The Tissue-Engineered Human Psoriatic Skin Substitute: A Valuable In Vitro Model to Identify Genes with Altered Expression in Lesional Psoriasis

 ,
, 


Abstract
:1. Introduction
2. Results
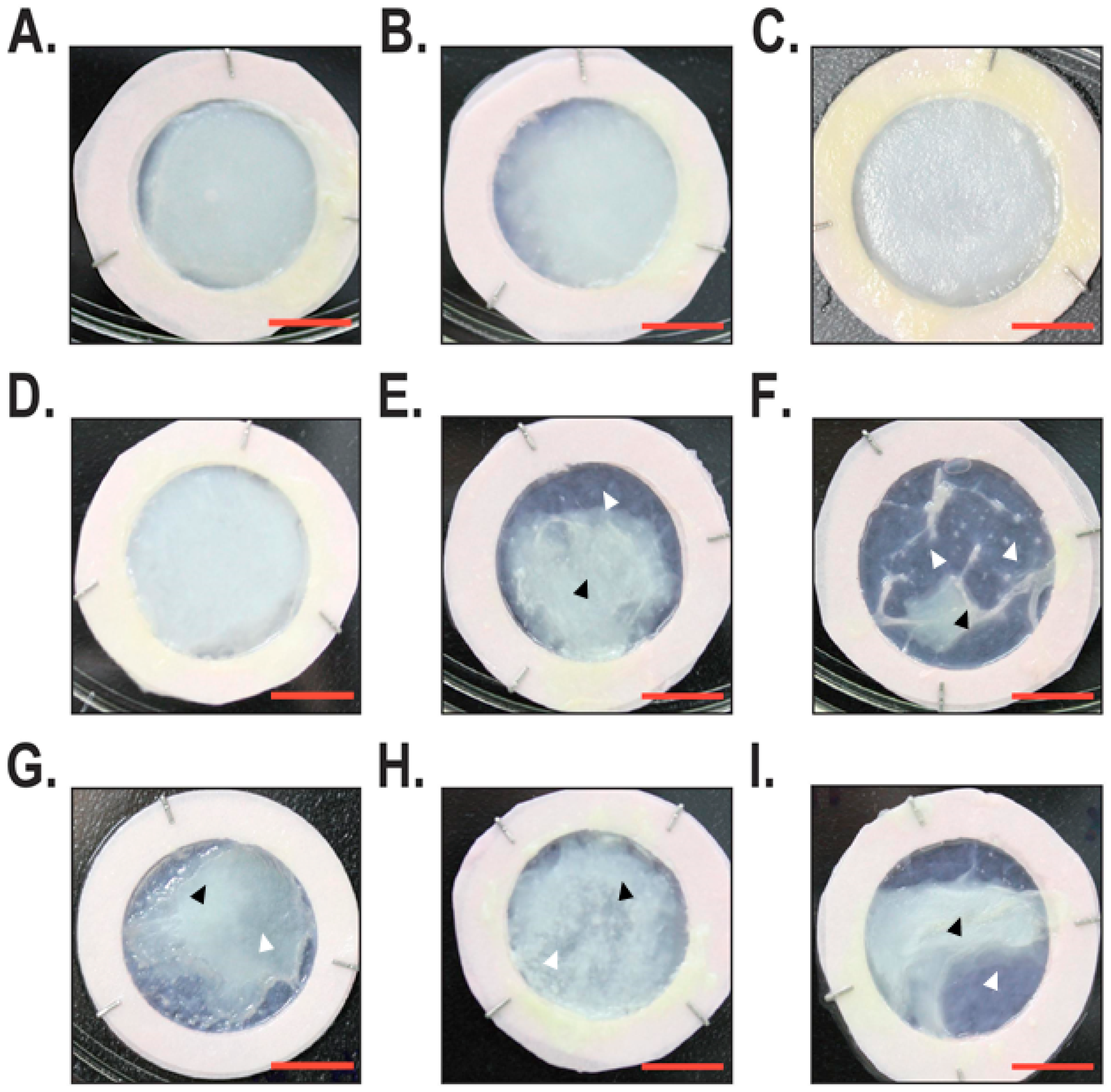
2.1. Macroscopic and Histological Analyses
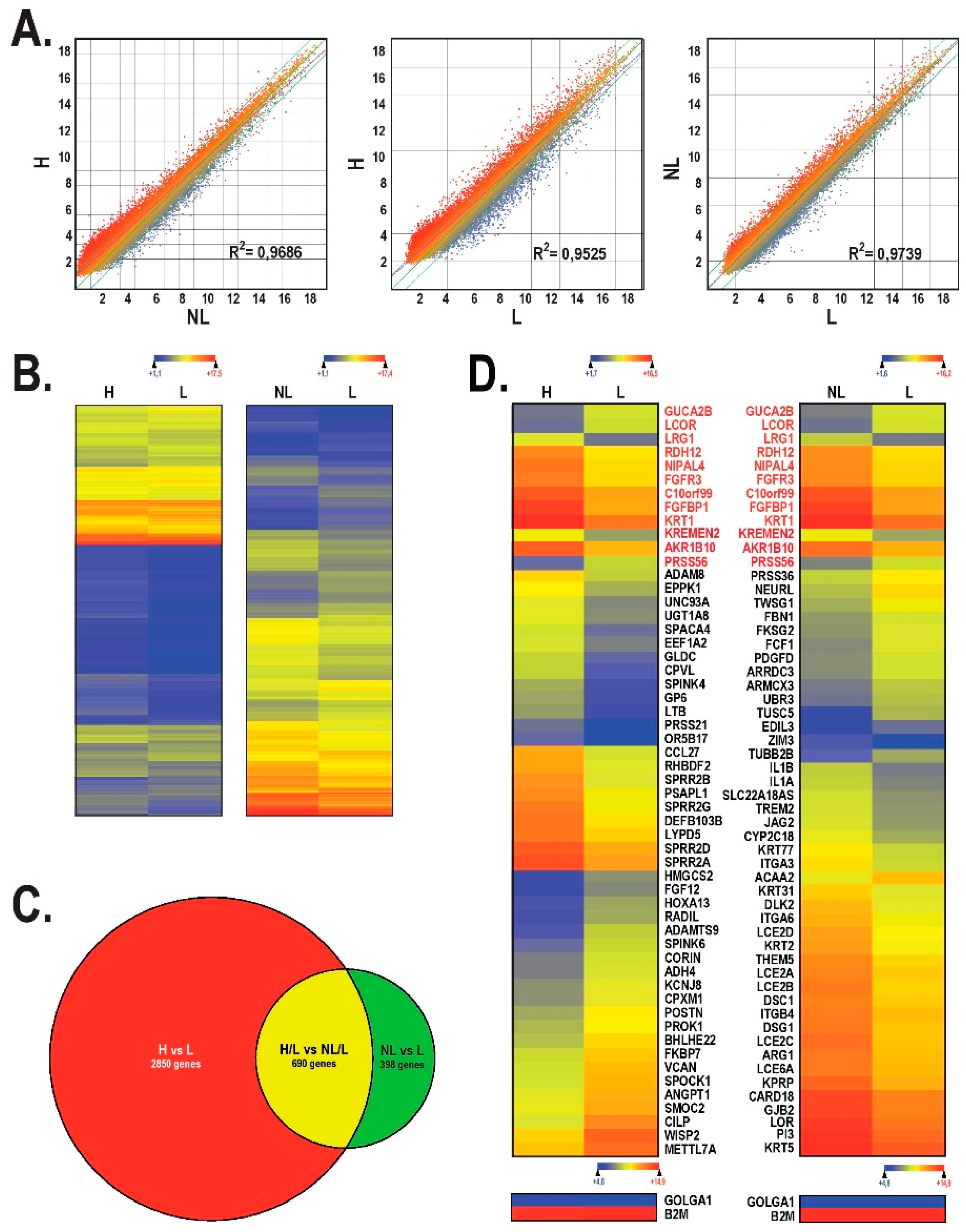
2.2. Gene Profiling Analysis of the Most Deregulated Genes between Healthy, Lesional and Non-Lesional Skin Substitutes
2.3. Alteration in Cytokines, Chemokines and Growth Factors Gene Expression in Healthy vs. Lesional Skin Substitutes
2.4. Gene Ontology Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Production of Tissue-Engineered Substitutes
4.3. Gene Expression Profiling
4.4. Histological Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and therapy of psoriasis. Nature 2007, 445, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.S. Psoriasis. Lancet 1997, 350, 349–353. [Google Scholar] [CrossRef]
- Smith, R.L.; Warren, R.B.; Griffiths, C.E.; Worthington, J. Genetic susceptibility to psoriasis: An emerging picture. Genome Med. 2009, 1, 72. [Google Scholar] [CrossRef] [PubMed]
- Henseler, T.; Christophers, E. Psoriasis of early and late onset: Characterization of two types of psoriasis vulgaris. J. Am. Acad. Dermatol. 1985, 13, 450–456. [Google Scholar] [CrossRef]
- Boyd, A.S.; Menter, A. Erythrodermic psoriasis. J. Am. Acad. Dermatol. 1989, 21, 985–991. [Google Scholar] [CrossRef]
- Menter, A.; Barker, J.N. Psoriasis in practice. Lancet 1991, 338, 231–234. [Google Scholar] [CrossRef]
- Naldi, L.; Gambini, D. The clinical spectrum of psoriasis. Clin. Dermatol. 2007, 25, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.; Korman, N.J.; Gelfand, J.M.; Lim, H.W.; Elmets, C.A.; Feldman, S.R.; Gottlieb, A.B.; Koo, J.Y.; Lebwohl, M.; Leonardi, C.L.; et al. Research gaps in psoriasis: Opportunities for future studies. J. Am. Acad. Dermatol. 2014, 70, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, T.D.; Schupp, C.W.; Armstrong, A.W. Psoriasis prevalence among adults in the United States. J. Am. Acad. Dermatol. 2014, 70, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Grozdev, I.; Korman, N.; Tsankov, N. Psoriasis as a systemic disease. Clin. Dermatol. 2014, 32, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.A.; Gupta, A.K. Psychiatric and Psychological Co-Morbidity in Patients with Dermatologic Disorders. J. Am. Acad. Dermatol. 2003, 4, 833–842. [Google Scholar] [CrossRef]
- Weigle, N.; Mcbane, S. Psoriasis. Am. Fam. Physician 2013, 87, 626–633. [Google Scholar] [PubMed]
- Nickoloff, B.J.; Qin, J.Z.; Nestle, F.O. Immunopathogenesis of psoriasis. Clin. Rev. Allergy Immunol. 2007, 33, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Zollner, T.M.; Renz, H.; Igney, F.H.; Asadullah, K. Animal models of T-cell-mediated skin diseases. Bioessays 2004, 26, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Schon, M.P. Animal Models of Psoriasis—What Can We Learn from Them? J. Investig. Dermatol. 1999, 112, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Jean, J.; Pouliot, R. In vivo and In vitro Models of Psoriasis. In Tissue Engineering; Eberli, D., Ed.; InTech Publishers: Novi Sad, Serbia, 2010; pp. 359–382. ISBN 978-953-307-079-7. [Google Scholar]
- Danilenko, D.M. Review paper: Preclinical models of psoriasis. Vet. Pathol. 2008, 45, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, H.; Yamanaka, K.; Konishi, H.; Murakami, T. Animal models of psoriasis and pustular psoriasis. Arch. Dermatol. Res. 2003, 295 (Suppl. 1), S67–S68. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Dauth, S.; Hassell, B.; Herland, A.; Jain, A.; Jang, K.J.; Karalis, K.; Kim, H.J.; MacQueen, L.; Mahmoodian, R.; et al. Engineered in vitro disease models. Annu. Rev. Pathol. 2015, 10, 195–262. [Google Scholar] [CrossRef] [PubMed]
- Jean, J.; Garcia-Perez, M.E.; Pouliot, R. Psoriatic Skin Models: A Need for the Pharmaceutical Industry. In Psoriasis; Soung, J., Ed.; InTech Publishers: Novi Sad, Serbia, 2011; pp. 47–62. ISBN 978-953-307-878-6. [Google Scholar]
- Jean, J.; Lapointe, M.; Soucy, J.; Pouliot, R. Development of an in vitro psoriatic skin model by tissue engineering. J. Dermatol. Sci. 2009, 53, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Jean, J.; Leroy, M.; Duque-Fernandez, A.; Bernard, G.; Soucy, J.; Pouliot, R. Characterization of a psoriatic skin model produced with involved or uninvolved cells. J. Tissue Eng. Regen. Med. 2012, 9, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Guerard, S.; Fortin, M.M.; Rusu, D.; Soucy, J.; Poubelle, P.E.; Pouliot, R. Pathological crosstalk in vitro between T lymphocytes and lesional keratinocytes in psoriasis: Necessity of direct cell-to-cell contact. Lab. Investig. 2012, 92, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Ayata, R.E.; Bouhout, S.; Auger, M.; Pouliot, R. Study of in vitro capillary-like structures in psoriatic skin substitutes. Biores. Open Access 2014, 3, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Yamasaki, K.; Muhleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin antimicrobial peptide LL-37 in psoriasis enables keratinocyte reactivity against TLR9 ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, J.E.; Ding, J.; Johnston, A.; Tejasvi, T.; Guzman, A.M.; Nair, R.P.; Voorhees, J.J.; Abecasis, G.R.; Elder, J.T. Assessment of the psoriatic transcriptome in a large sample: Additional regulated genes and comparisons with in vitro models. J. Investig. Dermatol. 2010, 130, 1829–1840. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.; Auger, M.; Soucy, J.; Pouliot, R. Physical characterization of the stratum corneum of an in vitro psoriatic skin model by ATR-FTIR and Raman spectroscopies. Biochim. Biophys. Acta 2007, 1770, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Lebwohl, M. Duration of remission of psoriasis therapies. J. Am. Acad. Dermatol. 1999, 41, 51–59. [Google Scholar] [CrossRef]
- Ragaz, A.; Ackerman, A.B. Evolution, maturation, and regression of lesions of psoriasis. Am. J. Dermatopathol. 1979, 1, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Niehues, H.; van den Bogaard, E.H. Past, present and future of in vitro 3D reconstructed inflammatory skin models to study psoriasis. Exp. Dermatol. 2018, 27, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, A.; Romanelli, M.; Panduri, S.; Donetti, E.; Prignano, F. Relevance of in vitro 3-D skin models in dissecting cytokine contribution to psoriasis pathogenesis. Histol. Histopathol. 2017, 32, 893–898. [Google Scholar] [PubMed]
- Zhou, X.; Krueger, J.G.; Kao, M.C.; Lee, E.; Du, F.; Menter, A.; Wong, W.H.; Bowcock, A.M. Novel mechanisms of T-cell and dendritic cell activation revealed by profiling of psoriasis on the 63,100-element oligonucleotide array. Physiol. Genom. 2003, 13, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oestreicher, J.; Walters, I.B.; Kikuchi, T.; Gilleaudeau, P.; Surette, J.; Schwertschlag, U.; Dorner, A.J.; Krueger, J.G.; Trepicchio, W.L. Molecular classification of psoriasis disease-associated genes through pharmacogenomic expression profiling. Pharmacogenomics 2001, 1, 272–287. [Google Scholar] [CrossRef] [Green Version]
- Henno, A.; Blacher, S.; Lambert, C.; Colige, A.; Seidel, L.; Noel, A.; Lapiere, C.; de la Brassinne, M.; Nusgens, B.V. Altered expression of angiogenesis and lymphangiogenesis markers in the uninvolved skin of plaque-type psoriasis. Br. J. Dermatol. 2009, 160, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Henno, A.; Blacher, S.; Lambert, C.A.; Deroanne, C.; Noel, A.; Lapiere, C.; de la Brassinne, M.; Nusgens, B.V.; Colige, A. Histological and transcriptional study of angiogenesis and lymphangiogenesis in uninvolved skin, acute pinpoint lesions and established psoriasis plaques: An approach of vascular development chronology in psoriasis. J. Dermatol. Sci. 2010, 57, 162–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickoloff, B.J.; Schroder, J.M.; von den Driesch, P.; Raychaudhuri, S.P.; Farber, E.M.; Boehncke, W.H.; Morhenn, V.B.; Rosenberg, E.W.; Schon, M.P.; Holick, M.F. Is psoriasis a T-cell disease? Exp. Dermatol. 2000, 9, 359–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.G.; Krane, J.F.; Carter, D.M.; Gottlieb, A.B. Role of growth factors, cytokines, and their receptors in the pathogenesis of psoriasis. J. Investig. Dermatol. 1990, 94 (Suppl. 6), 135s–140s. [Google Scholar] [CrossRef] [PubMed]
- McColl, A.; Thomson, C.A.; Nerurkar, L.; Graham, G.J.; Cavanagh, J. TLR7-mediated skin inflammation remotely triggers chemokine expression and leukocyte accumulation in the brain. J. Neuroinflamm. 2016, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Riis, J.L.; Johansen, C.; Vestergaard, C.; Bech, R.; Kragballe, K.; Iversen, L. Kinetics and differential expression of the skin-related chemokines CCL27 and CCL17 in psoriasis, atopic dermatitis and allergic contact dermatitis. Exp. Dermatol. 2011, 20, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Krueger, J.G.; Li, K.; Jabbari, A.; Brodmerkel, C.; Lowes, M.A.; Suarez-Farinas, M. Meta-analysis derived (MAD) transcriptome of psoriasis defines the “core” pathogenesis of disease. PLoS ONE 2012, 7, e44274. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Russell, C.B.; Martin, D.A.; Towne, J.E.; Krueger, J.G. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013, 34, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swindell, W.R.; Johnston, A.; Voorhees, J.J.; Elder, J.T.; Gudjonsson, J.E. Dissecting the psoriasis transcriptome: Inflammatory- and cytokine-driven gene expression in lesions from 163 patients. Genomics 2013, 14, 527. [Google Scholar] [CrossRef] [PubMed]
- Motta, S.; Monti, M.; Sesana, S.; Caputo, R.; Carelli, S.; Ghidoni, R. Ceramide composition of the psoriatic scale. Biochim. Biophys. Acta 1993, 1182, 147–151. [Google Scholar] [CrossRef]
- Van Smeden, J.; Janssens, M.; Gooris, G.S.; Bouwstra, J.A. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim. Biophys. Acta 2014, 1841, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Farwanah, H.; Raith, K.; Neubert, R.H.; Wohlrab, J. Ceramide profiles of the uninvolved skin in atopic dermatitis and psoriasis are comparable to those of healthy skin. Arch Dermatol. Res. 2005, 296, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ishii, I.; Kingsbury, M.A.; Chun, J. Lysophosphatidic acid as a novel cell survival/apoptotic factor. Biochim. Biophys. Acta 2002, 1585, 108–113. [Google Scholar] [CrossRef]
- Capestrano, M.; Mariggio, S.; Perinetti, G.; Egorova, A.V.; Iacobacci, S.; Santoro, M.; Di Pentima, A.; Iurisci, C.; Egorov, M.V.; Di Tullio, G.; et al. Cytosolic phospholipase A(2)epsilon drives recycling through the clathrin-independent endocytic route. J. Cell Sci. 2014, 127, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Ghosh, M.; Spencer, D.M.; Leslie, C.C. Enzymatic properties of human cytosolic phospholipase A(2)gamma. J. Biol. Chem. 2002, 277, 29526–29536. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Tucker, D.E.; Burchett, S.A.; Leslie, C.C. Properties of the Group IV phospholipase A2 family. Prog. Lipid Res. 2006, 45, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.K.; Cho, H.R.; Ju, W.C.; Cho, Y.; Kim, N.L. A Study on Altered Expression of Serine Palmitoyltransferase and Ceramidase in Psoriatic Skin Lesion. J. Korean Med. Sci. 2007, 22, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, A.; Michalak-Stoma, A.; Chodorowska, G.; Szepietowski, J.C. Lipid disturbances in psoriasis: An update. Mediat. Inflamm. 2010, 2010, 535612. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/gene/667 (accessed on 13 March 2018).
- Sonnenberg, A.; Liem, R.K. Plakins in development and disease. Exp. Cell Res. 2007, 313, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.; Begum, R.; Fong, K.; Pourreyron, C.; South, A.P.; McGrath, J.A.; Parsons, M. BPAG1-e restricts keratinocyte migration through control of adhesion stability. J. Investig. Dermatol. 2014, 134, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, A.; Boyer, J.G.; Kothary, R. Cellular and Molecular Biology of Neuronal Dystonin. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Elsevier: New York, NY, USA, 2013; pp. 88–95. [Google Scholar]
- Litjens, S.H.; Koster, J.; Kuikman, I.; van Wilpe, S.; de Pereda, J.M.; Sonnenberg, A. Specificity of binding of the plectin actin-binding domain to beta 4 integrin. Mol. Biol. Cell 2003, 14, 4039–4050. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Takahashi, K.; Kanaoka, M.; Okawa, T.; Tateishi, C.; Yasukochi, A.; Ishii, N.; Li, X.; Hashimoto, T.; Aihara, M. Case of subepidermal autoimmune bullous disease with psoriasis vulgaris reacting to both BP180 C-terminal domain and laminin gamma-1. J. Dermatol. 2015, 42, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Stoica, L.E.; Patrascu, V.; Dascalu, R.C.; Ciurea, M.E. Bullous pemphigoid associated with psoriasis, breast cancer and Parkinson’s disease. Curr. Health Sci. J. 2014, 40, 62–66. [Google Scholar] [PubMed]
- Nakayama, C.; Iwata, H.; Haga, N.; Hamade, Y.; Mizuno, O.; Nishie, W.; Shimizu, H. The different intensity of autoantibody deposits in bullous pemphigoid associated with psoriasis vulgaris. Eur. J. Dermatol. 2014, 25, 70–71. [Google Scholar]
- Bergboer, J.G.; Dulak, M.G.; van Vlijmen-Willems, I.M.; Jonca, N.; van Wijk, E.; Hendriks, W.J.; Zeeuwen, P.L.; Schalkwijk, J. Analysis of protein-protein interaction between late cornified envelope proteins and corneodesmosin. Exp. Dermatol. 2014, 23, 769–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cid, R.; Riveira-Munoz, E.; Zeeuwen, P.L.; Robarge, J.; Liao, W.; Dannhauser, E.N.; Giardina, E.; Stuart, P.E.; Nair, R.; Helms, C.; et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 2009, 41, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohl, D. Expression Patterns of Loricrin in Dermatological Disorders. Am. J. Dermatopathol. 1993, 15, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.; Rao, K.S.; Basavaraj, K.H. A comprehensive review of biomarkers in psoriasis. Clin. Exp. Dermatol. 2009, 34, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Tobin, D.J.; Theoharides, T.C.; Rivier, J. Key role of CRF in the skin stress response system. Endocr. Rev. 2013, 34, 827–884. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell Biol. 2012, 212, 1–115. [Google Scholar]
- Slominski, A.T.; Manna, P.R.; Tuckey, R.C. On the role of skin in the regulation of local and systemic steroidogenic activities. Steroids 2015, 103, 72–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef] [PubMed]
- Skobowiat, C.; Postlethwaite, A.E.; Slominski, A.T. Skin Exposure to Ultraviolet B Rapidly Activates Systemic Neuroendocrine and Immunosuppressive Responses. Photochem. Photobiol. 2017, 93, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Brozyna, A.A.; Tuckey, R.C. Cutaneous Glucocorticoidogenesis and Cortisol Signaling Are Defective in Psoriasis. J. Investig. Dermatol. 2017, 137, 1609–1611. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Luger, T.; Paus, R.; Solomon, S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev. 2000, 80, 979–1020. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Szczesniewski, A.; Wortsman, J. Cultured human dermal fibroblasts do produce cortisol. J. Investig. Dermatol. 2006, 126, 1177–1178. [Google Scholar] [CrossRef] [PubMed]
- Hannen, R.F.; Michael, A.E.; Jaulim, A.; Bhogal, R.; Burrin, J.M.; Philpott, M.P. Steroid synthesis by primary human keratinocytes; implications for skin disease. Biochem. Biophys. Res. Commun. 2011, 404, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Semak, I.; Sweatman, T.; Wortsman, J. CRH stimulates POMC activity and corticosterone production in dermal fibroblasts. J. Neuroimmunol. 2005, 162, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Szczesniewski, A.; Semak, I.; Kaminski, J.; Sweatman, T.; Wortsman, J. CRH stimulation of corticosteroids production in melanocytes is mediated by ACTH. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E701–E706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vukelic, S.; Stojadinovic, O.; Pastar, I.; Rabach, M.; Krzyzanowska, A.; Lebrun, E.; Davis, S.C.; Resnik, S.; Brem, H.; Tomic-Canic, M. Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J. Biol. Chem. 2011, 286, 10265–10275. [Google Scholar] [CrossRef] [PubMed]
- Jean, J.; Garci-Perez, M.E.; Pouliot, R. Bioengineered Skin: The Self-Assembly Approach. J. Tissue Sci. Eng. 2011, 3, 001. [Google Scholar] [CrossRef]
- Pouliot, R.; Larouche, D.; Auger, F.A.; Juhasz, J.; Xu, W.; Li, H.; Germain, L. Reconstructed human skin produced in vitro and grafted on athymic mice. Transplantation 2002, 73, 1751–1757. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Linear Signal | Fold Change (L/H) | |
|---|---|---|---|---|
| Healthy (H) | Lesional (L) | |||
| IL1R2 | Interleukin-1 receptor type 2, soluble form | 21.111 | 52.487 | 2.486 up |
| CXCL13 | C-X-C motif chemokine 13 | 8.956 | 60.844 | 6.793 up |
| CXCL14 | C-X-C motif chemokine 14 | 5475.355 | 26,650.308 | 4.867 up |
| CAMP | Cathelicidin antimicrobial peptide | 26.904 | 65.127 | 2.416 up |
| EPO | Erythropoietin | 23.081 | 94.42 | 4.090 up |
| INHBA | Inhibin beta A chain | 132.158 | 293.07 | 2.217 up |
| TNFSF9 | Tumor necrosis factor ligand superfamily member 9 | 110.858 | 332.436 | 2.998 up |
| CXCL2 | C-X-C motif chemokine 2 | 334.366 | 158.747 | 2.106 down |
| CCL20 | C-C motif chemokine 20 | 135.671 | 48.3 | 2.808 down |
| CXCL1 | Growth-regulated alpha protein | 1739.554 | 656.318 | 2.650 down |
| LTB | Lymphotoxin-beta | 57.051 | 16.161 | 3.530 down |
| TNFRSF10A | Tumor necrosis factor receptor superfamily member 10A | 179.232 | 84.848 | 2.112 down |
| IL24 | Interleukin-24 | 164.625 | 67.68 | 2.432 down |
| CCL27 | C-C motif chemokine 27 | 4802.716 | 731.651 | 6.564 down |
| IL15 | Interleukin-15 | 368.781 | 174.554 | 2.112 down |
| Pathways | Sample Frequency (n) | p-Value |
|---|---|---|
| Healthy against lesional condition | ||
| Keratinization (GO: 0031424) | 4 | 3.625 × 10−4 |
| Isoprenoid metabolic process (GO: 0006720) | 5 | 5.922 × 10−4 |
| Retinoid metabolic process (GO: 0001523) | 4 | 2.321 × 10−3 |
| Diterpenoid metabolic process (GO: 0016101) | 4 | 3.077 × 10−3 |
| Biological process (GO: 0008150) | 50 | 4.007 × 10−3 |
| Keratinocyte differentiation (GO: 0030216) | 4 | 5.312 × 10−3 |
| Non-lesional against lesional condition | ||
| Skin development (GO: 0043588) | 13 | 1.151 × 10−11 |
| Keratinization (GO: 0031424) | 5 | 7.952 × 10−6 |
| Epidermis development (GO: 0008544) | 8 | 1.088 × 10−5 |
| Single-organism developmental process (GO: 0044767) | 27 | 2.897 × 10−5 |
| Organ development (GO: 0048513) | 20 | 2.984 × 10−5 |
| Developmental process (GO: 0032502) | 27 | 3.619 × 10−5 |
| Gene | Protein Name | Linear Signals | Fold Change | ||||
|---|---|---|---|---|---|---|---|
| H | NL | L | H vs. NL | H vs. L | NL vs. L | ||
| AREG | Amphiregulin | 4556.114 | 3649.748 | 1740.864 | 1.248 down | 2.617 down | 2.096 down |
| CCL27 | C-C motif chemokine 27 | 4802.716 | 1196.792 | 327.991 | 4.012 down | 14.646 down | 3.649 down |
| CERS3 | Ceramide synthase 3 | 1205.298 | 1076.517 | 545.522 | 1.119 down | 2.209 down | 1.973 down |
| COL10A1 | Collagen alpha-1(X) chain, Collagen type X alpha 1 | 40.35 | 153.551 | 223.581 | 3.805 up | 5.540 up | 1.456 up |
| COL4A1 | Collagen alpha-1(IV) chain | 624.384 | 1325.645 | 1377.716 | 2.123 up | 2.206 up | 1.039 up |
| COL4A2 | Collagen type IV alpha 2, Collagen alpha-2(IV) chain | 3008.066 | 7008.3 | 7538.532 | 2.329 up | 2.506 up | 1.075 up |
| COL5A3 | Collagen alpha-3(V) chain | 81.195 | 201.437 | 272.105 | 2.480 up | 3.351 up | 1.350 up |
| COL6A3 | Collagen alpha-3(VI) chain, Uncharacterized protein | 216.945 | 264.225 | 666.849 | 1.217 up | 3.073 up | 2.523 up |
| COL8A1 | Collagen alpha-1(VIII) chain | 179.866 | 373.282 | 419.755 | 2.075 up | 2.333 up | 1.124 up |
| COL9A3 | Collagen alpha-3(IX) chain; Collagen, type IX, alpha 3 | 672.004 | 561.211 | 177.532 | 1.197 down | 3.785 down | 3.161 down |
| CXCL10 | C-X-C motif chemokine 10 | 18.223 | 16.471 | 6.664 | 1.106 down | 2.734 down | 2.471 down |
| DMRTA1 | Doublesex- and mab-3-related transcription factor A1 | 149.171 | 72.609 | 29.644 | 2.054 down | 5.032 down | 2.449 down |
| DST | Dystonin | 2666.323 | 2193.123 | 654.854 | 1.215 down | 4.071 down | 3.349 down |
| FABP6 | Gastrotropin | 396.131 | 270.887 | 105.903 | 1.462 down | 3.740 down | 2.557 down |
| LPL | Lipoprotein lipase | 354.755 | 560.341 | 1138.306 | 1.579 up | 3.208 up | 2.031 up |
| NOD2 | Nucleotide-binding oligomerization domain-containing protein 2 | 1002.491 | 893.358 | 445.888 | 1.122 down | 2.248 down | 2.003 down |
| PIK3R2 | Phosphatidylinositol 3-kinase regulatory subunit beta | 2620.755 | 5460.444 | 6354.303 | 2.083 up | 2.424 up | 1.163 up |
| PLA2G4C | Cytosolic phospholipase A2 gamma | 287.492 | 73.724 | 73.553 | 3.899 down | 3.908 down | 1.002 down |
| PLA2G4E | Cytosolic phospholipase A2 epsilon | 260.291 | 205.142 | 71.176 | 1.268 down | 3.657 down | 2.882 down |
| PNPLA5 | Patatin-like phospholipase domain-containing protein 5 | 15.78 | 81.347 | 98.806 | 5.155 up | 6.261 up | 1.214 up |
| POSTN | Periostin | 85.28 | 478.01 | 1044.007 | 5.605 up | 12.242 up | 2.184 up |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rioux, G.; Pouliot-Bérubé, C.; Simard, M.; Benhassine, M.; Soucy, J.; Guérin, S.L.; Pouliot, R. The Tissue-Engineered Human Psoriatic Skin Substitute: A Valuable In Vitro Model to Identify Genes with Altered Expression in Lesional Psoriasis. Int. J. Mol. Sci. 2018, 19, 2923. https://doi.org/10.3390/ijms19102923
Rioux G, Pouliot-Bérubé C, Simard M, Benhassine M, Soucy J, Guérin SL, Pouliot R. The Tissue-Engineered Human Psoriatic Skin Substitute: A Valuable In Vitro Model to Identify Genes with Altered Expression in Lesional Psoriasis. International Journal of Molecular Sciences. 2018; 19(10):2923. https://doi.org/10.3390/ijms19102923
Chicago/Turabian StyleRioux, Geneviève, Claudia Pouliot-Bérubé, Mélissa Simard, Manel Benhassine, Jacques Soucy, Sylvain L. Guérin, and Roxane Pouliot. 2018. "The Tissue-Engineered Human Psoriatic Skin Substitute: A Valuable In Vitro Model to Identify Genes with Altered Expression in Lesional Psoriasis" International Journal of Molecular Sciences 19, no. 10: 2923. https://doi.org/10.3390/ijms19102923