorders enhance this process. Several mechanisms mediate the protection of these telomeric regions in order to delay their degradation. The efficiency of these protective mechanisms depends on the length of telomeres, i.e., the number of TTAGGG repeats. Shelterin is a nucleoprotein complex that binds to those repeats and protects DNA from activation of DNA damage pathways. Ability of sheltering binding increases with telomeric length, which is modulated by telomerase activity. Telomerase replaces the missed repeats after each cell division, using an associated RNA molecule, TERC, as a template. However, aging-related telomerase activity decay leads to telomere shortening, resulting in a loss of shelterin’s ability to bind these shorter telomeres and activation of the DNA damage cascade and cell death [131,132].
The epigenetic machinery modulates DNA protection and chromatin structure and stability, which affects telomerase activity and the rate of telomere degradation. Histone hypoacetylation at telomeric regions induces a heterochromatin state which protects DNA at telomeric and subtelomeric regions from activation of DNA damaging pathways [133]. On the other hand, epigenetic alterations, including decreased histone trimethylation of H3K9 and H4K20 or reduced histone dimethylation of H3K79, as well as aberrant DNA methylation, and histone H3 and H4 acetylation at telomeric and subtelomeric regions, would disrupt chromatin stability in those regions and enhance telomere shortening [134,135,136].
Epigenetic mechanisms involving ncRNA also regulate telomere length. A number of subtelomeric loci express lnRNAs named telomeric repeat-containing RNAs (TERRAs) which control both chromatin remodeling at those regions and telomerase-mediated telomere elongation [137,138,139]. TERRA may be aberrantly upregulated by DNA methylation, histone acetylation, or reduced histone methylation at telomeric or subtelomeric regions, which may lead to interference with telomere replication.
4. Current Epigenetic-Based Strategies Targeting Neurodegeneration
Available treatments for complex disorders are mostly symptomatic and provide limited beneficial effects on the progression of the disease, and often with a payback of unacceptable side effects. Epigenetic mechanisms unveil many hidden pathological alterations of memory and learning impairment, synaptic loss, and cell death, involved in neurodegenerative processes. Many epigenetic alterations appear in early asymptomatic stages of the disease and are reversible. Thus, the new age attempts of treating these disorders involve the use of epigenetic-based drugs (epidrugs) targeting DNA methylation, chromatin remodeling, and non-coding RNAs as potential candidates for the treatment of these complex polygenic disorders. These drugs include activators and inhibitors of DNA methyltransferases, histone deacetylase inhibitors, sirtuin activators, modulators of histone acetylation and histone methylation, as well as RNA interference analogs.
4.1. DNA Methylation Modulators
4.1.1. DNA Methylation Activators
Strategies attempting to restore DNA methylation may re-establish the proper metabolic pathways disrupted by the global DNA hypomethylation associated with the progression of most prevalent neurodegenerative disorders, including AD and PD. In most cases, detrimental Vitamin B- and SAMe-mediated global hypomethylation associates with high levels of homocysteine (Hcy) and S-adenosylhomocysteine (SAH) [18,62,66,67,68,69,75,140], which promotes expression of pathogenic genes. Beneficial effects of some dietary interventions may address this issue, with special focus in diets with high contents of vitamin B complex (B2, B6, B12, and folic acid). Vitamin B and SAMe-mediated DNA methylation involves several signaling pathways affecting folate/methionine/homocystein metabolism, using folate, choline, betaine, methionine, and enzyme cofactors [18,65]. Vitamin B6-dependent serine-hydroxymethyltransferase catalyzes the conversion of tetrahydrofolate (THF) into 5,10-methylenetetrahydrofolate (MTHF), followed by the synthesis of 5-MTHF catalyzed by vitamin B2-dependent MTHF reductase (MTHFR). 5-MTHF provides the methyl groups for Hcy methylation by cobalamin-dependent methionine synthase, yielding methionine, which is converted to SAM by methionine adenosyltransferase. SAM is responsible for methylation of main macromolecules, including DNA, proteins, phospholipids neurotransmitters, and hormones. Donation of methyl group promotes the synthesis of SAH, which hydrolyzes to Hcy and adenosine by SAH hydrolase. According to the consensus statement, based on the Bradford Hill criteria, elevated levels of total Hcy is a recognizable risk factor for development of dementia and AD in older individuals [141]. Therefore, methylation restorage and brain protective properties of vitamin B, folic acid, and SAMe sites them as good diet supplements for treatment of these disorders [76,142,143,144] (Table 1). Indeed, Vitamin B6 and folate are currently submitted to clinical trials from which three of them are completed in phase II (ClinicalTrials.gov Identifier: NCT01320527), phase III (ClinicalTrials.gov Identifier: NCT00056225), and phase IV (ClinicalTrials.gov Identifier: NCT2457507) to determine whether reduction of homocysteine levels with these dietary interventions would reduce cognitive impairment in AD patients [145,146,147,148] (Table 1). In Sweden and UK, folate and Vitamin B6 are clinically prescribed in patients with elevated levels of total Hcy corresponding to high risk of dementia onset [149,150,151].
4.1.2. DNA Methylation Inhibitors
Hypermethylation of pathogenic genes also promotes neurodegeneration. Therefore, approaches using DNA methylation inhibitors may also be appropriate [2,19,26,53,54,152,153,154,155]. Indeed, several DNMT inhibitors are currently submitted to clinical trials for AD treatment [145,156,157,158,159,160]. DNMT inhibitors are often small molecules and natural products, although nucleoside analogs and ncRNAs also target DNMTs.
The epigallocatechin-3-gallate (EGCG) is the main polyphenol of the green tea (Camilla sinensis). EGCG prevents misfolded proteins from fibrillization [156] and restores respiratory rates and membrane potential in isolated mitochondria from hippocampus, cortex, and striatum [157]. In addition, ECGC activates the signaling pathway involving the α7 nicotinic acetylcholine receptor (α7 nAChR) and restores Bcl2 expression, preventing cell death in Aβ-treated neurons [158]. This DNMT inhibitor is currently under clinical trials in phases II and III to test the effects of this compound on the prevention of Aβ aggregation to toxic oligomers in AD through the direct binding to the unfolded peptide (ClinicalTrials.gov Identifier: NCT00951834) [145] (Table 1). Other natural products include non-nucleosides, such as curcumin derivatives RG-108 and SGI-1027 [54], psammaplins (inhibit both DNMT1 and HDACs [53]), catechins (catechin and epicatechin), and bioflavonoids (quercetin, genistein, and fisetin). Quercetin is one of the components of the Etanercept (Enbrel®), which is an approved drug for the treatment of several forms of arthritis when administered by injection. Some studies suggest that perispinally injected Etanercept may modulate certain aspects of the immune system and provide some beneficial effect for people with Alzheimer’s disease. Studies suggest that supplementation with specific nutrients may also have a positive effect in support of cognitive function [159,160]. Etanercept is currently under phase I clinical trials (ClinicalTrials.gov Identifier: NCT01716637) for treatment of mild to moderate AD [37] (Table 1).
Other DNMT inhibitors, such as the nucleoside analogs 5-aza-2′-deoxycytidine (Decitabine) and 5-azacytidine (Azacitidine) and the small molecules hydralazine and procainamide are also potential treatments for neurodegeneration, although they are not yet submitted to clinical trials. However, these epidrugs are currently FDA approved for other prevalent disorders including diverse types of cancer, myelodisplastic syndrome, thalasemia, hypertension, and cardiac arrhythmia [53].
4.2. Histone Deacetylase (HDAC) Modulators
4.2.1. Class I, II, and IV HDAC Inhibitors
HDAC inhibitors (HDACi) potentially restore global histone deacetylation, which is a common feature of most neurodegenerative processes. Most of HDACi under development provide beneficial effects at cognitive and memory levels in animal models of AD [2,46,53,55,71,156,161] and PD [162,163,164,165,166,167,168,169]. However, only valproic acid (VPA), nicotinamide, and sodium phenylbutyrate (4-PBA) are currently under clinical trials as epidrugs for treatment of neurodegeneration [145] (Table 1).
The most effective HDACi tested in those models are (i) the short-chain fatty acids, class I HDACi (valproic acid (VPA)) and class I and II HDACis (sodium butyrate (NaB) and sodium phenylbutyrate (NaPBA, 4-PBA)); (ii) the hydroxamic acids, class I and II HDACis (suberoylanilide hydroxamic acid (SAHA, vorinostat) and trichostatin (TSA)); (iii) some benzamides, class I and II HDACi (entinostat (MS-275), W2); (iv) miscellaneous compounds, class I and II HDACi (FRM-0334) and HDAC6 specific inhibitors (Tubacin, Tubastatin A, quinazolin-4-one, (E)-3-(2-Ethyl-7-fluoro-4-oxo-3-phenethyl-3,4-dihydroquinazolin-6-yl)-N-hydorxyacrylamide (4b), and N-hydroxy-3-(2-methyl-4-oxo-3-phenethyl-3,4-dihydro-quinazolin-7-yl)-acrylamide (3f)); and (v) the class III HDACi or SIRT inhibitor nicotinamide/niacinamide and SIRT activators as resveratrol and derivatives. Other benzamides, cyclic peptides, and ketones showed powerful HDAC inhibition properties and some of them are currently FDA approved for cancer treatment.
Low toxicity of NaB makes this drug tolerable for treatment in animals and humans [169,170,171]. NaB increases the peripheral levels of hypothalamic–pituitary–adrenal axis hormones and glucose. NaB administration reinstated memory and learning activities in transgenic AD mice. In addition, prolonged exposure to NaB improved associative learning and memory in APP/PS1-21-AD transgenic mice, even at a very advanced stage of pathology. This effect might be due to the NaB-mediated histone acetylation in the hippocampus, modifying chromatin structure and enhancing the transcription of genes involved in these tasks [161]. This compound may also be effective in reducing α-synuclein aggregation and toxicity and rescuing cognitive deficits associated with PD in animal models [172,173].
4-PBA constitutes one of the most promising HDACi-based therapeutic agents due to the successful results obtained in animal models. Histone acetylation mediated by 4-PBA, promotes transcription of genes associated with synaptic plasticity and promotes the active form of GSK-3β, preventing tau phosphorylation and restoring memory and learning activities in AD transgenic mice [174]. Among these effects, other studies show an additional Aβ clearance in alternative AD animal models [53]. Combination of 4-PBA with Tauroursodeoxycholic Acid is currently in phase II clinical trials (ClinicalTrials.gov Identifier: NCT03533257) [145] in order to evaluate diverse AD-relevant markers and produce an informative dataset that will allow for evaluation and correlation of imaging-based markers, neurobiological changes, functional measures, and cognitive outcomes. Treatment with 4-PBA also protects dopaminergic neurons, possibly through increased DJ-1 expression and activation of tyrosine hydroxylase promoter in the substantia nigra of mice exposed to the PD-promoting toxic agent 1-methyl-4-phenyl-pyridinium (MPTP) [172,175]. A current phase I clinical trial (ClinicalTrials.gov Identifier: NCT02046434) [145] is attempting to investigate the potential effects of phenylbutyrate on the removal of alpha-synuclein from the brain into the bloodstream.
The anticonvulsant VPA is a fatty acid originally used as treatment for epilepsy and as a mood stabilizing agent. This is one of the most studied compounds for as a potential treatment for neurodegeneration. VPA treatment promotes the expression of brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF), which play critical roles in the growth, survival, and synaptic plasticity of neurons. In addition, VPA induces the expression of the heat-shock protein Hsp70, accompanied by increased levels of H3 lysine di- and trimethylation (H3K4Me2 and H3K4Me3), which promote the recruitment of HAT p300 [166]. Several studies show the ability of VPA to reduce Aβ production and aggregation in AD cells and animal models, normally by inhibiting GSK-3β-mediated γ-secretase cleavage of APP [176,177]. VPA, in combination with NaB and SAHA, promotes histone H4 acetylation, which results in a mitigated memory impairment [178]. VPA is currently submitted to three clinical trials for AD and dementia patients: in phase I (ClinicalTrials.gov Identifier: NCT01729598) to test the effect of VPA on the expression of clusterin, which is a currently studied epigenetic biomarker of AD; in phase III (ClinicalTrials.gov Identifier: NCT00071721) to evaluate the effects of VPA in memory tasks of individuals with dementia; and in phase II (ClinicalTrials.gov Identifier: NCT00088387) using lithium alone or in combination with VPA (divalproex) in order to evaluate the potential decrease of altered tau protein in the spinal fluid of patients with Alzheimer’s disease. [145,179,180]. Diverse studies show that VPA enhanced H3 acetylation and consequently reduced α-synuclein-mediated toxicity and decreased pro-inflammatory mediators, in PD cell models and animals exposed to PD-promoting toxic agents, such as MPTP, rotenone, or lipopolysaccaride [162,163,164,165]. Furthermore, VPA, as well as NaB and TSA, were able to rescue dopaminergic neurons death induced by the toxic agents [167,181]. Combination of VPA with lithium enhances Ser 9 phosphorylation of GSK-3β in the lumbar spinal cord and brain. Despite the undergoing clinical trials and the beneficial effects of VPA in animal models, some human clinical studies revealed unsuccessful results or treatments required significantly high doses that result toxic and lead to unacceptable adverse effects [108,182,183]. In this regard, the pharmacogenetic profile of those patients would anticipate the interindividual tolerance levels of this treatment.
Hydroxamic acids constitute another important class of HDAC inhibitors. Among them, the antifungal protein synthesis inhibitor Trichostatin (TSA) and the HDAC6-specific inhibitor Vorinostat (SAHA) are the most widely explored compounds for the treatment of neurodegenerative diseases [48,108,109,110,111,112,113,114,116,167,168,184,185,186].
Trichostatin (TSA) is class I HDAC inhibitor that enhances the expression of genes involved in memory consolidation, possibly by promoting the acetylation of the histone H4 [108,109,110]. Some studies also reflect restorage of memory function in APP/PS1-AD transgenic mice [48,112]. TSA also reduces neurotoxicity in α-synuclein overexpressing Drosophila models of PD, improving locomotor impairment, and reducing early mortality rates [114,116], as well as promoting H3 acetylation-mediated GDNF upregulation in astrocytes [168,185,186].
The HDAC6-selective inhibitor Vorinostat (SAHA) is one of the most developed HDAC inhibitors and was approved by FDA in 2006 for the treatment of advanced cutaneous T-cell lymphoma. SAHA treatment enhances basal postsynaptic excitatory, but not inhibitory, synaptic function and restores memory function in animal models of impaired learning tasks [113] and in the transgenic APPswe/PS1dE9-AD mouse [111,184].
Some benzamides, such as entinostat and W2, provided promising results as HDACi in animal models of AD. The selective HDAC1 inhibitor entinostat (MS-275) improved behavioral activities in the AD-APPPS1-21 mouse model by reducing amyloid plaque deposition and neuroinflammatory processes [187], while the mercaptoacetamide-based class II HDACi (W2) improved memory tasks and reduced tau phosphorylation rates and Aβ deposition in triple transgenic 3xTg-AD mice [188].
Several miscellaneous HDAC inhibitors are under testing for neurodegenerative diseases, especially for those involving dementia. Among this group, one of the most promising epidrugs is the Forum Pharmaceutical compound (FRM-0334), specifically tested for dementia, which inhibits a subset of class I and II human HDACs with a high efficiency (nanomolar IC50 values) and addresses the issue of crossing the blood–brain barrier [189]. FRM-0334, also called EPV-0334, promotes histone 2A, 3, and 4 acetylation in the brain and exerts a potential neuroprotective role by restoring the levels of the growth factor progranulin, which results in a significant improvement of cognitive performance in mice and rat models of frontotemporal dementia [171]. This compound is currently under phase II clinical trials in individuals with mutations in the progranulin gene diagnosed with mild to moderate frontotemporal dementia [145,153].
HDACs, and specially HDAC6, correlate with neuronal impairment and cell death, normally via microtubule destabilization. These high levels of HDAC6 frequently lead to neurodegeneration in hippocampi of AD patients. Along with SAHA, other compounds including Tubacin, Tubastatin A, and quinazolin-4-one derivatives are HDAC6-selective inhibitors with valuable potential benefits on enhancing neurite extension and reducing cell death. Tubacin (EC50 = 2.5 μM in A549 cells) exhibits 70-fold higher selectivity for HDAC6 as compared to other HDAC inhibitors in alveolar basal epithelial A549 adenocarcinoma cells. Tubacin inhibits HDAC6-targeted α-tubulin deacetylation and migration in cancer cells expressing HDAC6. Furthermore, this compound attenuates tau phosphorylation in vitro [181,190,191]. Similar to Tubacin, the affinity of the hydroxamic acid derivative, Tubastatin A, for HDAC6 is 50 to 2000-fold higher compared to other isozymes [192]. The quinazolin-4-one derivative N-hydroxy-3-(2-methyl-4-oxo-3-phenethyl-3,4-dihydro-quinazolin-7-yl)-acrylamide (3f) is the synthetic compound with the highest affinity and efficiency to inhibit HDAC6 in vitro (IC50, 29 nM). This compound promoted the expression of the growth-associated protein 43 which favored neurite outgrowth and enhanced the synaptic activities of PC12 and SH-SY5Y neuronal cells without toxic or mitogenic effects, and decreased zinc-mediated β-amyloid aggregation without affecting membrane channel (IC50 >10 μM) or cytochrome P450 activity (IC50 > 6.5 μM) in vitro. In addition this quinazolin-4-one derivative enhanced memory performances in AD animal models with β-amyloid-induced hippocampal lesions. [193].
4.2.2. Class III HDAC (SIRT) Inhibitors
A number of studies demonstrate the beneficial effect of class III HDAC (SIRT) modulators for the treatment of several types of cancer; some of them are promising treatments for neurodegenerative disorders. Although SIRT inhibition often associates with activation of cell death pathways in multiple models of cancer, we will focus on those several SIRT inhibitors (SIRTi) that provide physiologically relevant benefits for neurodegenerative processes. Indeed, sirtuins, particularly SIRT2, favor the pathological progression of PD by promoting α-synuclein expression and aggregation. In this regard, SIRT2 inhibition rescued α-synuclein-mediated toxicity in several animal models of PD [115]. Some of these epidrugs, such as Nicotinamide, show pan-sirtuin inhibition properties, whereas other SIRT inhibitors target only SIRT1/2 or either one alone.
Among the pan inhibitors, Nicotinamide is the most widely tested for treatment of neurodegenerative diseases, especially Alzheimer’s and Huntington diseases (Table 1). Nicotinamide is a competitive and selective inhibitor of class III NAD+-dependent HDACs (SIRT inhibitor) used in gene regulation experiments [194]. This compound improves the stability of microtubules by reducing phosphorylated tau (at Thr231 level) and restored cognitive deficits in triple transgenic 3xTg-AD mice [195]. Nicotinamide is being currently tested, versus placebo, in two clinical trials as a potential treatment for mild to moderate AD. One of the trials completed the phase I (and phase II for placebo) (ClinicalTrials.gov Identifier: NTC00580931) and the other trial is recruiting patients for phase II (ClinicalTrials.gov Identifier: NTC03061474) [145,195,196].
SIRT2 inhibitors are also widely tested in animal models of neurodegeneration [117,197,198,199]. Among them, the brain-permeable inhibitor AK-7 displayed important neuroprotective properties in animal models of PD by improving motor functions, extending survival, and reducing alpha-synuclein aggregation [197,198].
The vinyl nitrile compound AGK2 significantly reduces tubulin deacetylation and formation of large α-synuclein inclusions, resulting in the rescue of dopaminergic neurons in vitro and in animal PD models [117]. AGK2 targets SIRT2 with a 10-fold higher selectivity as compared with the SIRTs 1 and 3 (IC50 of 3.5 μM), although SirReal2 is considered as the highest specific SIRT2 inhibitor, with an IC50 within the nM range [199]. Substrate competition chemical analyses demonstrated that this compound is able to bind and induce conformational changes in a previously unexploited binding pocket of SIRT2.
Other relevant SIRT inhibitors are sirtinol and selisistat [200,201,202,203,204]. Sirtinol is a SIRT1/2 inhibitor discovered in 2001 by a high-throughput cell-based screening and plays important roles in different physiological pathways, such as axonal protection following nerve injury or modulation of sirtuins in cardioprotection [200,201]. Selisistat was the first identified potent and cell permeable SIRT1-specific inhibitor [202,203].
4.2.3. Class III HDAC (SIRT) Activators
Stress signaling pathways resulting in DNA damage and impaired DNA repair mechanisms are common hallmarks of neurodegeneration. The brain protective roles of sirtuins (SIRT), which include response to stress and activation of DNA repair pathways, site SIRT activating compounds as good potential candidates for treatment of neurodegeneration [204,205,206,207,208,209]. Resveratrol and derivatives are the most widely-tested SIRT activators in animal models of neurodegeneration.
Resveratrol is a neuroprotective compound extracted from red grapes, with important antioxidant and anti-inflammatory roles which result in the inhibition of Aβ aggregation and Aβ-induced apoptosis [210,211]. This compound might reduce miR-124 and miR-134 expressions, which would enhance cAMP response element-binding protein (CBP) levels and promote BDNF synthesis [212]. All these effects result in increased cell viability through the stabilization of Ca2+ homeostasis, reduction of Aβ25–35 neurotoxicity, and Rho-associated kinase 1 downregulation [212]. Resveratrol belongs to the family of drugs regulating GABA receptors. Much research has corroborated the importance of GABA receptors in the regulation of the neuronal pathways involved in memory and learning and, therefore, the GABAergic system has come to be seen as a promising therapeutic target for AD [213,214]. Currently, for resveratrol, four clinical trials are underway to test the potential of resveratrol in the prevention of cognitive impairment and cerebrovascular dysfunction in AD. Of these, two studies have already been completed: in phase II (ClinicalTrials.gov Identifier: NCT01504854) and phase III (ClinicalTrials.gov Identifier: NCT00678431). Of the remaining two trials, one has been withdrawn (ClinicalTrials.gov Identifier: NCT00743743) and the other is still recruiting participants (ClinicalTrials.gov Identifier: NCT02502253) [147,214] (Table 1).
Some Resveratrol structural derivatives, such as the stilbene Piceatannol, the chalcones Butein and Isoliquiritigenin, and the flavones Fisetin and Quercetin [2,215,216,217], are SIRT1 deacetylase activators that significantly extended the lifespan of Saccharomyces cerevisiae [218] and in Drosophila melanogaster S2 cells [216]. Furthermore, a diet containing 100 μM Fisetin extended Drosophila lifespan at a rate of 23%, as compared with Resveratrol, which increased lifespan of the flies at a rate of 29% [216].
4.3. Histone Acetyltransferase (HAT) Modulators
HAT-activating compounds, targeting CBP, p300, and p300/PCAF, would be an alternative strategy of promoting histone acetylation levels, although the poor solubility and membrane permeability of these compounds make them rather unsuitable for this purpose [2,19,26].
Importantly, a variety of chemical modifications of different nonspecific HAT inhibitors in attempts to identify enzyme-specific inhibitors, came up with the synthesis of the N-(4-chloro-3-trifluoromethyl-phenyl)-2-ethoxy-6-pentadecyl benzamide (CTPB), which is considered as one of the unique p300-specific activator with the capability of crossing the blood brain barrier after intraperitoneal injection [53,54].
Alternative strategies also consider natural products as HAT inhibitors [218]. The most popular HAT-inhibiting compounds are curcumin and derivatives. Other HAT-specific inhibitors include Lys-Coa targeting p300 and H3-Coa-20 for PCAF. Other HAT inhibitors are less specific but capable of permeating cells in culture [53].
Curcumin is a phytochemical compound extracted from the rhizome of Curcuma longa, L., used for dyspepsia, stress, and mood disorders [219]. Curcumin is a cell-permeable compound and specific inhibitor for p300/CBP, having no effect on PCAF, HDAC, and DNMT [53]. This compound protects neurons from oxidation by enhancing phase II detoxification enzymes and heme oxygenase 1 and restored mitochondrial function in brains of animal models treated with aluminum [220]. Some studies associate curcumin with a behavioral improvement, prevention of neuroinflammation, and inhibition of signaling pathways leading to Aβ aggregation and tau phosphorylation [221]. The combination of curcumin with other derivatives, such as demethoxycurcumin and bisedethoxycurcumin, which constitute turmeric [222], enhances curcumin’s properties as a potential AD treatment [222,223]. Three completed clinical trials, in phases I and II (ClinicalTrials.gov Identifier: NCT00164749), phase II (ClinicalTrials.gov Identifier: NCT00099710), and phase I (ClinicalTrials.gov Identifier: NCT01716637) [224,225,226,227] are underway to test the properties of combinations of curcumin with other natural compounds as potential treatment for AD and mild cognitive impairment [145] (Table 1). Two additional trials (ClinicalTrials.gov Identifier: NCT01811381) and (ClinicalTrials.gov Identifier: NCT02114372) are recruiting patients [147,228,229] (Table 1).
4.4. Modulators of Histone Methylation
Histone Methyltransferase Inhibitors
This subgroup of epidrugs includes histone methyltransferase and histone demethylase inhibitors. The first ones modulate gene expression and promote DNA repair by inducing histone acetylation. Despite of their potential activity, these compounds are not often good candidates for preclinical studies due to their high toxicity and low specificity in different cell lines. SAMe, one of the most important methyl donors in the body, along with l-methylfolate, also used as a DNA methylation activator, was the first HMT inhibitor used for treatment of cancer. Importantly, this compound is also submitted to a phase II clinical trial (ClinicalTrials.gov Identifier: NCT01320527) as an additive for a nutraceutical compound versus placebo for treatment of mild to moderate AD [145,147,148] and, also, in phases II and III for the treatment of depression in PD patients (ClinicalTrials.gov Identifier: NCT00070941) [145] (Table 1). Some studies indicate that SAMe induces PSEN1 promoter methylation resulting in gene downregulation, which meliorate AD symptoms [142,143,230].
The most analyzed histone demethylase family is the Lysine-specific demethylase 1 (LSD1), which is a flavin-dependent monoamine oxidase (MAO) that can demethylate mono- and dimethylated lysines, specifically histone 3 and lysines 4 and 9 (H3K4 and H3K9) and shares catalytic sites with MAO-A and MAO-B [231]. Inhibition of these MAO catalytic sites is a current strategy for treatment of anxiety and depressive disorders, as well as neurodegenerative PD progression [232]. Tranylcypromine (2-PCPA) is the most widely analyzed and relatively potent LSD1 inhibitor in vivo (IC50 20.7) that irreversibly blocks MAO A and MAO B with IC50 values of 2.3 and 0.95 μM and Ki values of 101.9 and 16 μM, respectively [233].
4.5. Non-Coding RNAs
Non-coding RNAs (ncRNAs) modulate the expression of genes involved in brain development and function. A number of diseases link with aberrant expression of those ncRNAs, which requires the implementation of new strategies that regulate ncRNA expression and function. Indeed several approaches include RNA interference as a novel and promising therapeutic strategy for the treatment of neurodegenerative diseases. These ncRNA-based treatment strategies include the use or modulation of miRNA analogs, miRNA precursors, and anti-miRNAs.
One of the most popular strategies to reduce the detrimental effects involves downregulation of pathogenic genes. This may be achieved posttranscriptional levels by RNA interference mediated by small interference RNAs (siRNAs), short-hairpin RNAs (shRNAs), and micro-RNAs (miRNAs). Altering the expression of ncRNAs targeting pathogenic genes associated with the disease may be an acceptable strategy. However, the extremely high number of gene targets and associated ncRNAs would make the approach rather difficult. In this regard, different studies suggest more specific targets that may be suitable as potential treatments. In this regard, overexpression of miR-124 and miR-195 reduce Aβ levels by targeting BACE1 [234,235], or miR-323-3p, might reduce AD-related neuroinflammation [236]. Other ncRNAs, such as miR-34b/c, miR-132 [237,238,239,240,241], and miR-221 [237,242], are also potential biomarkers and therapeutic targets for PD. Inhibition of miR-34b/c leads to parkin and DJ-1 downregulation in SH-SY5Y cells [238], while downregulation of miR-132 results in α-synuclein accumulation [239,240]. In addition, serum levels of miR-29c, miR-146b, miR-214, and miR-221 were significantly downregulated in patients, resulting in miR-221 being a potential predictor and therapeutic target of disease [237,242]. A recent study supports the consideration of miR-221 as a potential treatment for PD due to its protective role by regulating PC12 cell viability and apoptosis by targeting phosphatase and tensin homolog (PTEN) [243].
Other strategies deal with the regulation of other ncRNAs involved in cell growth, development, and homeostasis, such as miR-485 and miR-26a [244,245]. The synaptic vesicle glycoprotein SV2A, along with miR-485, regulates neuron homeostasis by controlling the number of dendritic spines and the establishment of synapses, as well as miR-26a. Interestingly, high miR-485 levels reduce spontaneous synaptic responses, which might have implications in AD progression [245], whereas miR-26a overexpression enhances synaptic plasticity and regulates neuronal morphogenesis [244]. Indeed, miR-26a inhibition via PTEN attenuates neuronal outgrowth. Thus, PTEN suppression by miR-26a may enhance synaptic plasticity and regulate neuronal morphogenesis [244]. Another important ncRNA involved in neurodevelopment and neurodegeneration, miR-132, is considered a potential biomarker for diagnosis and treatment of PD [237,241].
Other miRNA-based approaches target the components of the epigenetic machinery and exert direct control in DNA methylation and chromatin remodeling processes [246]. These miRNAs may target DNMT inhibitors, alternatively or synergistically, such as miRNAs targeting DNMT3A (miR-29, miR-29c, miR370, and miR-450A) and DNMT3B (miR-29, miR-148, and miR-29b) induced hypomethylation-mediated enhanced expression of tumor suppression genes, which may also be achieved by miRNAs targeting EZH2 (miR-26a, miR-101, miR138, and miR-124) and decreasing histone methylation. Other miRNAs target HDACs, such as miR-449 and miR-874 for HDAC1, and miR-1 and miR-155 target HDAC4 reducing transcriptional activity of B-cell lymphoma 6. Other miRNAs, such as miR-155 and miRNA-627, reduced histone dimethylation and hypoxic gene expression [246].
4.6. Other Potential Epigenetic Treatments
Other promising epigenetic-based therapeutic approaches, currently submitted to preclinical studies [2,145,152,153,247,248], include the following, (i) small molecule inhibitors to chromatin-associated proteins, especially those targeting histone methyltransferases and histone demethylases; (ii) bromodomain/chromodomain inhibitors, which regulate chromatin structure and inhibit targeting gene transcription, respectively; and (iii) dietary regimens based on B vitamins and folate, in order to restore global methylation by increasing the SAMe levels in the organism, or low caloric-based regimes that might promote SIRT-mediated DNA repair mechanisms.
5. Neurodegeneration-Mediated DNA Methylation Patterns of Genes Involved in Drug Metabolism and Transport
Drug pharmacodynamics and pharmacokinetics influence drug response in terms of efficiency, required dosage, and toxicity. The variability of genetic and epigenetic profiles, as well as disease determinants, explain the individual differences in drug response. Pharmacogenomics accounts for 30 to 90% of the variability in pharmacokinetics and pharmacodynamics. This variability depends on polymorphic variants of five different categories of genes: (i) pathogenic genes associated with disease development or potential risk. Not all individuals carrying the same disease present the same affected pathogenic genes; (ii) genes associated with the mechanism of action of drugs (enzymes, receptors, messengers, etc.); (iii) genes associated with drug metabolism. This category includes genes associated with Phase I enzymes, such as, alcohol dehydrogenases (ADHs), monoamine oxidases (MAOs), cytochrome p450 family genes (CYPs), and Phase II enzymes, which include UDP glucuronosyltransferases (UGTs), gluthatione S-transferase family genes (GSTs), N-acetyltransferase (NATs), and sulfonotransferases (SULTs); (iv) genes encoding drug transporters (Phase III), such as ATP-binding cassette family members (ABCs), solute carrier superfamily (SLCs), and the solute carrier organic transporter family (SLCOs); and (v) pleiotropic genes involved in multiple pathways and metabolic reactions [2,3,13,15,16,19,26]. The efficiency of drug metabolizing products is influenced by genetic and epigenetic modifications on these genes [3,13,105,106].
Pharmacoepigenomics deals with the influence of epigenetic modifications on the pharmacogenomic network responsible for the pharmacokinetics and pharmacodynamics of drugs, as well as with the effects that drugs may have on the epigenetic machinery. Despite of the scarce available information on the pharmacoepigenomics of most drugs, growing evidence indicates that epigenetic changes are determinant in the pathogenesis of many medical conditions and in drug response and drug resistance [2,249,250]. The drug response varies according to polymorphic variants of genes involved in the pharmacogenomic response, as well as by epigenetic modifications in these genes that alter their expression patterns. The acquisition of drug resistance involves post-transcriptional regulators, such as RNA-binding proteins (RBPs) and miRNAs, which alter the stability and expression of genes and gene clusters involved in cell survival, proliferation, and drug metabolism [2,249,250].
The implication of cytochrome P450 enzymes (CYPs) in PD pathophysiology is fairly well-demonstrated [251], although there is no clear-cut evidence whether polymorphisms in these genes confer susceptibility to PD or what could be the effects of these polymorphisms on enzyme activity [252,253]. In the CNS, CYP2E1 colocalizes to tyrosine hydroxylase-positive neurons in the substantia nigra [254,255]. Enhanced CYP2E1 activity promoted ROS production, inhibited dopamine release in animal models, and facilitated the production of isoquinolines, structurally related to the PD-inducing toxicant MPTP, which may thus contribute to dopaminergic neurodegeneration in PD [251,252,253,256]. A genome-wide methylation analysis of PD with quantitative DNA methylation levels of 27,500 CpG sites corresponding to 14,495 genes showed a significant methylation decrease of the CYP2E1 gene with the corresponding mRNA overexpression in brains from PD patients, suggesting that epigenetic variants of this cytochrome contribute to PD susceptibility [257] (Table 2). These results suggest that altered methylation of CYP2E1 in PD may contribute to the individual susceptibility and help to explain the conflicting findings with regard to environmental toxins and genetic vulnerability [257].
Genome-wide DNA methylation analyses in brain and blood samples from PD patients displayed a number of epigenetic biomarkers associated with the pathological mechanisms of the disease. Importantly, authors identified concordant methylation alterations in brain and blood, suggesting that the blood might hold promise as a surrogate for brain tissue to detect DNA methylation in PD and as a source for biomarker discovery [10]. Among these biomarkers, they found altered methylation and gene expression levels in the phase II metabolizing genes encoding glutathione S-transferase GSTT1 [10,258,259,260], GSTTP1 [10,258,259,260], and GSTTP2 [10,258,261], involved in conjugation of electrophiles and protection against reactive oxygen species (Table 2). Polymorphisms in these genes have been previously associated with increased risk to PD after exposure to the herbicide Paraquat [10]. Other studies found correlation in brain and blood biomarkers associated with PD progression, reinforcing the idea that detection of differential methylation events pertinent to PD pathology is feasible from blood samples [10,261,262,263,264]. Among these biomarkers, they found significant hypomethylation levels at the promoter region of the drug ABC and solute carrier organic transporter genes ABCA3 [10,261,262], SLC12A5 [10,263,265], and SLC25A24 [10,264,265] (Table 2).
Several transporter genes are involved in the control of cholesterol homeostasis and influence the pathogenesis of neurodegenerative diseases. ABCA1, ABCB1, and ABCG2 influence AD and Aβ deposition in extracellular senile plaques [266,267,268,269,270,271] (Table 2). Brain ABCA1 mediates cholesterol and phospholipid efflux and lipidates APOE to allow its interaction with Aβ and inhibits formation of Aβ deposits [272]. ABCA2, the most abundant ABC transporter in human and rodents, may regulate esterification of plasma membrane-derived cholesterol by modulation of the sphingolipid metabolism. Some authors suggest that dysregulation of ABCA2 gene may be involved in AD pathogenesis [273]. The epigenetic machinery controls the expression of these ABC transporter genes through interaction with miRNAs, such as miR-33a/b-5p, miR-106b, and miR-758–5p, regulating ABCA1 gene expression [274].
Brain DNA methylation and mRNA expression patterns in the genes coding for the ABC transporter ABCA7 and the solute carrier organic transporter SLC24A4 also associated with AD progression in a study with 740 autopsied participants older than 66 years old [262] (Table 2). The authors suggest that altered methylation in these loci might involve both Aβ and tau tangle pathologies [264]. Previous studies associate ABCA7 with the regulation of APP processing in vitro, inhibition of Aβ secretion in cultured cells, and regulation of Aβ homeostasis in the brain and deletion of ABCA7 doubles cerebral Aβ accumulation in transgenic mouse models [275,276]. The functional relationship between SLC24A4 and AD is rather indirect. Some authors suggest that SLC24A4 may be involved in neural development [277] or may interact with genes directly involved in AD progression, such as BIN1 [278]. A recent study also concluded that the ABCA7 mRNA expression level in peripheral blood may be an early diagnostic and disease progression AD biomarker regardless of the genetic polymorphism and the promoter methylation level [265].
6. Conclusions and Future Directions
Neurodegenerative diseases, as well as other complex multifactorial disorders, result from a complex interplay of genetic and epigenetic factors, influenced by environmental factors, which makes understanding the molecular mechanisms underlying their pathological progression difficult. Neurodegeneration involves premature events affecting cell metabolism, growth, and development only detected after several years of disease progression, when the rate of cell loss is high enough to hinder treatment possibilities. Therefore, the attempts to achieve effective treatments for those diseases have been rather unsuccessful, expensive, and limited to a symptomatic relief. It is therefore important to find diagnostic strategies for detection of neurodegenerative diseases during early, preferably asymptomatic stages, when a pharmacological intervention is still possible.
The analysis of epigenetic alterations during disease progression allows the detection of early diagnostic biomarkers of the disease. Furthermore, assuming that these epigenetic modifications are reversible and potentially targeted by pharmacological interventions, a number of epigenetic-based drugs (epidrugs), are opening a novel, promising approach for the treatment of such complex diseases [2,13,18,19,20]. However, in spite of the promising results of these epidrugs in vitro, in cell, and in animal models of neurodegeneration, only a few of them are currently submitted to clinical trials for the treatment of these diseases and none of them are yet approved by any of the main regulatory agencies [145]. A number of these epidrugs, like some DNMT inhibitors or pan HDAC inhibitors, induce toxicity and cell death due to global hypomethylation or histone acetylation, respectively, the high doses required, or the incapability to cross the blood–brain barrier [2,248,249]. Furthermore, the wide range of epigenetic alterations directly and indirectly linked to these diseases hinders the accurate selection of specific targets for effective treatments. Specific gene targeting by using ncRNAs may be a potential strategy to overcome these issues, especially by their relatively easy detection as cell-free biomarkers using noninvasive techniques that allow the tracking of the patient’s disease progression. However, most ncRNAs are not gene-specific but recognize several targets. In addition, the major problem with ncRNA approaches includes delivery systems and off-target effects. Exosomes and conjugation with cholesterol molecules may help delivery of ncRNA across the blood–brain barrier, although the detrimental effects of the moiety of these molecules to the human brain still needs to be tested [2,279,280,281,282].
Alternative strategies in order to minimize clinical complications include the development of novel natural bioproducts with neuroprotective properties and additional benefits including antioxidant, anti-inflammatory, and neurotrophic effects. E-PodoFavalin-15999 (Atremorine) is an example of a novel bioproduct obtained by means of nondenaturing biotechnological procedures from structural components of Vicia faba L., for the prevention and treatment of PD [17,283,284,285,286,287]. The high content of natural L-DOPA (average concentration 20 mg/g) in the composition of Atremorine provides its high dopaminergic effect on dopaminergic neurons [17,283,286,287], whereas the neuroprotective effect of this compound relies on other intrinsic constituents, such as selective neurotrophic factors [17,283]. The combination of Atremorine with conventional antiparkinsonian drugs minimizes the “wearing off” effect, as it extends the beneficial effects of the last ones with a dose reduction of 25 to 50%, which minimizes the adverse effects of the conventional antiparkinsonian compounds [17,283,286,287].
Since therapeutic outcomes are highly dependent upon the individual genomic and epigenomic profiles, personalized treatments should rely on pharmacogenetic and pharmacoepigenetic procedures to optimize therapeutics. Versatility of the epigenetic machinery allows the manipulation of epigenetic aberrations leading to drug resistance. Thus, routine procedures should incorporate pharmacoepigenetic studies for the proper evaluation of efficacy and safety issues in drug development and clinical trials. However, there is not information about long-term effects of epigenetic-based using targets without any particular cell specificity. Thus, despite substantial progress, the field of pharmacoepigenetics and the role of epigenetic modifications in human health and the treatment of disease require further investigation.
Acknowledgments
The authors appreciate colleagues at EuroEspes Biomedical Research Center and EuroEspes Biotechnology for their technical support.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Jim, J.E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation statespecific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetic drug discovery for Alzheimer’s disease. Expert Opin. Drug Discov. 2014, 9, 1059–1086. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenomics in Alzheimer’s disease. Methods Mol. Biol. 2008, 448, 213–357. [Google Scholar] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and aging. Methods Find. Exp. Clin. Pharmacol. 2005, 27 (Suppl. A), 1–573. [Google Scholar] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Feng, Y.; Jankovic, J.; Wu, Y.C. Epigenetic mechanisms in Parkinson’s disease. J. Neurol. Sci. 2015, 349, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ammal Kaidery, N.; Tarannum, S.; Thomas, B. Epigenetic landscape of Parkinson’s disease: Emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics 2013, 10, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. Genetics and epigenetics of Parkinson’s disease. Sci. World J. 2012, 2012, 489830. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; McKnight, A.J.; Craig, D.; O’Neill, F. Epigenome-wide association study for Parkinson’s disease. Neuromol. Med. 2014, 16, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goeder, M. Alpha-synuclein in Lewi bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Mallory, M.; Sundsmo, M.; Honer, W.; Hansen, L.; Masliah, E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am. J. Pathol. 1998, 152, 367–372. [Google Scholar] [PubMed]
- Cacabelos, R.; Cacabelos, P.; Torrellas, C.; Tellado, I.; Carril, J.C. Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Methods Mol. Biol. 2014, 1175, 323–556. [Google Scholar] [PubMed]
- Cacabelos, R. The path to personalized medicine in mental disorders. In The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes, 1st ed.; Ritsner, M., Ed.; Springer: Dordrecht, The Netherlands, 2011; Volume 4, pp. 3–63. ISBN 978-0-12-811060-7. [Google Scholar]
- Cacabelos, R. Pharmacogenetic basis for therapeutic optimization in Alzheimer’s disease. Mol. Diagn. Ther. 2007, 11, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenomics and therapeutic prospects in dementia. Eur. Arch. Psychiatry Clin. Neurosci. 2008, 258 (Suppl. A), 1–553. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, J.T.; Tan, M.S.; Jiang, T.; Tan, L. Epigenetic mechanisms in Alzheimer’s disease: Implications for pathogenesis and therapy. Ageing Res. Rev. 2013, 12, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Epigenomic networking in drug development: From pathogenic mechanisms to pharmacogenomics. Drug Dev. Res. 2014, 75, 348–365. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Takae, H.; Miyake, K. Epigenetic mechanisms and therapeutic perspectives for neurodevelopmental disorders. Pharmaceuticals 2012, 5, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Teijido, O.; Cacabelos, R. Interrogating the epigenome to unveil the secrets of neurodegeneration: Promising epigenetic therapies. J. Genom. Med. Pharmacogenom. 2016, 1, 95–150. [Google Scholar]
- Takeda, M.; Martínez, R.; Kudo, T.; Tanaka, T.; Okochi, M.; Tagami, S.; Morihara, T.; Hashimoto, R.; Cacabelos, R. Apolipoprotein E and central nervous system disorders: Reviews of clinical findings. Psychiatry Clin. Neurosci. 2010, 64, 592–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, R.; Fernández-Novoa, L.; Martínez-Bouza, R.; McKay, A.; Carril, J.C.; Lombardi, V.; Corzo, L.; Carrera, I.; Tellado, I.; Nebril, L.; et al. Future trends in the pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6 variants. Pharmaceuticals 2010, 3, 3040–3100. [Google Scholar] [CrossRef]
- Cacabelos, R.; Takeda, M. Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Future 2006, 31 (Suppl. B), 5–146. [Google Scholar]
- Cacabelos, R.; Martínez, R.; Fernández-Novoa, L.; Carril, J.C.; Lombardi, V.; Carrera, I.; Corzo, L.; Tellado, I.; Lescek, J.; McKay, A.; et al. Genomics of Dementia: APOE- and CYP2D6-Related Pharmacogenetics. Int. J. Alzheimer’s Dis. 2012, 2012, 518901. [Google Scholar]
- Cacabelos, R. World Guide for Drug Use and Pharmacogenomics, 1st ed.; EuroEspes Publishing: Corunna, Spain, 2012; pp. 1–3466. ISBN 978-84-940770-0-5. [Google Scholar]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dang, L.M.; Salvesen, G.S.; Pericak Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–80102. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, J.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D. Pharmacogenetics and drug development: The path to safer and more effective drugs. Nat. Rev. Genet. 2004, 5, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Kása, P.; Rakonczay, Z.; Gulya, K. The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 1977, 52, 511–535. [Google Scholar] [CrossRef]
- Qizilbash, N.; Birks, J.; López-Arrieta, J.; Lewington, S.; Szeto, S. Tacrine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2000, 2, CD000202. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Jörg Möbius, H. Memantine in moderater-to-severe Alzheimer’s disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Dagerman, K.S.; Higgins, J.P.; McShane, R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch. Neurol. 2011, 68, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Yang, R.; Guo, J.C.; Ren, H.M.; Zha, X.L.; Chen, J.S.; Cai, D.F. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Cole, N.B.; Dieuliis, D.; Leo, P.; Mitchell, D.C.; Nussbaum, R.L. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 2008, 314, 2076–2089. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, C.; Zhu, Y.; Cai, Q.; Chan, P.; Uéda, K.; Yu, S.; Yang, H. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: An immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008, 1244, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode) generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Elkon, H.; Don, J.; Melamed, E.; Ziv, I.; Shirvan, A.; Offen, D. Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. J. Mol. Neurosci. 2002, 18, 229–238. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein Shows High Affinity Interaction with Voltage dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Latrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, A.A.; Hof, P.R.; van den Hove, D.L.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Jin, P. Integrating DNA methylation dynamics into a framework for understanding epigenetic codes. Bioessays 2014, 36, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Tsai, L.H. The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Mikaelsson, M.A.; Miller, C.A. The path to epigenetic treatment of memory disorders. Neurobiol. Learn. Mem. 2011, 96, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Hove, D.L.; Kompotis, K.; Lardenoije, R.; Kenis, G.; Mill, J.; Steinbusch, H.W.; Lesch, K.P.; Fitzsimons, C.P.; De Strooper, B.; Rutten, B.P. Epigenetically regulated microRNAs in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6809. [Google Scholar] [CrossRef] [PubMed]
- Gavery, M.R.; Roberts, S.B. Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 2013, 1, e215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbioso, A.; Carafa, V.; Benedetti, R.; Altucci, L. Trials with epigenetic drugs: An update. Mol. Oncol. 2012, 6, 657–682. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Oyarzabal, J.; Lucas, M.P.; Franco, R.; García-Osta, A. Epigenetic drugs in Alzheimer’s disease. BioMol. Concepts 2013, 4, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Landgrave-Gómez, J.; Mercado-Gómez, O.; Guevara-Guzmán, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Faltraco, F.; Lista, S.; Garaci, F.G.; Hampel, H. Epigenetic mechanisms in Alzheimer’s disease: State-of-the-art. Eur. J. Neurodegener. Dis. 2012, 1, 1–19. [Google Scholar]
- Lu, H.; Liu, X.; Deng, Y.; Qing, H. DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 2013, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Lovrečić, L.; Maver, A.; Zadel, M.; Peterlin, B. The Role of Epigenetics in Neurodegenerative Diseases. Chapter 14. 2013. Available online: http://dx.doi.org/10.5772/54744 (accessed on 2 September 2018).[Green Version]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockeinstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed]
- O’Suilleabhain, P.E.; Sung, V.; Hernandez, C.; Lacritz, L.; Dewey, B.R.; Bottiglieri, T.; Diaz.Arrastia, R. Elevated plasma homocysteine level in patients with Parkinson disease. Arch. Neurol. 2004, 61, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Brattstrom, L. Plasma homocysteine and MTHFR C677T genotype in levodopa-treated patients with PD. Neurology 2001, 56, 281. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Ladenheim, B.; Cutler, R.G.; Kruman, I.I.; Cadet, J.L.; Mattson, M.P. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J. Neurochem. 2002, 80, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.; van den Hove, D.L. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T.; Godfrey, P.; Flynn, T.; Carney, M.W.; Toone, B.K.; Reynolds, E.H. Cerebrospinal fluid S-adenosyl-methionine in depression and dementia: Effects of treatment with parenteral and oral S-adenosyl-methionine. J. Neurol. Neurosurg. Psychiatry 1990, 53, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J. Neurochem. 1996, 67, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Serot, J.M.; Christmann, D.; Dubost, T.; Bene, M.C.; Faure, G.C. CSF-folate levels are decreased in late-onset AD patients. J. Neural. Transm. 2001, 108, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Tannorella, P.; Pezzini, I.; Migheli, F.; Ricci, G.; Caldarazzo lenco, E.; Piaceri, I.; Polini, A.; Nacmias, B.; Monzani, F.; et al. Folate, homocysteine, vitamin B12, and polymorphisms of genes participating in one-carbon metabolism in lateonset Alzheimer’s disease patients and healthy controls. Antioxid. Redox Signal. 2012, 17, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.C.; Lemos, R.; Ferreiro, E.; Martins, M.; de Mendonca, A.; Santana, I.; Outeiro, T.F.; Pereira, C.M. Epigenetic regulation of BACE1 in Alzheimer’s disease patients and in transgenic mice. Neuroscience 2012, 220, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent dysregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Schadt, A.; Dillmann, U.; Kostopoulos, P.; Fassbender, K.; Herrmann, W. Methylation status and neurodegenerative markers in Parkinson disease. Clin. Chem. 2009, 55, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Kalbe, E.; Kessler, J.; Calabrese, P.; Smith, R.; Passmore, A.P.; Brand, M.; Bullock, R. DemTect: A new, sensitive cognitive screening test to support the diagnosis of mild cognitive impairment and early dementia. Int. J. Geriatr. Psychiatry 2004, 19, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of Epigenetically Altered Genes in Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Paez-Colasante, X.; Figueroa-Romero, C.A.; Sakowski, S.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2010, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Yamagata, K.; Hong, K.; Wakayama, T.; Zhang, Y. A role for the elongator complex in zygotic paternal genome demethylation. Nature 2010, 463, 554–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van der Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region −224~−101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Brohede, J.; Rinde, M.; Winblad, B.; Graff, C. A DNA methylation study of the amyloid precursor protein gene in several brain regions from patients with familial Alzheimer disease. J. Neurogenet. 2010, 24, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wu, X.; Ren, L.; Liu, G.; Li, L. Epigenetic mechanisms of amyloid-betaproduction in anisomycin-treated SH-SY5Y cells. Neuroscience 2011, 194, 272–281. [Google Scholar] [CrossRef] [PubMed]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.; Ferrer, I. DNA methylation of Alzheimer disease and tautopathy-related genes in postmortem brain. J. Neuropathol. Exp. Neurol. 2009, 68, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Fuso, A.; Cavallaro, R.A.; Di Luzio, A.; Scarpa, S. B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A. J. Alzheimer’s Dis. 2010, 19, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; van den Hove, D.L.; Kenis, G.; Keitel, S.; Hof, P.R.; van Os, J.; Steinbusch, H.W.; Schmitz, C.; Rutten, B.P. Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Curr. Alzheimer Res. 2012, 9, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Aso, E.; Panayotis, N.; Lott, I.; Dierssen, M.; Rabano, A.; Urdinguio, R.G.; Fernandez, A.F.; Astudillo, A.; Martin-Subero, J.I.; et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013, 136, 3018–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caesar, I.; Gandy, S. Evidence that an APOE 4 ‘double whammy’ increases risk for Alzheimer’s disease. BMC Med. 2012, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Pihlstrøm, L.; Berge, V.; Rengmark, A.; Toft, M. Parkinson’s disease correlates with promoter methylation in the α-synuclein gene. Mov. Disord. 2015, 30, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, S.; Sothern, R.B.; Xu, S.; Chan, P. Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease. Eur. J. Neurol. 2010, 17, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Hood, S.; Cassidy, P.; Cossette, M.P.; Weigl, Y.; Verwey, M.; Robisnson, B.; Stewart, J.; Amir, S. Endogenous dopamine regulates the rhythm of expression of the clock protein PER2 in the rat dorsal striatum via daily activation of D2 dopamine receptors. J. Neurosci. 2010, 30, 14046–14058. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Ding, H.; Zheng, Z.; Gu, Z.; Ma, J.; Chen, L.; Chan, P.; Cai, Y. Promoter methylation analysis of seven clock genes in Parkinson’s disease. Neurosci. Lett. 2012, 507, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Van Heesbeen, H.J.; Mesman, S.; Veenvliet, J.V.; Smidt, M.P. Epigenetic mechanisms in the development and maintenance of dopaminergic neurons. Development 2013, 140, 1159–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrer, I.A.; Cupples, L.A.; Kiely, D.K.; Conneally, P.M.; Myers, R.H. Inverse relationship between age at onset of Huntington disease and paternal age suggests involvement of genetic imprinting. Am. J. Hum. Genet. 1992, 50, 528–535. [Google Scholar] [PubMed]
- Behnkrappa, A.; Doerfler, W. Enzymatic amplification of synthetic oligodeoxyribonucleotides: Implication for triplet repeat expansions in the human genome. Hum. Mutat. 1994, 3, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mittelman, D.; Wilson, J.H. Genome-wide demethylation destabilizes CTG.CAG trinucleotide repeats in mammalian cells. Hum. Mol. Genet. 2004, 13, 2979–2989. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modification. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.L.; Casaccia, P. Epigenetic mechanisms in multiple sclerosis: Implications for pathogenesis and treatment. Lancet Neurol. 2013, 12, 195–206. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Konsoula, Z.; Barile, F.A. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicol. Methods 2012, 66, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Dai, X.L.; Huang, H.C.; Jiang, Z.F. Targeting HDACs: A promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 2011, 143269. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Ann. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Stilling, R.M.; Fischer, A. The role of histone acetylation in age-associated memory impairment and Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Francis, Y.I.; Fa, M.; Ashraf, H.; Zhang, H.; Staniszewski, A.; Latchman, D.S.; Arancio, O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lei, J.X.; Luo, C.; Lan, X.; Chi, L.; Deng, P.; Lei, S.; Ghribi, O.; Liu, Q.Y. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2012, 45, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Maldonado, M.A.; Bokov, A.F.; Majumder, S.; Oddo, S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 22687–22692. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintas, A.; de Solis, A.J.; Diez-Guerra, F.J.; Carrascosa, J.M.; Bogonez, E. Age-associated decrease of SIRT1 expression in rat hippocampus: Prevention by late onset caloric restriction. Exp. Gerontol. 2012, 47, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.; Poliak, N.; Upadhyay, S.; Ratovitski, E.; Nelkin, B.D.; Donehower, L.A.; Sidransky, D. DeltaNp63alpha overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle 2006, 5, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Maier, B.; Bartke, A.; Scrable, H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell 2006, 5, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Balazs, R. Epigenetic mechanisms in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2014, 4, 85–102. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Reversing histone methylation. Nature 2005, 436, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Lithner, C.U.; Lacor, P.N.; Zhao, W.Q.; Mustafiz, T.; Klein, W.L.; Sweatt, J.D.; Hernandez, C.M. Disruption of neocortical histone H3 homeostasis by soluble Abeta: Implications for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, T.; Li, J.; Yang, J.; Liu, H.; Zhang, X.J.; Le, W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014, 21, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, A.P.; Lubin, F.D.; Hallett, P.J.; Vattem, P.; Ravenscroft, P.; Bezard, E.; Zhou, S.; Fox, S.H.; Brotchie, J.M.; Sweatt, J.D.; et al. Striatal histone modifications in models of levodopa-induced dyskinesia. J. Neurochem. 2008, 106, 486–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCord, R.A.; Broccoli, D. Telomeric chromatin: Roles in aging, cancer and hereditary disease. Mutat. Res. 2008, 647, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrod, S.; Gasser, S.M. Long-range silencing and position effects at telomeres and centromeres: Parallels and differences. Cell. Mol. Life Sci. 2003, 60, 2303–2318. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Benetti, R.; Garcia-Cao, M.; Blasco, M.A. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007, 39, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Jaco, I.; Fraga, M.F.; Chen, T.; Li, E.; Esteller, M.; Blasco, M.A. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat. Cell Biol. 2006, 8, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Luke, B.; Lingner, J. TERRA: Telomeric repeat-containing RNA. EMBO J. 2009, 28, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Schoeftner, S.; Blasco, M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008, 10, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Nunbhakdi-Craig, V.; Sontag, J.M.; Diaz-Arrastia, R.; Ogris, E.; Dayal, S.; Lentz, S.R.; Arning, E.; Bottiglieri, T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 2007, 27, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and dementia: An international consensus statement. J. Alzheimer’s Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.L.; Hardy, T.M.; Tollefsbol, T.O. Medicinal chemistry of the epigenetic diet and caloric restriction. Curr. Med. Chem. 2013, 20, 4050–4059. [Google Scholar] [CrossRef] [PubMed]
- Dauncey, M.J. Genomic and epigenomic insights into nutrition and brain disorders. Nutrients 2013, 5, 887–914. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, H.; Jin, P. Epigenetics-Based Therapeutics for Neurodegenerative Disorders. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 2012, 1, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. US National Institutes of Health. Available online: http://clinicaltrials.gov/ (accessed on 1 August 2018).
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2008, 300, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A Phase II Randomized Clinical Trial of a Nutritional Formulation for Cognition and Mood in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Page, R.; Morrell, C.; Shea, T.B. Maintenance of Cognitive Performance and Mood for Individuals with Alzheimer’s Disease Following Consumption of a Nutraceutical Formulation: A One-Year, Open-Label Study. J. Alzheimer’s Dis. 2016, 51, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Lökk, J. B-vitaminer kan prövas vid kognitiv svikt. Lakartidningen 2013, 110, 1528. [Google Scholar]
- Tsiachristas, A.; Smith, A.D. B-vitamins are potentially a cost-effective population health strategy to tackle dementia: Too good to be true? Alzheimers Dement. 2016, 2, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Whalley, L.J.; Duthie, S.J.; Collins, A.R.; Starr, J.M.; Deary, I.J.; Lemmon, H.; Duthie, A.C.; Murray, A.D.; Staff, R.T. Homocysteine, antioxidant micronutrients and late onset dementia. Eur. J. Nutr. 2014, 53, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Lötsch, J.; Schneider, G.; Reker, D.; Parnham, M.J.; Schneider, P.; Geisslinger, G.; Doehring, A. Common non-epigenetic drugs as epigenetic modulators. Trends Mol. Med. 2013, 19, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.P.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Bouffard, D.Y.; Momparler, L.F.; Dionne, J.; Belanger, K.; Ayoub, J. Pilot phase I-II study on 5-aza-2’-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs 1997, 8, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J. Natural compounds may open new routes to treatment of amyloid diseases. Neurotherapeutics 2013, 10, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2011, 26, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, M.; Lu, F.; Luo, N.; He, Z.P.; Yang, H. Involvement of alpha7 nAChR signaling cascade in epigallocatechin gallate suppression of beta-amyloid-induced apoptotic cortical neuronal insults. Mol. Neurobiol. 2014, 49, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E. Perispinal etanercept for treatment of Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.H. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.K.; Schneider, J.S. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1;2;3;6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 2011, 194, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Wang, C.C.; Bortner, C.D.; Peng, G.S.; Wu, X.; Pang, H.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; Hong, J.S. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 2007, 149, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.S.; Li, G.; Tzeng, N.S.; Chen, P.S.; Chuang, D.M.; Hsu, Y.D.; Yang, S.; Hong, J.S. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: Role of microglia. Brain Res. Mol. Brain Res. 2005, 134, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Monti, B.; Gatta, V.; Piretti, F.; Raffaelli, S.S.; Virgili, M.; Contestabile, A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: Involvement of alpha-synuclein. Neurotox. Res. 2010, 17, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Marinova, Z.; Ren, M.; Wendland, J.R.; Leng, Y.; Liang, M.H.; Yasuda, S.; Leeds, P.; Chuang, D.M. Valproic acid induces functional heat-shock protein 70 via class I histone deacetylase inhibition in cortical neurons: A potential role of Sp1 acetylation. J. Neurochem. 2009, 111, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Peng, G.S.; Li, G.; Yang, S.; Wu, X.; Wang, C.C.; Wilson, B.; Lu, R.B.; Gean, P.W.; Cuang, D.M.; et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurothrophic factors from astrocytes. Mol. Psychiatry 2006, 11, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, P.S.; Dallas, S.; Wilson, B.; Block, M.L.; Wang, C.C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng, Y.; et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 2008, 11, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Brazier, M.; Cerutti, I.; Pieri, F.; Tardivel, I.; Desmet, G.; Baillet, J.; Chany, C. Pharmacokinetic study of butyric acid administered in vivo as sodium and arginine butyrate salts. Clin. Chim. Acta 1989, 181, 255–263. [Google Scholar] [CrossRef]
- Egorin, M.J.; Yuan, Z.M.; Sentz, D.L.; Plaisance, K.; Eiseman, J.L. Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother. Pharmacol. 1999, 43, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Kurschel, E.; Osieka, R.; Schmidt, C.G. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur. J. Cancer Clin. Oncol. 1987, 23, 1283–1287. [Google Scholar] [CrossRef]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate-up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef] [PubMed]
- Rane, P.; Shields, J.; Heffernan, M.; Guo, Y.; Akbarian, S.; King, J.A. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 2012, 62, 2409–2412. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.G. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J. Periodontol. 2008, 79, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Yang, L.; Cleren, C.; Calingasan, N.Y.; Klivenyi, P.; Beal, M.F. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromol. Med. 2004, 5, 235–241. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Wang, S.S.; Yang, Y.Y.; Yuang, R.Y.; Chen, R.M.; Hu, C.J. The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem. Biophys. Res. Commun. 2009, 378, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Rephaeli, A.; Zhuk, R.; Nudelman, A. Prodrugs of butyric acid from bench to bedside: Synthetic design, mechanisms of action, and clinical applications. Drug Dev. Res. 2000, 50, 379–391. [Google Scholar] [CrossRef]
- Tariot, P.N.; Schneider, L.S.; Cummings, J.; Thomas, R.G.; Raman, R.; Jakimovich, L.J.; Loy, R.; Bartocci, B.; Fleisher, A.; Ismail, M.S.; et al. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D. Pharmacotherapy for Alzheimer’s disease: 2002. Clin. Neuropharmacol. 2003, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollati, V.; Galimberti, D.; Pergoli, L.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Bertazzi, P.A.; Baccarelli, A. DNA methylation in repetitive elements and Alzheimer disease. Brain Behav. Immun. 2011, 25, 1078–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegmund, K.D.; Connor, C.M.; Campan, M.; Long, T.; Weisenberg, D.J.; Biniszkiewicz, D.; Jaenisch, R.; Laird, P.W.; Akbarian, S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE 2007, 2, e895. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.K.; Schneider, J.S. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res. 2010, 1354, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Dzitoyeva, S.; Manev, H. Effect of valproic acid on mitochondrial epigenetics. Eur. J. Pharmacol. 2012, 690, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Schluesener, Y.H.J. Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J. Neuropathol. Exp. Neurol. 2013, 72, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Lee, T.; Yoon, H.; DiBattista, A.M.; Song, J.; Sohn, Y.; Moffat, E.I.; Turner, R.S.; Jung, M.; Kim, J.; et al. Mercaptoacetamide-based class II HDAC inhibitor lowers Aβ levels and improves learning and memory in a mouse model of Alzheimer’s disease. Exp. Neurol. 2013, 239, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, J.V.W. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.C.; Hong, R.; Schreiber, S.L. Structural basis for in-cell histone deacetylase paralog selectivity. J. Am. Chem. Soc. 2003, 125, 5586–5587. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.W.; Chang, P.T.; Hsin, L.W.; Chern, J.W. Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 6775–6791. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.T.; Smith, B.C.; Jackson, M.D.; Denu, J.M. Coenzyme specificity of Sir2 protein deacetylases: Implications for physiological regulation. J. Biol. Chem. 2004, 279, 40122–40129. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattsson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wales, P.; Quinti, L.; Zuo, F.; Moniot, S.; Herisson, F.; Rauf, N.A.; Wang, H.; Silverman, R.B.; Ayata, C.; et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS ONE 2015, 10, e0116919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcendor, R.R.; Kirshenbaum, L.A.; Imai, S.; Vatner, S.F.; Sadoshima, J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 2004, 95, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear NAD biosynthesis and SirT1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Wang, D.; Cohen, D.E.; Guarente, L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell 2010, 142, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Bordone, L.; Guarente, L. Calorie restriction, SIRT1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell Biol. 2005, 6, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Zatta, P. Resveratrol acts not through anti-aggregative pathways but mainly via its scavenging properties against Abeta and Abeta-metal complexes toxicity. PLoS ONE 2011, 6, e21565. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liang, N.; Zhu, D.; Gao, Q.; Peng, L.; Dong, H.; Yue, Q.; Liu, H.; Bao, L.; Zhang, J.; et al. Resveratrol inhibits beta-amyloid-induced neuronal apoptosis through regulation of SIRT1-ROCK1 signaling pathway. PLoS ONE 2013, 8, e59888. [Google Scholar]
- Zhao, Y.Q.; Jordan, I.K.; Lunyak, V.V. Epigenetics components of aging in the central nervous system. Neurotherapeutics 2013, 10, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Tuerner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Alcaín, F.J.; Villalba, J.M. Sirtuin activators. Expert Opin. Ther. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.J.; Dragunow, M. High content analysis of histone acetylation in human cells and tissues. J. Neurosci. Methods 2010, 193, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Witkin, J.M.; Li, X. Curcumin, an active constituent of the ancient medicinal herb Curcuma longa L.: Some uses and the establishment and biological basis of medical efficacy. CNS Neurol. Disord. Drug Targets 2013, 12, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sood, P.K.; Nahar, U.; Nehru, B. Curcumin attenuates aluminum-induced oxidative stress and mitocondrial dysfunction in rat brain. Neurotox. Res. 2011, 20, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, J.B.; Coradini, K.; Frozza, R.L.; Oliveira, C.M.; Meneghetti, A.B.; Bernardi, A.; Pires, E.S.; Beck, R.C.; Salbego, C.G. Free and nanoencapsulated curcumin suppress beta-amyloid –induced cognitive impairments in rats: Involvement of BDNF and Akt/GSK-3beta signaling pathway. Neurobiol. Learn. Mem. 2013, 106, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Gilani, A.H. Therapeutic potential of turmeric in Alzheimer’s disease: Curcumin or curcuminoids? Phytother. Res. 2013, 28, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, N.; Takahashi, Y.; Amakusa, Y.; Tanno, Y.; Tuji, Y.; Niwa, H.; Murakami, N.; Krishna, U.K. Effects of turmeric on Alzheimer’s disease with behavioral and psychological symptoms of dementia. Ayu 2012, 33, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Cheung, S.K.; Mok, V.C.; Lam, L.C.; Leung, V.P.; Hui, E.; Ng, C.C.; Chow, M.; Ho, P.C.; Lam, S.; et al. Curcumin effects on blood lipid profile in a 6-month human study. Pharmacol. Res. 2007, 56, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.; Cheung, S.K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention Website. CDC Features: Alzheimer’s Disease. Available online: http://www.cdc.gov/Features/Alzheimers/ (accessed on 4 October 2018).
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Peedicayil, J. Epigenetic drugs in cognitive disorders. Curr. Pharm. Des. 2014, 20, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Battaglioli, E.; Mattevi, A. Human histone demethylase LSD1 reads the histone code. J. Biol. Chem. 2005, 280, 41360–41365. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Schmidt, D.M.; McCafferty, D.G.; Shiekhattar, R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.M.; McCafferty, D.G. Trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid- production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.R.; Wang, J.; Zhang, X.B.; Geng, Y.; Hu, Z.Y.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Li, L.; Huang, C.; Li, X.; Peng, Y.; Li, J. MicroRNA-323-3p with clinical potential in rheumatoid arthritis, Alzheimer’s disease and ectopic pregnancy. Expert Opin. Ther. Targets 2014, 18, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sen, D. MicroRNAs in Parkinson’s disease. Exp. Brain Res. 2017, 235, 2359–2374. [Google Scholar] [CrossRef] [PubMed]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of miR-34b and miR-34c enhances α synuclein expression in Parkinson’s disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Mack, M.; Rist, W.; Schnack, C.; Lenter, M.; Hildebrandt, T.; Hengerer, B. MicroRNA and proteome expression profiling in early-symptomatic α-synuclein(A30P)-transgenic mice. Proteom. Clin. Appl. 2008, 2, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Tacheny, A.; Arnould, T.; Renard, P. miR-212/132 expression and functions: Within and beyond the neuronal compartment. Nucleic Acids Res. 2012, 40, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Li, T.; Wang, Y.; Tang, Y.; Cui, H.; Tang, Y.; Zhang, X.; Chen, D.; Shen, N.; Le, W. miR-132 regulates the differentiation of dopamine neurons by directly targeting Nurr1 expression. J. Cell. Sci. 2012, 125, 1673–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Li, Y.; Wang, C.; Xu, F.; Wang, M.; Liu, Y. Serum miR-221 serves as a biomarker for Parkinson’s disease. Cell Biochem. Funct. 2016, 34, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, J.; Wu, M.; Hu, J.M. Protective role of microRNA-221 in Parkinson’s disease. Bratisl. Lek. Listy 2018, 119, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Sun, H. MiR-26a promotes neurite outgrowth by repressing PTEN expression. Mol. Med. Rep. 2013, 8, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.E.; Lee, P.R.; Chen, S.; Li, W.; Fields, R.D. MicroRNA regulation of homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 11650–11655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, M.A.; Roberts, T.C.; Wood, M.J. Epigenetics and ncRNAs in brain function and disease: Mechanisms and prospects for therapy. Neurotherapeutics 2013, 10, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Helin, K.; Dhanak, D. Chromatin proteins and modifications as drug targets. Nature 2013, 502, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, T.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, R.; Torrellas, C.; Teijido, O.; Carril, J.C. Pharmacogenetic considerations in the treatment of Alzheimer’s disease. Pharmacogenomics 2016, 17, 1041–1074. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, A.G.; Watts, P.M.; Jenner, P.; Marsden, C.D. P450 enzymes and Parkinson’s disease: The story so far. Mov. Disord. 1998, 13, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, H.N.; Westberg, L.; Melke, J.; Håkansson, A.; Belin, A.C.; Sydow, O.; Olson, L.; Holmberg, B.; Nissbrandt, H. Cytochrome P450 2E1 gene polymorphisms/haplotypes and Parkinson’s disease in a Swedish population. J. Neural. Transm. 2009, 116, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Kessova, I.; Cederbaum, A.I. CYP2E1: Biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Curr. Mol. Med. 2003, 3, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Watts, P.M.; Riedl, A.G.; Douek, D.C.; Edwards, R.J.; Boobis, A.R.; Jenner, P.; Marsden, C.D. Co-localization of P450 enzymes in the rat substantia nigra with tyrosine hydroxylase. Neuroscience 1998, 86, 511–519. [Google Scholar] [CrossRef]
- Howard, L.A.; Miksys, S.; Hoffmann, E.; Mash, D.; Tyndale, R.F. Brain CYP2E1 is induced by nicotine and ethanol in rat and is higher in smokers and alcoholics. Br. J. Pharmacol. 2003, 138, 1376–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahabi, H.N.; Andersson, D.R.; Nissbrandt, H. Cytochrome P450 2E1 in the substantia nigra: Relevance for dopaminergic neurotransmission and free radical production. Synapse 2008, 62, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Kaut, O.; Schmitt, I.; Wüllner, U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 2012, 13, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Roth, R. Human Body Index—Transcriptional Profiling; Gene Expression Omnibus (GEO) NCBI 2007, Series GSE7307; Neurocrine Biosciences, Inc.: San Diego, CA, USA, 2007. [Google Scholar]
- Lewandowski, N.M.; Ju, S.; Verbitsky, M.; Ross, B.; Geddie, M.L.; Rockenstein, E.; Adame, A.; Muhammad, A.; Vonsattel, J.P.; Ringe, D.; et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16970–16975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 137B, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, R.; van der Brug, M.; Vandrovcova, J.; Ding, J.; Sharma, S.; Renton, A.; Dillman, A.; Lees, A.; Cookson, M.R.; Bandopadhyay, R. Gene Expression Changes across Multiple Regions of the Parkinson’s Disease Brain; Gene Expression Omnibus (GEO) NCBI 2011, Series GSE28894; NIA, NIH: Bethesda, MD, USA, 2011. [Google Scholar]
- Moran, L.B.; Duke, D.C.; Deprez, M.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics 2006, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, K.; Yoshino, Y.; Mori, T.; Yoshida, T.; Ozaki, Y.; Sao, T.; Mori, Y.; Ochi, S.; Iga, J.I.; Ueno, S.I. Gene Expression and Methylation Analysis of ABCA7 in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; López-Muñoz, F. The ABCB1 transporter in Alzheimer’s disease. Clin. Exp. Pharmacol. 2014, 4, e128. [Google Scholar] [CrossRef]
- Abuznait, A.H.; Kaddoumi, A. Role of ABC transporters in the pathogenesis of Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Bauer, B.; Hartz, A.M. ABC transporters and the Alzheimer’s disease enigma. Front. Psychiatry 2012, 3, 54. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Van Assema, D.M.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2012, 135, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, G.D.; Messier, A.A.; Miller, M.C.; Machan, J.T.; Majmudar, S.S.; Stopa, E.G.; Donahue, J.E.; Johanson, C.E. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J. Neuropathol. Exp. Neurol. 2010, 69, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim. Biophys. Acta 2010, 1801, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Davis, W., Jr. The ATP-binding cassette transporter-2 (ABCA2) regulates esterification of plasma membrane cholesterol by modulation of sphingolipid metabolism. Biochim. Biophys. Acta 2014, 1841, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedeke, L.; Fernández-Hernando, C. MicroRNAs: A connection between cholesterol metabolism and neurodegeneration. Neurobiol. Dis. 2014, 72 Pt A, 48–53. [Google Scholar] [CrossRef]
- Chan, S.L.; Kim, W.S.; Kwok, J.B.; Hill, A.F.; Cappai, R.; Rye, K.A.; Garner, B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 2008, 106, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of Abca7 increases cerebral amyloid-β accumulation in the J20 mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Duffy, D.L.; Zhu, G.; Liu, J.Z.; Macgregor, S.; McRae, A.F.; Wright, M.J.; Sturm, R.A.; Mackey, D.A.; Montgomery, G.W.; et al. GWAS findings for human iris patterns: Associations with variants in genes that influence normal neuronal pattern development. Am. J. Hum. Genet. 2011, 89, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kajiho, H.; Saito, K.; Tsujita, K.; Kontani, K.; Araki, Y.; Kurosu, H.; Katada, T. RIN3: A novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 2003, 116, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Lakhal, S.; Andaloussi, S.E.; O’Loughlin, A.J.; Li, J.; Wood, M.J.A. RNAi therapeutic delivery by exosomes. In RNA Interference from Biology to Therapeutics, 1st ed.; Howard, K.N., Ed.; Springer: London, UK, 2013; pp. 185–207. ISBN 978-1-4614-4743-6. [Google Scholar]
- Kuhn, D.E.; Nuovo, G.J.; Terry, A.V., Jr.; Martin, M.M.; Malana, G.E.; Sansom, S.E.; Pleister, A.P.; Beck, W.D.; Head, E.; Feldman, D.S.; et al. Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human Down Syndrome brains. J. Biol. Chem. 2013, 288, 4228. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Mourdian, M.M. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 2012, 133, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, R.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Alcaraz, M.; Nebril, L.; Cacabelos, P.; Fraile, C.; Carrera, I.; Carril, J.C. E-PodoFavalin-15999 (Atremorine®)-induced dopamine response in Parkinson’s Disease: Pharmacogenetics-related effects. J. Genom. Med. Pharmacogenom. 2016, 1, 1–26. [Google Scholar]
- Cacabelos, R.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Cacabelos, P.; Fraile, C.; Carrera, I.; Carril, J.C. E-PodoFavalin-15999 (Atremorine®)-induced neurotransmitter and hormonal response in Parkinson’s Disease. J. Exp. Res. Pharmacol. 2016, 1, 1–12. [Google Scholar]
- Cacabelos, R.; Carrera, I.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Nebril, L.; Casas, A.; Fraile, C.; et al. Parkinson’s Disease: New solutions to old problems. EuroEspes J. 2017, 11, 74–96. [Google Scholar]
- Carrera, I.; Fernández-Novoa, L.; Sampedro, C.; Cacabelos, R. Neuroprotective effect of atremorine in an experimental model of Parkinson’s disease. Curr. Pharm. Des. 2017, 23, 2673–2684. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Bioactive Extract Obtained from Vicia Faba and Its Use in the Treatment and/or Prevention of Neurodegenerative Diseases. Eur. Pat. EP16382138, 29 March 2016. [Google Scholar]

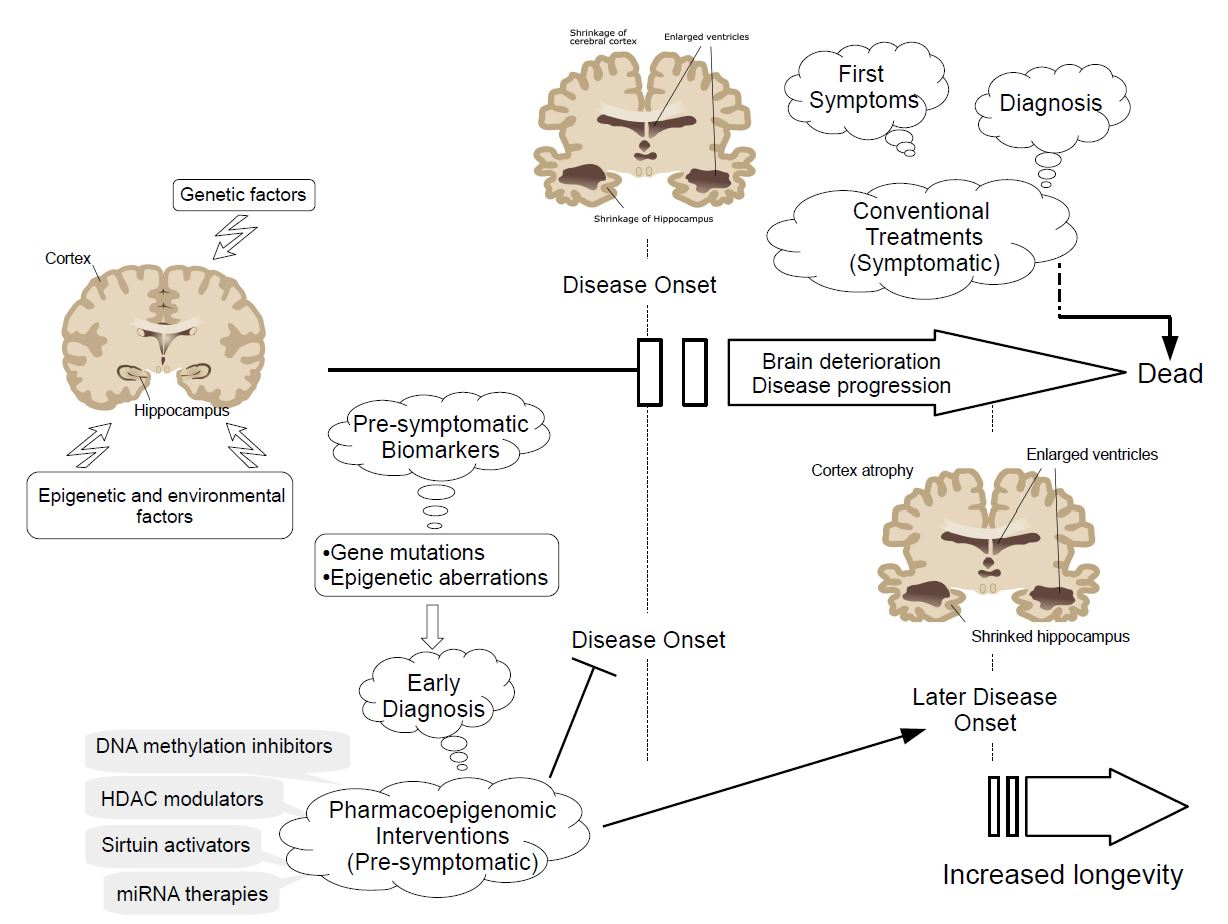{kind=link}
Table 1.
Pharmacogenetic profiles of epigenetic-based compounds currently submitted to clinical trials for the treatment of Alzheimer’s and Parkinson’s diseases.
Table 1.
Pharmacogenetic profiles of epigenetic-based compounds currently submitted to clinical trials for the treatment of Alzheimer’s and Parkinson’s diseases.
| Drug | Compound | Pharmacogenetics | Mechanisms of action | ClinicalTrials.gov ID |
|---|---|---|---|---|
 | Name: Vitamin B9; Folic acid; Folate; 59-30-3; Folacin; Pteroylglutamic acid IUPAC name: (2S)-2-[[4-[(2-amino-4-oxo-1H-pteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid Molecular formula: C19H19N7O6 Molecular Weight: 441.40 g/mol Category: SAMe methyl donors Targets: SAMe | Pathogenic genes: ADORA2A, AOX1, APOB, CDKN2A, COMPT Mechanistic genes: ALDH1A1, GSTA1, GSTP1, IL2, IL6, NAT2, SOD3, TNF, VGFA Metabolic genes: Substrate:ABCG2, MTHFR Inhibitor:ABCB1, ERCC2, MTHFR Inducer:CYP2C9 Transporter genes: ABCC1, ABCC2, ABCC3, SLC19A1, SLC22A8, SLC28A2, SLCOB1 Pleiotropic genes: PPAR, TNF, TP53, VCAM1 |
| NCT00056225-Phase III NCT01320527-Phase II NCT02457507-Phase IV |
 | Name: EGCG, (−)-epigallocatechin gallate, epigallocatechin 3-gallate, tea catechin, teavigo, catechin deriv., 989-51-5 IUPAC name: [(2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-3-yl] 3,4,5-trihydroxybenzoate Molecular formula: C22H18O11 Molecular Weight: 458.37 g/mol Category: DNMT inhibitors Targets: DNMT1 | Pathogenic genes: APP, BACE1, CDX2, EGFR, FAS PIK3CA, ROS1 Mechanistic genes: APP, BACE1, BMP2, CDX2, CHRNA7, ECEs, EGFR, IRS1, PIK3CA, ROS1 Metabolic genes: Inhibitor:SOD Transporter genes: CD36, SLC5A1, SLC27A4, SLCO1B1, SLCO1B3 Pleiotropic genes: ACACA, CHRNA7, SCD |
| NTC00951834-Phases II, III |
 | Name: Quercetin; Sophoretin; Quercetol; Meletin; Xanthaurine; Quercitin; 3,3′,4′,5,7-Pentahydroxyflavone IUPAC name: 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one Molecular formula: C15H10O7 Molecular Weight: 302.24 g/mol Category: DNMT inhibitors Targets: DNMT1 | Pathogenic genes: IL1R, NFkB, Ccl8, IKK, STAT3, CD4, CDK2, IL2 Mechanistic genes: MTND4, CDKN2A, PRDX4, DIO2, HSD17B1, MSH2, GSS, COMT, FOS, CRP, NR1I3, PON1 Metabolic genes: Substrate:UGT1A1, UGT1A3, GSTT1, CYP2J2, GSTK1, CYP2C8, CYP1A1, CYP1A2, CYP1B1, GSTA1, CYP19A1 Inhibitor:SULT1E1 Transporter genes: ABCB1, ABCG2 |
| NCT01716637-Phase I |
 | Name: Valproic acid, 2-propylpentanoic acid, depakene, depakine, ergenyl, dipropylacetic acid, mylproin, convulex, myproic acid IUPAC name: 2-propylpentanoic acid Molecular formula: C8H16O2 Molecular Weight: 144.21 g/mol Category: HDAC inhibitors Targets: Class I HDAC; Class II HDAC | Pathogenic genes: CREB1, IL6, LEP, SCN2A, TGFB1, TNF, TRNK Mechanistic genes: ABAT, CDK5, GSK3B, HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, LEP, LEPR, SCNs, SMN2 Metabolic genes: Substrate:CYP2A6 (major), CYP2C9 (major), CYP4B1 (major), CYP1A1 (minor), CYP2B6 (minor), CYP2C19 (minor), CYP2E1 (minor), CYP3A4 (minor), CYP4F2 (minor), ABCB1 (minor), UGT1A4, UGT1A6, UGT1A8, UGT1A10, UGT2B7 Inhibitor: ABCB1, ACADSB, AKR1A1, CYP2C9 (strong), CYP2A6 (moderate), CYP2C19 (moderate), CYP3A4 (moderate), CYP2D6 (weak), HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, UGT1A9, UGT2B1, UGT2B7 Inducer: ABCB1, AKR1C4, CASR, CYP2A6, CYP2B6, CYP3A4, CYP7A1, MAOA, NR1I2, SLC5A5, SLC6A2, SLC12A3, SLC22A16 Transporter genes: ABCB1, ABCC2, ABCG1, ABCG2, SCNs, SLC5A5, SLC6A2, SLC12A3, SLC22A16 Pleiotropic genes: ABL2, AGPAT2, ASL, ASS1, CDK4, CHRNA1, COL1A1, CPS1, CPT1A, DRD4, FMR1, FOS, HBB, HFE, HLA-A, HLA-B, ICAM1, IFNG, IL6, IL10, LEPR, NAGS, NR3C1, OTC, PTGES, STAT3, TGFB1, TNF, TP53 |
| NTC01729598-Phase I NTC00088387-Phase II NTC00071721-Phase III |
 | Name: sodium phenylbutyrate, buphenyl, 4-phenylbutiric acid, 4-phenylbutonoic acid, benzenebutanoic acid, benzenebutyric acid, butyric acid IUPAC name: sodium;4-phenylbutanoate Molecular formula: C10H11NaO2 Molecular Weight: 186.182909 g/mol Category: HDAC inhibitors Targets: Class I HDAC, Class IIa HDAC, Class IIb HDAC | Pathogenic genes: ARG1, ASS1, BCL2, CPS1, NAGS, OTC Mechanistic genes: BCL2, BDNF, EDN1, HDACs, HSPA8, ICAM1, NFKB2, NT3, VCAM1 Metabolic genes: Inhibitor:HDACs Inducer:ARG1, CFTR, CYP2B6, NFKB2 Transporter genes: CFTR Pleiotropic genes: ASL, BDNF, VCAM1 |
| NCT03533257-Phase II NCT02046434-Phase I |
 | Name: Nicotinamide, niacinamide, vitamin PP, aminicotin, nicotinic acid amide, amixicotyn, 3-pyridinecarboxamide, papulex, nicotylamide IUPAC name: pyridine-3-carboxamide Molecular formula: C6H6N2O Molecular Weight: 122.12 g/mol Category: SIRT inhibitors Targets: class III HDAC (SIRT1-7) | Pathogenic genes: IL6, IL8, PTGS2, TNF Mechanistic genes: ARTs, CAT, CLOCK, FOXO3, GPXs, IL6, IL8, PARP1, PTGS2, SIRT1, SOD1, TNF Metabolic genes: Inhibitor:CYP2D6, CYP3A4, CYP2E1, SIRT1-7 Pleiotropic genes: CAT, PARP1 |
| NTC00580931-Phases I, II NTC03061474-Phase II |
 | Name: Resveratrol, trans-resveratrol, 501-36-0, 3,4′,5-trihydroxystilbene, (E)-resveratrol, resvida IUPAC name: 5-[(E)-2-(4-hydroxyphenyl)ethenyl]benzene-1,3-diol Molecular formula: C14H12O3 Molecular Weight: 228.24 g/mol Category: SIRT inhibitors Targets: class III HDAC (SIRT1) | Pathogenic genes: BCL2, CAV1, ESR1, ESR2, GRIN2B, NOS3, PTGS2, TNFRSF10A, TNFRSF10B Mechanistic genes: APP, ATF3, BAX, BAK1, BBC3, BCL2, BCL2L1, BCL2L11, BIRC5, CASP3, CAV1, CFTR, ESR1, ESR2, GRIN1, GRIN2B, HTR3A, NFKB1, NOS3, PMAIP1, PTGS1, PTGS2, SIRT1, SIRT3, SIRT5, SRC, TNFRSF10A, TNFRSF10B, TRPs Metabolic genes: Substrate:CYP1A1, CYP1A2, CYP1B1, CYP2E1, GSTP1, PTGS1, PTGS2 Inhibitor:CYP1A1, CYP1B1, CYP2C9, CYP2D6, CYP3A4, NQO2 Inducer:CYP1A2, SIRT1 Transporter genes: ABCC1, ABCC2, ABCC3, ABCC4, ABCC8, ABCG1, ABCG2, CFTR, TRPs |
| NCT01504854-Phase II NCT00678431-Phase III NCT00743743-withdrown NCT02502253-Phase I |
 | Name: Curcumin, diferuloylmethane, turmeric yellow, turmeric, gelbwurz, kacha haldi, curcuma, haldar, souchet IUPAC name: (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione Molecular formula: C21H20O6 Molecular Weight: 368.38 g/mol Category: HAT inhibitors Targets: HATs | Pathogenic genes: BACE1, CCND1, CDH1, GSK3B, IL1A, IL6, JUN, MSR1, PSEN1, PTGS2, SNCA, SREBF1, TNF Mechanistic genes: AKT1, PRKAs, BACE1, CCND1, CDH1, CDKs, CRM1, CTNNB1, EGF, GSK3B, HDACs, HIF1A, IL1A, IL6, JUN, MMPs, MSR1, NFKB1, NOS2, PDGFRs, PSEN1, PTGS2, SNCA, SOCS1, SOCS3, SREBF1, STAT3, TNF, VEGFA Metabolic genes: Inhibitor:CYP2C8, CYP2C9, EP300 Inducer:CYP2C8, CYP2C9, CYP2D6, CYP3A4 Transporter genes: ABCA1, SNCA Pleiotropic genes: CTNNB1, MSR1 |
| NTC00164749-Phases I, II NTC00099710-Phase II NTC01716637-Phase I NTC01811381-Recruting NTC02114372-Recruting |
 | Name: S-adenosylmethionine, ademetionine, AdoMet, donamet, methioninyladenylate, S-adenosyl-l-methionine, SAM-e IUPAC name: [(3S)-3-amino-3-carboxypropyl]-[[(2S,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl]-methylsulfanium Molecular formula: C15H23N6O5S+ Molecular Weight: 399.45 g/mol Category: HMT inhibitors Targets: HMTs | Pathogenic genes: AKT, ERK, GNMT, MAT1A, PSEN1 Mechanistic genes: AMD1, CAT, CBS, GCLC, GNMT, GSS, NOS2, ROS1, STAT1, TNF Metabolic genes: Substrate:COMT, GNMT, TPMT, SRM Inhibitor:ABCB1, CYP2E1, NOS2 Transporter genes: SLC25A26 Pleiotropic genes: CAT, TNF |
| NTC01320527-Phase II NTC00070941-Phases II, III |
ABAT: 4-aminobutyrate aminotransferase; ABCA1: ATP-binding cassette, subfamily A, member 1; ABCB1: ATP-binding cassette, subfamily B, member 1; ABCC1: ATP-binding cassette, subfamily C, member 1; ABCC2: ATP-binding cassette, subfamily C, member 2; ABCC3: ATP-binding cassette, subfamily C, member 3; ABCC4: ATP-binding cassette, subfamily C, member 4; ABCC8: ATP-binding cassette, subfamily C, member 8; ABCG1: ATP-binding cassette, subfamily G, member 1; ABCG2: ATP-binding cassette, subfamily G, member 2; ABL2: c-abl proto-oncogene 2, non-receptor tyrosine kinase; ACACA: acetyl-CoA carboxylase alpha; ACADSB: acyl-CoA dehydrogenase, short/branched chain; ADORA2A: adenosine 2A2 receptor; AGPAT2: 1-acylglycerol-3-phosphate O-acyltransferase 2; AKR1A1: aldo-keto reductase family 1 member A1; AKT1: AKT serine/threonine kinase 1; ALDH1A1: Aldehyde dehydrogenase 1 family, member A1; AMD1: adenosylmethionine decarboxylase 1; AOX1: aldehyde oxidase 1; APOB: apolipoprotein B; APP: amyloid beta precursor protein; ARG1: arginase 1; ARTS: ADP ribosyltransferases; ASL: argininosuccinate lyase; ASS1: argininosuccinate synthase 1; ATF3: activating transcription factor 3; BACE1: beta-secretase 1; BAK1: BCL2 antagonist/killer 1; BAX: BCL2 associated X, apoptosis regulator; BBC3: BCL2 binding component 3; BCL2: B-cell lymphoma 2, apoptosis regulator; BCL2L1: BCL2 like 1; BCL2L11: BCL2 like 11; BDNF: brain-derived neurotrophic factor; BIRC5: baculoviral IAP repeat containing 5; BMP2: bone morphogenetic protein 2; CASP3: caspase 3; CASR: calcium sensing receptor; CAT: catalase; CAV1: caveolin 1; CBS: cystathionine-beta-synthase; Ccl8: C-C motif chemokine ligand 8; CCND1: cyclin D1; CD36: CD36 molecule; CD4: CD4 molecule; CDH1: cadherin 1; CDK: cyclin-dependent kinase; CDK2: cyclin-dependent kinase 2; CDK4: cyclin-dependent kinase 4; CDK5: cyclin-dependent kinase 5; CDKN2A: cyclin-dependent kinase inhibitor 2A; CDX2: caudal type homeobox 2; CFTR: cystic fibrosis transmembrane conductance regulator; CHRNA1: cholinergic receptor nicotinic alpha 1 subunit; CHRNA7: cholinergic receptor nicotinic alpha 7 subunit; CLOCK: circadian locomotor output cycles kaput; COL1A1: collagen type I alpha 1 chain; COMT: catechol-O-methyltransferase; CPS1: carbamoyl-phosphate synthase 1; CPT1A: carnitine palmitoyltransferase 1A; CREB1: cAMP responsive element binding protein 1; CRM1: exportin CRM1; CRP: C-reactive protein; CTNNB1: catenin beta 1; CYP19A1: cytochrome P450 family 19 subfamily A member 1; CYP1A1: cytochrome P450 family 1 subfamily A member 1; CYP1A2: cytochrome P450 family 1 subfamily A member 2; CYP1B1: cytochrome P450 family 1 subfamily B member 1; CYP2A6: cytochrome P450 family 2 subfamily A member 6; CYP2B6: cytochrome P450 family 2 subfamily B member 6; CYP2C19: cytochrome P450 family 2 subfamily C member 19; CYP2C8: cytochrome P450 family 2 subfamily C member 8; CYP2C9: cytochrome P450 family 2 subfamily C member 9; CYP2D6: cytochrome P450 family 2 subfamily D member 6; CYP2E1: cytochrome P450 family 2 subfamily E member 1; CYP2J2: cytochrome P450 family 2 subfamily J member 2; CYP3A4: cytochrome P450 family 3 subfamily A member 4; CYP4B1: cytochrome P450 family 4 subfamily B member 1; CYP4F2: cytochrome P450 family 4 subfamily F member 2; CYP7A1: cytochrome P450 family 7 subfamily A member 1; DIO2: iodothyronine deiodinase 2; DR4: drought-repressed 4; ECEs: endothelin converting enzymes; EDN1: endothelin 1; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; EP300: E1A binding protein p300; ERCC2: excision repair, complementing defective, in Chinese hamster, 2; ERK: extracellular regulated MAP kinase; ESR1: estrogen receptor 1; ESR2: estrogen receptor 1; FAS: Fas (TNF receptor superfamily member 6); FMR1: fragile X mental retardation 1; FOS: FBJ osteosarcoma oncogene; FOXO3: forkhead box O3; GCLC: glutamate-cysteine ligase catalytic subunit; GNMT: glycine N-methyltransferase; GPX: phage tail protein; GRIN1: glutamate ionotropic receptor NMDA type subunit 1; GRIN2B: glutamate ionotropic receptor NMDA type subunit 2B; GSK3B: glycogen synthase kinase 3 beta; GSS: glutathione synthetase; GSTA1: glutathione S-transferase alpha 1; GSTK1: glutathione S-transferase kappa 1; GSTP1: glutathione S-transferase pi 1; GSTT1: glutathione S-transferase theta 1; HATs: Histone acetyltransferases; HBB: hemoglobin subunit beta; HDACs: histone deacetylases; HDAC 1-9: histone deacetylases 1–9; HFE: hemochromatosis; HIF1A: hypoxia inducible factor 1 alpha subunit; HLA-A: major histocompatibility complex, class I, A; HLA-B: major histocompatibility complex, class I, B; HMTs: Histone Methyl Transferases; HSD17B1: hydroxysteroid 17-beta dehydrogenase 1; HSPA8: heat-shock 70-KD protein 8; HTR3A: 5-hydroxytryptamine receptor 3A; ICAM1: intercellular adhesion molecule 1; IFNG: interferon gamma; IKK: I-kappaB kinase beta; IL10: interleukin 10; IL1A: interleukin 1A; IL1R: interleukin receptor; IL2: interleukin 2; IL6: interleukin 6; IL8: interleukin 8; IRS1: insulin receptor substrate 1; JUN: Jun proto-oncogene, AP-1 transcription factor subunit; LEP: leptin; LEPR: leptin receptor; MAOA: monoamine oxidase A; MAT1A: methionine adenosyltransferase 1A; MMP: matrix metalloproteinase; MSH2: mutS homolog 2; MSR1: macrophage scavenger receptor 1; MTHF: 5,10 methylenetetrahydrofolate; MTHFR: 5,10 methylenetetrahydrofolate receptor; MTND4: mitochondrially encoded NADH dehydrogenase 4; NAGS: N-acetylglutamate synthase; NAT2: N-actyltransferase 2; NFkB: nuclear factor kappa-B; NFkB1: nuclear factor kappa B subunit 1; NFkB2: nuclear factor kappa B subunit 2; NOS2: nitric oxide synthase 2; NOS3: nitric oxide synthase 3; NQO2: N-ribosyldihydronicotinamide:quinone reductase 2; NR1I2: nuclear receptor subfamily 1 group I member 2; NR1I3: nuclear receptor subfamily 1 group I member 3; NR3C1: nuclear receptor subfamily 3 group C member 1; NT3: neurotrophin 3; OTC: ornithine carbamoyltransferase; PARP1: poly(ADP-ribose) polymerase 1; PDGFR: platelet derived growth factor receptor; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PMAIP1: phorbol-12-myristate-13-acetate-induced protein 1; PON1: paraoxonase 1; PPAR: peroxisome proliferator-activated receptor; PRDX4: peroxiredoxin 4; PRKA: serine protein kinase PrkA; PSEN1: presenilin 1; PTGS1: prostaglandin-endoperoxide synthase 1; PTGS2: prostaglandin-endoperoxide synthase 2; ROS1: ROS proto-oncogene 1, receptor tyrosine kinase; SAMe: S-adenosylmethionine; SCD: stearoyl-CoA desaturase; SCN2A: sodium voltage-gated channel alpha subunit 2; SIRT1-7: sirtuins 1–7; SLC12A3: solute carrier family 12 member 3; SLC22A16: solute carrier family 22 member 16; SLC25A26: solute carrier family 25 member 26; SLC27A4: solute carrier family 27 member 4; SLC5A1: solute carrier family 5 member 1; SLC5A5: solute carrier family 5 member 5; SLC6A2: solute carrier family 6 member 2; SLC19A1: solute carrier family 19 member 1; SLC22A8: solute carrier family 22 member 8; SLC28A2: solute carrier family 28 member 8; SLCO1B1: solute carrier organic anion transporter family member 1B1; SLCO1B3: solute carrier organic anion transporter family member 1B3; SMN2: survival of motor neuron 2, centromeric; SNCA: synuclein alpha; SOCS1: suppressor of cytokine signaling 1; SOCS3: suppressor of cytokine signaling 3; SOD1: superoxide dismutase 1; SOD3: superoxide dismutase 3; SRC: SRC proto-oncogene, non-receptor tyrosine kinase; SREBF1: sterol regulatory element binding transcription factor 1; SRM: spermidine synthase; STAT1: signal transducer and activator of transcription 1; STAT3: signal transducer and activator of transcription 3; SULT1E1: sulfotransferase family 1E member 1; TGFB1: transforming growth factor beta 1; THF: Tetrahydrofolate; TNF: tumor necrosis factor; TNFRSF10A: TNF receptor superfamily member 10A; TNFRSF10B: TNF receptor superfamily member 10B; TP53: tumor protein p53; TPMT: thiopurine S-methyltransferase; TRNK: mitochondrially encoded tRNA lysine; UGT1A1: UDP glucuronosyltransferase family 1 member A1; UGT1A10: UDP glucuronosyltransferase family 1 member A10; UGT1A3: UDP glucuronosyltransferase family 1 member A3; UGT1A4: UDP glucuronosyltransferase family 1 member A4; UGT1A6: UDP glucuronosyltransferase family 1 member A6; UGT1A8: UDP glucuronosyltransferase family 1 member A8; UGT1A9: UDP glucuronosyltransferase family 1 member A9; UGT2B1: UDP glucuronosyltransferase family 2 member B1; UGT2B7: UDP glucuronosyltransferase family 2 member B7; VCAM1: vascular cell adhesion molecule 1; VEGFA: vascular endothelial growth factor A.
Table 2.
Epigenetic modifications in genes involved in the pharmacogenomic response to drugs associated with the onset and/or progression of Alzheimer’s and Parkinson’s diseases.
Table 2.
Epigenetic modifications in genes involved in the pharmacogenomic response to drugs associated with the onset and/or progression of Alzheimer’s and Parkinson’s diseases.
| Category | Gene | Locus | OMIM | Pathology | Epigenetic changes |
|---|---|---|---|---|---|
| Phase I Drug Metabolizers | CYP2E1 | 10q26.3 | 124,040 | Parkinson’s Disease | Hypomethylated Up-regulated mRNA |
| Phase II Drug Metabolizers | GSTT1 | 22q11.23 | 600,436 | Parkinson’s Disease | Hypomethylated Upregulated mRNA |
| GSTTP1 | 22q11.23 | 600,436 | Parkinson’s Disease | Hypermethylated Downregulated mRNA | |
| GSTTP2 | 22q11.23 | 600,436 | Parkinson’s Disease | Hypermethylated Downregulated mRNA | |
| Phase III Drug Transporters | ABCA1 | 9q31.1 | 600,046 | Alzheimer’s Disease | Hypermethylated Downregulated mRNA |
| ABCB1 | 7q21.12 | 171,050 | Alzheimer’s Disease | Hypermethylated Downregulated mRNA | |
| ABCG2 | 4q22.1 | 603,756 | Parkinson’s Disease | Hypermethylated Downregulated mRNA | |
| ABCA3 | 16p13.3 | 601,615 | Parkinson’s Disease | Hypomethylated Upregulated mRNA | |
| ABCA7 | 19p13.3 | 605,414 | Alzheimer’s Disease | Hypomethylated Upregulated mRNA | |
| SLC12A5 | 20q13.12 | 606,726 | Parkinson’s Disease | Hypomethylated Upregulated mRNA | |
| SLC24A4 | 14q32.12 | 609,840 | Alzheimer’s Disease | Hypomethylated Upregulated mRNA | |
| SLC25A24 | 1p13.3 | 608,744 | Parkinson’s Disease | Hypomethylated Upregulated mRNA |
ABCA1: ATP-binding cassette, subfamily A (ABCA), member 1; ABCA3: ATP-binding cassette, subfamily A (ABCA), member 3; ABCA7: ATP binding cassette subfamily A member 7; ABCB1: ATP-binding cassette, subfamily B (ABCB), member 1; ABCG2: ATP-binding cassette, subfamily G (ABCG), member 2; CYP2E1: cytochrome P450 family 2 subfamily E member 1; GSTT1: Glutathione S-transferase theta 1; GSTTP1: Glutathione S-transferase theta pseudogene 1; GSTTP2: Glutathione S-transferase theta pseudogene 2; SLC12A5: Solute carrier family 12 (potassium/chloride transporter), member 5; SLC24A4: solute carrier family 24 member 4; SLC25A24: solute carrier family 25 member 24.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Teijido, O.; Cacabelos, R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2018, 19, 3199. https://doi.org/10.3390/ijms19103199
AMA Style
Teijido O, Cacabelos R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. International Journal of Molecular Sciences. 2018; 19(10):3199. https://doi.org/10.3390/ijms19103199
Chicago/Turabian StyleTeijido, Oscar, and Ramón Cacabelos. 2018. "Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases" International Journal of Molecular Sciences 19, no. 10: 3199. https://doi.org/10.3390/ijms19103199
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.