Exploring the Role of Fallopian Ciliated Cells in the Pathogenesis of High-Grade Serous Ovarian Cancer


Abstract
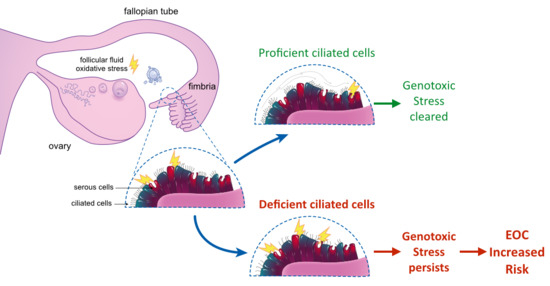
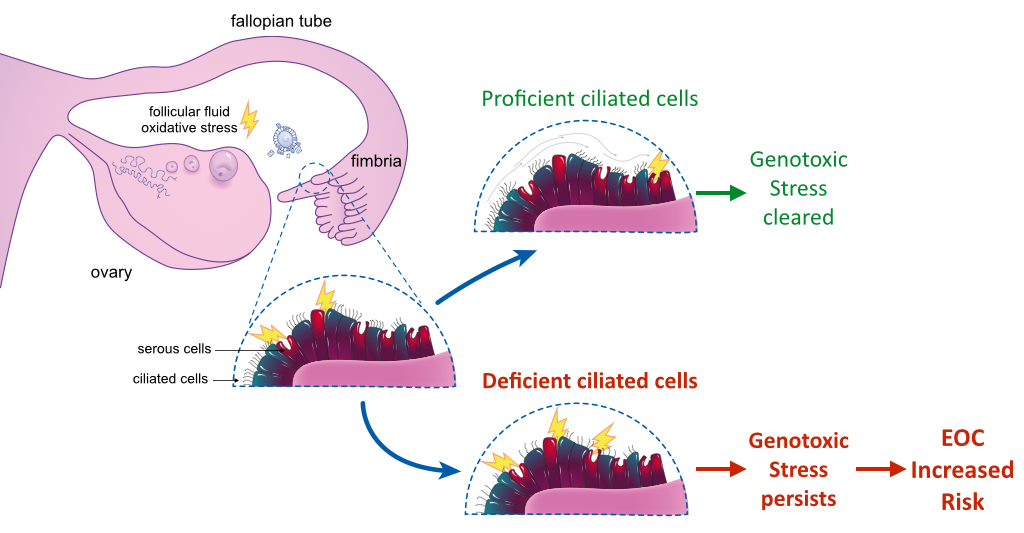
:
1. Introduction
2. Epithelial Ovarian Cancers: Classification and Cell of Origin
3. HGSOC Predisposition
4. Novel Candidate Genes Associated with HGSOC Predisposition
5. Ciliated Cells in the Fallopian Tube: Function and Tumor Predisposition
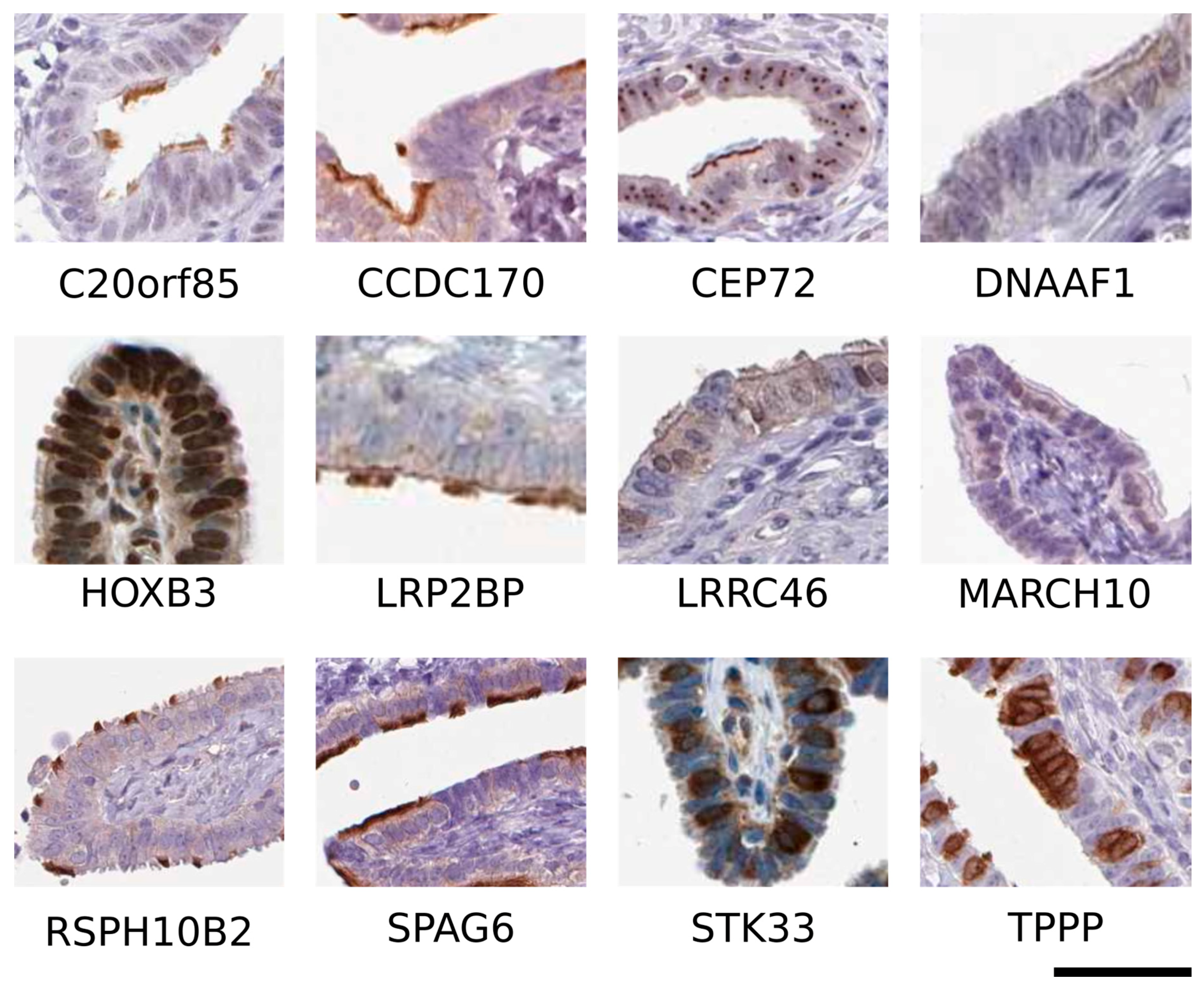
6. Overview of Candidate Genes Expressed in Ciliated Cells
6.1. Chromosome 20 Open Reading Frame 85 (C20orf85)
6.2. Coiled-Coil Domain-Containing Protein 170 (CCDC170)
6.3. Centrosomal Protein 72 (CEP72)
6.4. Dynein Axonemal Assembly Factor 1 (DNAAF1)
6.5. Homeobox B3 (HOXB3)
6.6. Low-Density Lipoprotein (LDL) Receptor-Related Protein 2 Binding Protein (LRP2BP)
6.7. Leucine-Rich Repeat Containing 46 (LRRC46)
6.8. Membrane-Associated RING-CH-Type Finger 10 (MARCH10)
6.9. Radial Spoke Head 10 Homolog B2 (RSPH10B2)
6.10. Sperm-Associated Antigen 6 (SPAG6)
6.11. Serine/Threonine Kinase 33 (STK33)
6.12. Tubulin Polymerization-Promoting Protein (TPPP)
7. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HGSOC | High Grade Serous Epithelial Ovarian Cancer |
| EOC | Epithelial Ovarian Cancer |
References
- Ferlay, J.S.I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11; International Agency for Research on Cancer: Lyon, France, 2013; Available online: http://globocan.iarc.fr (accessed on 13 July 2018).
- Rare Cancers Europe. Available online: https://www.rarecancerseurope.org/About-Rare-Cancers (accessed on 14 July 2018).
- SEER Cancer Statistics Review 1975–2015. Available online: https://seer.cancer.gov/csr/1975_2015/results_merged/sect_21_ovary.pdf (accessed on 13 July 2018).
- Dubeau, L. Pathogenesis of serous, extra-uterine Mullerian epithelial cancer and therapeutic implications. Transl. Cancer Res. 2015, 4, 3–13. [Google Scholar] [PubMed]
- Lim, D.; Oliva, E. Precursors and pathogenesis of ovarian carcinoma. Pathology 2013, 45, 229–242. [Google Scholar] [CrossRef] [PubMed]
- May, T.; Shoni, M.; Crum, C.P.; Xian, W.; Vathipadiekal, V.; Birrer, M.; Rosen, B.; Tone, A.; Murphy, K.J. Low-grade and high-grade serous Mullerian carcinoma: Review and analysis of publicly available gene expression profiles. Gynecol. Oncol. 2013, 128, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PDQ Adult Treatment Editorial Board. Ovarian Epithelial, Fallopian Tube, and Primary Peritoneal Cancer Treatment (PDQ®): Health Professional Version; PDQ Cancer Information Summaries [Internet]; National Cancer Institute: Bethesda, MD, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK66007/?report=cla ssic (accessed on 19 July 2018).
- PDQ Cancer Genetics Editorial Board. Genetics of Breast and Gynecologic Cancers (PDQ®): Health Professional Version; PDQ Cancer Information Summaries [Internet]; National Cancer Institute: Bethesda, MD, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK65767/ (accessed on 20 July 2018).
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.L.; Jameson, C.; Gore, M.E. Mucinous ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Padilla, I.; Malpica, A.L.; Minig, L.; Chiva, L.M.; Gershenson, D.M.; Gonzalez-Martin, A. Ovarian low-grade serous carcinoma: A comprehensive update. Gynecol. Oncol. 2012, 126, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Schmeler, K.M.; Sun, C.C.; Bodurka, D.C.; Deavers, M.T.; Malpica, A.; Coleman, R.L.; Ramirez, P.T.; Gershenson, D.M. Neoadjuvant chemotherapy for low-grade serous carcinoma of the ovary or peritoneum. Gynecol. Oncol. 2008, 108, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Levanon, K.; Crum, C.; Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P. Intercepting pelvic cancer in the distal fallopian tube: Theories and realities. Mol. Oncol. 2009, 3, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Folkins, A.K.; Saleemuddin, A.; Garrett, L.A.; Garber, J.E.; Muto, M.G.; Tworoger, S.S.; Crum, C.P. Epidemiologic correlates of ovarian cortical inclusion cysts (CICs) support a dual precursor pathway to pelvic epithelial cancer. Gynecol. Oncol. 2009, 115, 108–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarboe, E.; Folkins, A.; Nucci, M.R.; Kindelberger, D.; Drapkin, R.; Miron, A.; Lee, Y.; Crum, C.P. Serous carcinogenesis in the fallopian tube: A descriptive classification. Int. J. Gynecol. Pathol. 2008, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dubeau, L. The cell of origin of ovarian epithelial tumours. Lancet Oncol. 2008, 9, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Coffey, D.M.; Creighton, C.J.; Yu, Z.; Hawkins, S.M.; Matzuk, M.M. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 3921–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubeau, L.; Drapkin, R. Coming into focus: The nonovarian origins of ovarian cancer. Ann. Oncol. 2013, 24 (Suppl. 8), viii28–viii35. [Google Scholar] [CrossRef]
- Sorensen, R.D.; Schnack, T.H.; Karlsen, M.A.; Hogdall, C.K. Serous ovarian, fallopian tube and primary peritoneal cancers: A common disease or separate entities—A systematic review. Gynecol. Oncol. 2015, 136, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sacchetti, A.; van Dijk, M.R.; van der Zee, M.; van der Horst, P.H.; Joosten, R.; Burger, C.W.; Grootegoed, J.A.; Blok, L.J.; Fodde, R. Identification of quiescent, stem-like cells in the distal female reproductive tract. PLoS ONE 2012, 7, e40691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chene, G.; Dauplat, J.; Radosevic-Robin, N.; Cayre, A.; Penault-Llorca, F. Tu-be or not tu-be: That is the question… about serous ovarian carcinogenesis. Crit. Rev. Oncol. Hematol. 2013, 88, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Barker, N. Ovary and fimbrial stem cells: Biology, niche and cancer origins. Mol. Cell. Biol. 2015, 16, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Liu, Q.; Shattuck-Eidens, D.; Cochran, C.; Harshman, K.; Tavtigian, S.; Bennett, L.M.; Haugen-Strano, A.; Swensen, J.; Miki, Y.; et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science 1994, 266, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.; Rothermundt, C.; Thomas, K.; Bancroft, E.; Eeles, R.; Shanley, S.; Ardern-Jones, A.; Norman, A.; Kaye, S.B.; Gore, M.E. “BRCAness” syndrome in ovarian cancer: A case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J. Clin. Oncol. 2008, 26, 5530–5536. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol. Cell 1998, 2, 317–328. [Google Scholar] [CrossRef]
- Chen, J.J.; Silver, D.; Cantor, S.; Livingston, D.M.; Scully, R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999, 59, 1752s–1756s. [Google Scholar] [PubMed]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Feng, L.P.; Chen, H.L.; Shen, M.Y. Breastfeeding and the risk of ovarian cancer: A meta-analysis. J. Midwifery Women’s Health 2014, 59, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Engeland, A.; Tretli, S.; Bjorge, T. Height, body mass index, and ovarian cancer: A follow-up of 1.1 million Norwegian women. J. Natl. Cancer Inst. 2003, 95, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Schouten, L.J.; Goldbohm, R.A.; van den Brandt, P.A. Height, weight, weight change, and ovarian cancer risk in the Netherlands cohort study on diet and cancer. Am. J. Epidemiol. 2003, 157, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Pearce, C.L.; Templeman, C.; Rossing, M.A.; Lee, A.; Near, A.M.; Webb, P.M.; Nagle, C.M.; Doherty, J.A.; Cushing-Haugen, K.L.; Wicklund, K.G.; et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: A pooled analysis of case-control studies. Lancet Oncol. 2012, 13, 385–394. [Google Scholar] [CrossRef]
- Mogensen, J.B.; Kjaer, S.K.; Mellemkjaer, L.; Jensen, A. Endometriosis and risks for ovarian, endometrial and breast cancers: A nationwide cohort study. Gynecol. Oncol. 2016, 143, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.M.; Lin, W.T.; Kvaskoff, M.; De Vivo, I.; Terry, K.L.; Missmer, S.A. Endometriosis and risk of ovarian and endometrial cancers in a large prospective cohort of U.S. nurses. Cancer Causes Control 2017, 28, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Ovarian, Fallopian Tube, and Primary Peritoneal Cancer Prevention (PDQ®). Available online: https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0032587/ (accessed on 22 June 2018).
- Fathalla, M.F. Incessant ovulation and ovarian cancer—A hypothesis re-visited. Facts Views Vis. ObGyn 2013, 5, 292–297. [Google Scholar] [PubMed]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer; Beral, V.; Doll, R.; Hermon, C.; Peto, R.; Reeves, G. Ovarian cancer and oral contraceptives: Collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet 2008, 371, 303–314. [Google Scholar] [CrossRef]
- Stewart, S.L.; Querec, T.D.; Ochman, A.R.; Gruver, B.N.; Bao, R.; Babb, J.S.; Wong, T.S.; Koutroukides, T.; Pinnola, A.D.; Klein-Szanto, A.; et al. Characterization of a carcinogenesis rat model of ovarian preneoplasia and neoplasia. Cancer Res. 2004, 64, 8177–8183. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.A.; Giles, J.R. The hen as a model of ovarian cancer. Nat. Rev. Cancer 2013, 13, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Fathalla, M.F. Incessant ovulation—A factor in ovarian neoplasia? Lancet 1971, 2, 163. [Google Scholar] [CrossRef]
- Murdoch, W.J.; Townsend, R.S.; McDonnel, A.C. Ovulation-induced DNA damage in ovarian surface epithelial cells of ewes: Prospective regulatory mechanisms of repair/survival and apoptosis. Biol. Reprod. 2001, 65, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Hilliard, T.S.; Wu, L.Y.; Jaffe, R.C.; Fazleabas, A.T.; Burdette, J.E. The impact of ovulation on fallopian tube epithelial cells: Evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr. Relat. Cancer 2011, 18, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Donnez, J.; Casanas-Roux, F.; Caprasse, J.; Ferin, J.; Thomas, K. Cyclic changes in ciliation, cell height, and mitotic activity in human tubal epithelium during reproductive life. Fertil. Steril. 1985, 43, 554–559. [Google Scholar] [CrossRef]
- Kar, S.P.; Berchuck, A.; Gayther, S.A.; Goode, E.L.; Moysich, K.B.; Pearce, C.L.; Ramus, S.J.; Schildkraut, J.M.; Sellers, T.A.; Pharoah, P.D.P. Common Genetic Variation and Susceptibility to Ovarian Cancer: Current Insights and Future Directions. Cancer Epidemiol. Biomark. Prev. 2018, 27, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Moller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Goode, E.L.; Chenevix-Trench, G.; Song, H.; Ramus, S.J.; Notaridou, M.; Lawrenson, K.; Widschwendter, M.; Vierkant, R.A.; Larson, M.C.; Kjaer, S.K.; et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet. 2010, 42, 874–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesaratto, L.; Grisard, E.; Coan, M.; Zandona, L.; De Mattia, E.; Poletto, E.; Cecchin, E.; Puglisi, F.; Canzonieri, V.; Mucignat, M.T.; et al. BNC2 is a putative tumor suppressor gene in high-grade serous ovarian carcinoma and impacts cell survival after oxidative stress. Cell Death Dis. 2016, 7, e2374. [Google Scholar] [CrossRef] [PubMed]
- Lawrenson, K.; Li, Q.; Kar, S.; Seo, J.H.; Tyrer, J.; Spindler, T.J.; Lee, J.; Chen, Y.; Karst, A.; Drapkin, R.; et al. Cis-eQTL analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat. Commun. 2015, 6, 8234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Ning, G.; Howitt, B.E.; Mehra, K.; Wu, L.; Wang, X.; Hong, Y.; Kern, F.; Wei, T.S.; Zhang, T.; et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J. Pathol. 2016, 238, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tone, A.A.; Begley, H.; Sharma, M.; Murphy, J.; Rosen, B.; Brown, T.J.; Shaw, P.A. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin. Cancer Res. 2008, 14, 4067–4078. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/about/antibody+validation#ih (accessed on 22 August 2018).
- Li, J.; Ning, Y.; Abushahin, N.; Yuan, Z.; Wang, Y.; Wang, Y.; Yuan, B.; Cragun, J.M.; Chambers, S.K.; Hatch, K.; et al. Secretory cell expansion with aging: Risk for pelvic serous carcinogenesis. Gynecol. Oncol. 2013, 131, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Lyons, R.A.; Saridogan, E.; Djahanbakhch, O. The reproductive significance of human Fallopian tube cilia. Hum. Reprod. Update 2006, 12, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satir, P. Mechanisms of ciliary movement: Contributions from electron microscopy. Scanning Microsc. 1992, 6, 573–579. [Google Scholar] [PubMed]
- Lyons, R.A.; Saridogan, E.; Djahanbakhch, O. The effect of ovarian follicular fluid and peritoneal fluid on Fallopian tube ciliary beat frequency. Hum. Reprod. 2006, 21, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Raidt, J.; Werner, C.; Menchen, T.; Dougherty, G.W.; Olbrich, H.; Loges, N.T.; Schmitz, R.; Pennekamp, P.; Omran, H. Ciliary function and motor protein composition of human fallopian tubes. Hum. Reprod. 2015, 30, 2871–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagoory, O.; Braiman, A.; Priel, Z. The mechanism of ciliary stimulation by acetylcholine: Roles of calcium, PKA, and PKG. J. Gen. Physiol. 2002, 119, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, P. Ca2+-dependent hormonal stimulation of ciliary activity. Nature 1980, 283, 764–765. [Google Scholar] [CrossRef] [PubMed]
- Villalon, M.; Verdugo, P. Hormonal regulation of ciliary function in the oviduct: The effect of β-adrenergic agonists. Prog. Clin. Biol. Res. 1982, 80, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, P.; Rumery, R.E.; Tam, P.Y. Hormonal control of oviductal ciliary activity: Effect of prostaglandins. Fertil. Steril. 1980, 33, 193–196. [Google Scholar] [CrossRef]
- Saridogan, E.; Djahanbakhch, O.; Puddefoot, J.R.; Demetroulis, C.; Collingwood, K.; Mehta, J.G.; Vinson, G.P. Angiotensin II receptors and angiotensin II stimulation of ciliary activity in human fallopian tube. J. Clin. Endocrinol. Metab. 1996, 81, 2719–2725. [Google Scholar] [PubMed]
- Mahmood, T.; Saridogan, E.; Smutna, S.; Habib, A.M.; Djahanbakhch, O. The effect of ovarian steroids on epithelial ciliary beat frequency in the human Fallopian tube. Hum. Reprod. 1998, 13, 2991–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, R.A.; Djahanbakhch, O.; Mahmood, T.; Saridogan, E.; Sattar, S.; Sheaff, M.T.; Naftalin, A.A.; Chenoy, R. Fallopian tube ciliary beat frequency in relation to the stage of menstrual cycle and anatomical site. Hum. Reprod. 2002, 17, 584–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comer, M.T.; Leese, H.J.; Southgate, J. Induction of a differentiated ciliated cell phenotype in primary cultures of Fallopian tube epithelium. Hum. Reprod. 1998, 13, 3114–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saed, G.M.; Diamond, M.P.; Fletcher, N.M. Updates of the role of oxidative stress in the pathogenesis of ovarian cancer. Gynecol. Oncol. 2017, 145, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Kvaskoff, M.; Mu, F.; Terry, K.L.; Harris, H.R.; Poole, E.M.; Farland, L.; Missmer, S.A. Endometriosis: A high-risk population for major chronic diseases? Hum. Reprod. Update 2015, 21, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Kvaskoff, M.; Horne, A.W.; Missmer, S.A. Informing women with endometriosis about ovarian cancer risk. Lancet 2017, 390, 2433–2434. [Google Scholar] [CrossRef]
- Xia, W.; Zhang, D.; Ouyang, J.; Liang, Y.; Zhang, H.; Huang, Z.; Liang, G.; Zhu, Q.; Guan, X.; Zhang, J. Effects of pelvic endometriosis and adenomyosis on ciliary beat frequency and muscular contractions in the human fallopian tube. Reprod. Biol. Endocrinol. 2018, 16, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levanon, K.; Ng, V.; Piao, H.Y.; Zhang, Y.; Chang, M.C.; Roh, M.H.; Kindelberger, D.W.; Hirsch, M.S.; Crum, C.P.; Marto, J.A.; et al. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 2010, 29, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- George, S.H.; Milea, A.; Sowamber, R.; Chehade, R.; Tone, A.; Shaw, P.A. Loss of LKB1 and p53 synergizes to alter fallopian tube epithelial phenotype and high-grade serous tumorigenesis. Oncogene 2016, 35, 59–68. [Google Scholar] [CrossRef] [PubMed]
- McConnell, A.M.; Yao, C.; Yeckes, A.R.; Wang, Y.; Selvaggio, A.S.; Tang, J.; Kirsch, D.G.; Stripp, B.R. p53 Regulates Progenitor Cell Quiescence and Differentiation in the Airway. Cell Rep. 2016, 17, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.M.; Yang, S.H.; Chowdhuri, S.R.; Player, A.; Hames, M.; Fukuoka, J.; Meerzaman, D.; Dracheva, T.; Sun, Z.; Yang, P.; et al. Inactivation of LLC1 gene in nonsmall cell lung cancer. Int. J. Cancer 2007, 120, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Choi, Y.B.; Hwang, H.L.; Lee, J.H.; Park, S.Y.; Kim, H.K.; Poojan, S.; Koh, J.S.; Kim, H.S.; Hong, K.M. Immunohistochemical localization of LLC1 in human tissues and its limited expression in non-small cell lung cancer. Histol. Histopathol. 2015, 30, 1111–1120. [Google Scholar] [PubMed]
- Jiang, P.; Li, Y.; Poleshko, A.; Medvedeva, V.; Baulina, N.; Zhang, Y.; Zhou, Y.; Slater, C.M.; Pellegrin, T.; Wasserman, J.; et al. The Protein Encoded by the CCDC170 Breast Cancer Gene Functions to Organize the Golgi-Microtubule Network. EBioMedicine 2017, 22, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, Y.; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; Jones, S.; et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, A.M.; Michailidou, K.; Kuchenbaecker, K.B.; Thompson, D.; French, J.D.; Beesley, J.; Healey, C.S.; Kar, S.; Pooley, K.A.; Lopez-Knowles, E.; et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 2016, 48, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeraraghavan, J.; Tan, Y.; Cao, X.X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.; Edwards, D.P.; Contreras, A.; et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014, 5, 4577. [Google Scholar] [CrossRef] [PubMed]
- Kodani, A.; Yu, T.W.; Johnson, J.R.; Jayaraman, D.; Johnson, T.L.; Al-Gazali, L.; Sztriha, L.; Partlow, J.N.; Kim, H.; Krup, A.L.; et al. Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshimori, N.; Li, X.; Ohsugi, M.; Yamamoto, T. Cep72 regulates the localization of key centrosomal proteins and proper bipolar spindle formation. EMBO J. 2009, 28, 2066–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowe, T.R.; Wilkinson, C.J.; Iqbal, A.; Stearns, T. The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol. Biol. Cell 2012, 23, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Slaats, G.G.; Saldivar, J.C.; Bacal, J.; Zeman, M.K.; Kile, A.C.; Hynes, A.M.; Srivastava, S.; Nazmutdinova, J.; den Ouden, K.; Zagers, M.S.; et al. DNA replication stress underlies renal phenotypes in CEP290-associated Joubert syndrome. J. Clin. Investig. 2015, 125, 3657–3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q. CpG methylation patterns are associated with gene expression variation in osteosarcoma. Mol. Med. Rep. 2017, 16, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Fitzgerald, R.C.; Vaughan, T.L.; Palles, C.; Gockel, I.; Tomlinson, I.; Buas, M.F.; May, A.; Gerges, C.; Anders, M.; et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: A large-scale meta-analysis. Lancet Oncol. 2016, 17, 1363–1373. [Google Scholar] [CrossRef]
- Kang, J.U.; Koo, S.H.; Kwon, K.C.; Park, J.W.; Kim, J.M. Gain at chromosomal region 5p15.33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer Genet. Cytogenet. 2008, 182, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Luddecke, S.; Ertych, N.; Stenzinger, A.; Weichert, W.; Beissbarth, T.; Dyczkowski, J.; Gaedcke, J.; Valerius, O.; Braus, G.H.; Kschischo, M.; et al. The putative oncogene CEP72 inhibits the mitotic function of BRCA1 and induces chromosomal instability. Oncogene 2016, 35, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Loges, N.T.; Olbrich, H.; Becker-Heck, A.; Haffner, K.; Heer, A.; Reinhard, C.; Schmidts, M.; Kispert, A.; Zariwala, M.A.; Leigh, M.W.; et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009, 85, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Levy, M.; Dudakia, D.; Proszek, P.; Shipley, C.; Basten, S.; Rapley, E.; Bishop, D.T.; Reid, A.; Huddart, R.; et al. Rare disruptive mutations in ciliary function genes contribute to testicular cancer susceptibility. Nat. Commun. 2016, 7, 13840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, N.; Jee, B.K.; Chae, S.W.; Jeon, Y.W.; Lee, K.H.; Rha, H.K. HOX gene analysis of endothelial cell differentiation in human bone marrow-derived mesenchymal stem cells. Mol. Biol. Rep. 2009, 36, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.; Charboneau, A.; Cheung, I.; Hanks, D.; Boudreau, N. Sustained expression of homeobox D10 inhibits angiogenesis. Am. J. Pathol. 2002, 161, 2099–2109. [Google Scholar] [CrossRef]
- Bjornsson, J.M.; Larsson, N.; Brun, A.C.; Magnusson, M.; Andersson, E.; Lundstrom, P.; Larsson, J.; Repetowska, E.; Ehinger, M.; Humphries, R.K.; et al. Reduced proliferative capacity of hematopoietic stem cells deficient in Hoxb3 and Hoxb4. Mol. Cell. Biol. 2003, 23, 3872–3883. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Homeobox genes in normal hematopoiesis and leukemogenesis. Int. J. Hematol. 1998, 68, 343–353. [Google Scholar] [CrossRef]
- Ko, K.H.; Lam, Q.L.; Zhang, M.; Wong, C.K.; Lo, C.K.; Kahmeyer-Gabbe, M.; Tsang, W.H.; Tsang, S.L.; Chan, L.C.; Sham, M.H.; et al. Hoxb3 deficiency impairs B lymphopoiesis in mouse bone marrow. Exp. Hematol. 2007, 35, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.J.; Sauvageau, G.; Humphries, R.K.; Largman, C. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells 1996, 14, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.; Brun, A.C.; Lawrence, H.J.; Karlsson, S. Hoxa9/hoxb3/hoxb4 compound null mice display severe hematopoietic defects. Exp. Hematol. 2007, 35, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, G.; Thorsteinsdottir, U.; Eaves, C.J.; Lawrence, H.J.; Largman, C.; Lansdorp, P.M.; Humphries, R.K. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 1995, 9, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, G.; Thorsteinsdottir, U.; Hough, M.R.; Hugo, P.; Lawrence, H.J.; Largman, C.; Humphries, R.K. Overexpression of HOXB3 in hematopoietic cells causes defective lymphoid development and progressive myeloproliferation. Immunity 1997, 6, 13–22. [Google Scholar] [CrossRef]
- Bi, L.; Zhou, B.; Li, H.; He, L.; Wang, C.; Wang, Z.; Zhu, L.; Chen, M.; Gao, S. A novel miR-375-HOXB3-CDCA3/DNMT3B regulatory circuitry contributes to leukemogenesis in acute myeloid leukemia. BMC Cancer 2018, 18, 182. [Google Scholar] [CrossRef] [PubMed]
- Drabkin, H.A.; Parsy, C.; Ferguson, K.; Guilhot, F.; Lacotte, L.; Roy, L.; Zeng, C.; Baron, A.; Hunger, S.P.; Varella-Garcia, M.; et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia 2002, 16, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindblad, O.; Chougule, R.A.; Moharram, S.A.; Kabir, N.N.; Sun, J.; Kazi, J.U.; Ronnstrand, L. The role of HOXB2 and HOXB3 in acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2015, 467, 742–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.C.; Shih, L.Y.; May Chen, M.J.; Wang, C.C.; Yeh, T.C.; Lin, T.H.; Chen, C.Y.; Lin, C.J.; Liang, D.C. Expression of HOXB genes is significantly different in acute myeloid leukemia with a partial tandem duplication of MLL vs. a MLL translocation: A cross-laboratory study. Cancer Genet. 2011, 204, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Pineault, N.; Abramovich, C.; Ohta, H.; Humphries, R.K. Differential and common leukemogenic potentials of multiple NUP98-Hox fusion proteins alone or with Meis1. Mol. Cell. Biol. 2004, 24, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Davison, J.; Du, L.; Storer, B.; Stirewalt, D.L.; Heimfeld, S.; Estey, E.; Appelbaum, F.R.; Fang, M. Identification of differentially methylated markers among cytogenetic risk groups of acute myeloid leukemia. Epigenetics 2015, 10, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Zeng, C.; Baron, A.; Gadgil, S.; Gemmill, R.M.; Tigaud, I.; Thomas, X.; Drabkin, H.A. Hox expression in AML identifies a distinct subset of patients with intermediate cytogenetics. Leukemia 2004, 18, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsteinsdottir, U.; Kroon, E.; Jerome, L.; Blasi, F.; Sauvageau, G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol. Cell. Biol. 2001, 21, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Starkova, J.; Zamostna, B.; Mejstrikova, E.; Krejci, R.; Drabkin, H.A.; Trka, J. HOX gene expression in phenotypic and genotypic subgroups and low HOXA gene expression as an adverse prognostic factor in pediatric ALL. Pediatr. Blood Cancer 2010, 55, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Bodey, B.; Bodey, B., Jr.; Siegel, S.E.; Kaiser, H.E. Immunocytochemical detection of the homeobox B3, B4, and C6 gene products in breast carcinomas. Anticancer Res. 2000, 20, 3281–3286. [Google Scholar] [PubMed]
- Fu, H.; Fu, L.; Xie, C.; Zuo, W.S.; Liu, Y.S.; Zheng, M.Z.; Yu, J.M. miR-375 inhibits cancer stem cell phenotype and tamoxifen resistance by degrading HOXB3 in human ER-positive breast cancer. Oncol. Rep. 2017, 37, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, F.; Chen, P. miR-7 and miR-218 epigenetically control tumor suppressor genes RASSF1A and Claudin-6 by targeting HoxB3 in breast cancer. Biochem. Biophys. Res. Commun. 2012, 424, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, I.; Dominguez, D.; Kjeldsen, T.E.; Kristensen, L.S.; Hager, H.; Wojdacz, T.K.; Hansen, L.L. Identification and validation of candidate epigenetic biomarkers in lung adenocarcinoma. Sci. Rep. 2016, 6, 35807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, F.C.; Destro, M.F.; Duarte, C.M.; Nunes, F.D. Epigenetic repression of HOXB cluster in oral cancer cell lines. Arch. Oral Biol. 2014, 59, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, N.; Morita, K.; Kobayashi, N.; Tada, M.; Itoh, T.; Saitoh, S.; Kondo, M.; Takahashi, N.; Kataoka, A.; Nakanishi, K.; et al. Array comparative genomic hybridization analysis revealed four genomic prognostic biomarkers for primary gastric cancers. Cancer Genet. Cytogenet. 2010, 201, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.U.; Marques, I.J.; Woltering, J.M.; Vlecken, D.H.; Aghdassi, A.; Partecke, L.I.; Heidecke, C.D.; Lerch, M.M.; Bagowski, C.P. Retinoic acid receptor antagonists inhibit miR-10a expression and block metastatic behavior of pancreatic cancer. Gastroenterology 2009, 137, 2136-45.e1–2136-45.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yan, R.; Zhang, X.; Zhu, Z.; Wang, C.; Liang, C.; Zhang, X. Deregulation of MicroRNA-375 inhibits cancer proliferation migration and chemosensitivity in pancreatic cancer through the association of HOXB3. Am. J. Transl. Res. 2016, 8, 1551–1559. [Google Scholar] [PubMed]
- Xu, K.; Qiu, C.; Pei, H.; Mehmood, M.A.; Wang, H.; Li, L.; Xia, Q. Homeobox B3 promotes tumor cell proliferation and invasion in glioblastoma. Oncol. Lett. 2018, 15, 3712–3718. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.R.; Patel, J.N.; Zhang, Q.; Norris, E.J.; Symanowski, J.; Michener, C.; Sehouli, J.; Braicu, I.; Destephanis, D.D.; Sutker, A.P.; et al. HOXA4/HOXB3 gene expression signature as a biomarker of recurrence in patients with high-grade serous ovarian cancer following primary cytoreductive surgery and first-line adjuvant chemotherapy. Gynecol. Oncol. 2018, 149, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ji, C.; Zheng, H.; Fei, X.; Zheng, M.; Dai, J.; Gu, S.; Xie, Y.; Mao, Y. Molecular cloning and characterization of a novel human gene containing 4 ankyrin repeat domains. Cell. Mol. Biol. Lett. 2005, 10, 185–193. [Google Scholar] [PubMed]
- Hao, L.; Fu, J.; Tian, Y.; Wu, J. Systematic analysis of lncRNAs, miRNAs and mRNAs for the identification of biomarkers for osteoporosis in the mandible of ovariectomized mice. Int. J. Mol. Med. 2017, 40, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starnawska, A.; Hansen, C.S.; Sparso, T.; Mazin, W.; Olsen, L.; Bertalan, M.; Buil, A.; Bybjerg-Grauholm, J.; Baekvad-Hansen, M.; Hougaard, D.M.; et al. Differential DNA methylation at birth associated with mental disorder in individuals with 22q11.2 deletion syndrome. Transl. Psychiatry 2017, 7, e1221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, L.; Xu, B.M.; Tang, W.H.; Ye, Z.D.; Huang, C.; Ma, X.; Zhao, J.J.; Guo, F.X.; Kang, C.M.; et al. LncRNA-RP11-714G18.1 suppresses vascular cell migration via directly targeting LRP2BP. Immunol. Cell Biol. 2018, 96, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.M.; Zuhlke, K.A.; Plotts, C.; McDonnell, S.K.; Middha, S.; Riska, S.M.; Schaid, D.J.; Thibodeau, S.N.; Douglas, J.A.; Cooney, K.A. Mutational landscape of candidate genes in familial prostate cancer. Prostate 2014, 74, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebhan, M.; Chalifa-Caspi, V.; Prilusky, J.; Lancet, D. GeneCards: Integrating information about genes, proteins and diseases. Trends Genet. 1997, 13, 163. [Google Scholar] [CrossRef]
- Kott, E.; Duquesnoy, P.; Copin, B.; Legendre, M.; Dastot-Le Moal, F.; Montantin, G.; Jeanson, L.; Tamalet, A.; Papon, J.F.; Siffroi, J.P.; et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, P.V.; Hirota, T.; Hirose, S.; Nakamura, N. Membrane-associated RING-CH 10 (MARCH10 protein) is a microtubule-associated E3 ubiquitin ligase of the spermatid flagella. J. Biol. Chem. 2011, 286, 39082–39090. [Google Scholar] [CrossRef] [PubMed]
- Pigino, G.; Ishikawa, T. Axonemal radial spokes: 3D structure, function and assembly. Bioarchitecture 2012, 2, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Roy, S. SnapShot: Motile Cilia. Cell 2015, 162, 224–224.e221. [Google Scholar] [CrossRef] [PubMed]
- Castleman, V.H.; Romio, L.; Chodhari, R.; Hirst, R.A.; de Castro, S.C.; Parker, K.A.; Ybot-Gonzalez, P.; Emes, R.D.; Wilson, S.W.; Wallis, C.; et al. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009, 84, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Zietkiewicz, E.; Bukowy-Bieryllo, Z.; Voelkel, K.; Klimek, B.; Dmenska, H.; Pogorzelski, A.; Sulikowska-Rowinska, A.; Rutkiewicz, E.; Witt, M. Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS ONE 2012, 7, e33667. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, K.; Chen, D.; Nishida, T.; Misaki, K.; Yonemura, S.; Hamada, H. Absence of Radial Spokes in Mouse Node Cilia Is Required for Rotational Movement but Confers Ultrastructural Instability as a Trade-Off. Dev. Cell 2015, 35, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neilson, L.I.; Schneider, P.A.; Van Deerlin, P.G.; Kiriakidou, M.; Driscoll, D.A.; Pellegrini, M.C.; Millinder, S.; Yamamoto, K.K.; French, C.K.; Strauss, J.F., 3rd. cDNA cloning and characterization of a human sperm antigen (SPAG6) with homology to the product of the Chlamydomonas PF16 locus. Genomics 1999, 60, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jones, B.H.; Tang, W.; Moss, S.B.; Wei, Z.; Ho, C.; Pollack, M.; Horowitz, E.; Bennett, J.; Baker, M.E.; et al. Dissecting the axoneme interactome: The mammalian orthologue of Chlamydomonas PF6 interacts with sperm-associated antigen 6, the mammalian orthologue of Chlamydomonas PF16. Mol. Cell. Proteom. 2005, 4, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, E.; Zhang, Z.; Jones, B.H.; Moss, S.B.; Ho, C.; Wood, J.R.; Wang, X.; Sammel, M.D.; Strauss, J.F., 3rd. Patterns of expression of sperm flagellar genes: Early expression of genes encoding axonemal proteins during the spermatogenic cycle and shared features of promoters of genes encoding central apparatus proteins. Mol. Hum. Reprod. 2005, 11, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Sapiro, R.; Kostetskii, I.; Olds-Clarke, P.; Gerton, G.L.; Radice, G.L.; Strauss, I.J. Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm-associated antigen 6. Mol. Cell. Biol. 2002, 22, 6298–6305. [Google Scholar] [CrossRef] [PubMed]
- Teves, M.E.; Sears, P.R.; Li, W.; Zhang, Z.; Tang, W.; van Reesema, L.; Costanzo, R.M.; Davis, C.W.; Knowles, M.R.; Strauss, J.F., 3rd; et al. Sperm-associated antigen 6 (SPAG6) deficiency and defects in ciliogenesis and cilia function: Polarity, density, and beat. PLoS ONE 2014, 9, e107271. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, B.; Wang, L.; Chen, L.; Luo, X.; Liu, L. SPAG6 regulates cell apoptosis through the TRAIL signal pathway in myelodysplastic syndromes. Oncol. Rep. 2017, 37, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Silina, K.; Zayakin, P.; Kalnina, Z.; Ivanova, L.; Meistere, I.; Endzelins, E.; Abols, A.; Stengrevics, A.; Leja, M.; Ducena, K.; et al. Sperm-associated antigens as targets for cancer immunotherapy: Expression pattern and humoral immune response in cancer patients. J. Immunother. 2011, 34, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, D.; Schramm, A.; Eggert, A.; Onda, M.; Dawczynski, K.; Rump, A.; Pastan, I.; Wittig, S.; Pfaffendorf, N.; Voigt, A.; et al. Identification of a set of seven genes for the monitoring of minimal residual disease in pediatric acute myeloid leukemia. Clin. Cancer Res. 2006, 12, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, L.; Luo, X.; Chen, L.; Yang, Z.; Liu, L. SPAG6 silencing inhibits the growth of the malignant myeloid cell lines SKM-1 and K562 via activating p53 and caspase activation-dependent apoptosis. Int. J. Oncol. 2015, 46, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Mujica, A.O.; Brauksiepe, B.; Saaler-Reinhardt, S.; Reuss, S.; Schmidt, E.R. Differential expression pattern of the novel serine/threonine kinase, STK33, in mice and men. FEBS J. 2005, 272, 4884–4898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujica, A.O.; Hankeln, T.; Schmidt, E.R. A novel serine/threonine kinase gene, STK33, on human chromosome 11p15.3. Gene 2001, 280, 175–181. [Google Scholar] [CrossRef]
- Martins, L.R.; Bung, R.K.; Koch, S.; Richter, K.; Schwarzmuller, L.; Terhardt, D.; Kurtulmus, B.; Niehrs, C.; Rouhi, A.; Lohmann, I.; et al. Stk33 is required for spermatid differentiation and male fertility in mice. Dev. Biol. 2018, 433, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Scholl, C.; Frohling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Babij, C.; Zhang, Y.; Kurzeja, R.J.; Munzli, A.; Shehabeldin, A.; Fernando, M.; Quon, K.; Kassner, P.D.; Ruefli-Brasse, A.A.; Watson, V.J.; et al. STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res. 2011, 71, 5818–5826. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.D.; Ma, S.P.; Liu, F.; Chen, Y.Z. Role of serine/threonine kinase 33 methylation in colorectal cancer and its clinical significance. Oncol. Lett. 2018, 15, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Sun, T.; Kong, X.; Xie, D.; Li, Z.; Xie, K. Kruppel-like Factor 4 Suppresses Serine/Threonine Kinase 33 Activation and Metastasis of Gastric Cancer through Reversing Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2018, 24, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tang, J.; Zhang, W.; Shen, C.; Xu, L.; Yang, D. Correlation between STK33 and the pathology and prognosis of lung cancer. Oncol. Lett. 2017, 14, 4800–4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.; Kong, X.; Du, Y.; Chen, Y.; Deng, X.; Zhu, J.; Du, J.; Li, L.; Jia, Z.; Xie, D.; et al. STK33 Promotes Growth and Progression of Pancreatic Cancer as a Critical Downstream Mediator of HIF1α. Cancer Res. 2017, 77, 6851–6862. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Song, B.; Zhang, J.; Yang, G.S.; Zhang, H.; Yu, W.F.; Wu, M.C.; Lu, J.H.; Shen, F. STK33 promotes hepatocellular carcinoma through binding to c-Myc. Gut 2016, 65, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Hlavanda, E.; Kovacs, J.; Olah, J.; Orosz, F.; Medzihradszky, K.F.; Ovadi, J. Brain-specific p25 protein binds to tubulin and microtubules and induces aberrant microtubule assemblies at substoichiometric concentrations. Biochemistry 2002, 41, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Olah, J.; Bertrand, P.; Ovadi, J. Role of the microtubule-associated TPPP/p25 in Parkinson’s and related diseases and its therapeutic potential. Expert Rev. Proteom. 2017, 14, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Szenasi, T.; Olah, J.; Szabo, A.; Szunyogh, S.; Lang, A.; Perczel, A.; Lehotzky, A.; Uversky, V.N.; Ovadi, J. Challenging drug target for Parkinson’s disease: Pathological complex of the chameleon TPPP/p25 and α-synuclein proteins. Biochim. Biophys. Acta 2017, 1863, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Orosz, F.; Ovadi, J. TPPP orthologs are ciliary proteins. FEBS Lett. 2008, 582, 3757–3764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inokawa, Y.; Sonohara, F.; Kanda, M.; Hayashi, M.; Nishikawa, Y.; Sugimoto, H.; Kodera, Y.; Nomoto, S. Correlation Between Poor Prognosis and Lower TPPP Gene Expression in Hepatocellular Carcinoma. Anticancer Res. 2016, 36, 4639–4645. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cheng, Y.; Zhu, M.; Wen, Y.; Wang, C.; Wang, Y.; Geng, L.; Shen, W.; Liu, J.; Li, Z.; et al. Fine mapping of chromosome 5p15.33 identifies novel lung cancer susceptibility loci in Han Chinese. Int. J. Cancer 2017, 141, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Chochi, Y.; Matsuyama, H.; Eguchi, S.; Kawauchi, S.; Furuya, T.; Oga, A.; Kang, J.J.; Naito, K.; Sasaki, K. Gain of 5p15.33 is associated with progression of bladder cancer. Oncology 2007, 72, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Olah, J.; Tokesi, N.; Lehotzky, A.; Orosz, F.; Ovadi, J. Moonlighting microtubule-associated proteins: Regulatory functions by day and pathological functions at night. Cytoskeleton 2013, 70, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Mukherjee, A.; Wu, J.; Zhang, L.; Teves, M.E.; Li, H.; Nambiar, S.; Henderson, S.C.; Horwitz, A.R.; Strauss, J.F., III; et al. Sperm Associated Antigen 6 (SPAG6) Regulates Fibroblast Cell Growth, Morphology, Migration and Ciliogenesis. Sci. Rep. 2015, 5, 16506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Thompson, J.; Yates, J.R., 3rd; Marshall, W.F. Proteomic analysis of mammalian primary cilia. Curr. Biol. 2012, 22, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, H.; Ohwada, N.; Aoki, T.; Suzuki, T.; Takata, K. The primary cilia of secretory cells in the human oviduct mucosa. Med. Mol. Morphol. 2008, 41, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Ciliopathies: Replication stress-induced DNA damage in renal ciliopathies. Nat. Rev. Nephrol. 2015, 11, 632. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, M. Ciliopathies and DNA damage: An emerging nexus. Curr. Opin. Nephrol. Hypertens. 2015, 24, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| GWAS_Block | Coordinate_GWAS_Block | Gene a | HPA Validation b | HPA Localization | Subcellular Localization |
|---|---|---|---|---|---|
| GWAS_EOC_54 | chr9:135138764-137155444 | AK8 | Enhanced | Ambiguous | NA |
| GWAS_EOC_52 | chr9:15913285-17915021 | BNC2 | Approved | Ambiguous | NA |
| GWAS_EOC_25 | chr19:16389703-40732752 | BST2 | Approved | Ambiguous | NA |
| GWAS_EOC_30 | chr20:56330568-58330569 | C20orf85 | Enhanced | Ciliated cells | Brush border |
| GWAS_EOC_46 | chr6:150405376-152405377 | CCDC170 | Enhanced | Ciliated cells | Brush border |
| GWAS_EOC_52 | chr9:15913285-17915021 | CCDC171 | Uncertain | Ambiguous | NA |
| GWAS_EOC_12 | chr11:85642871-87642872 | CCDC81 | Uncertain | Ambiguous | NA |
| GWAS_EOC_38 | chr5:279789-2279790 | CEP72 | Approved | Serous and ciliated cells | NA |
| GWAS_EOC_20 | chr16:83537526-85537527 | DNAAF1 | Enhanced | Ciliated cells | Cytoplasm and brush border |
| GWAS_EOC_5 | chr1:243240447-245240448 | EFCAB2 | Approved | Ambiguous | NA |
| GWAS_EOC_42 | chr5:174418048-176418049 | FAM153B | Uncertain | Ambiguous | NA |
| GWAS_EOC_34 | chr3:155397748-157435952 | GMPS | NA | Not available | NA |
| GWAS_EOC_23 | chr17:42516401-47500673 | HOXB3 | Approved | Serous and ciliated cells | NA |
| GWAS_EOC_26 | chr19:38732751-40732752 | KCNK6 | Uncertain | Ambiguous | NA |
| GWAS_EOC_16 | chr14:41173640-43173641 | LRFN5 | Uncertain | Not detected | NA |
| GWAS_EOC_37 | chr4:184470585-186470586 | LRP2BP | Approved | Ciliated cells | Brush border |
| GWAS_EOC_23 | chr17:42516401-47500673 | LRRC46 | Enhanced | Ciliated cells | Nucleus, cytoplasm and brush border |
| GWAS_EOC_24 | chr17:58880645-61480968 | MARCH10 | Enhanced | Ciliated cells | Nucleus and blefaroplast |
| GWAS_EOC_11 | chr11:35386754-37386755 | PAMR1 | NA | Not available | NA |
| GWAS_EOC_35 | chr4:118949959-120949960 | PDE5A | Approved | Ambiguous | NA |
| GWAS_EOC_35 | chr4:118949959-120949960 | PRSS12 | Uncertain | Ambiguous | NA |
| GWAS_EOC_48 | chr7:6108187-8108188 | RSPH10B2 | Supported | Ciliated cells (not all) | Cytoplasm and brush border |
| GWAS_EOC_6 | chr10:20827795-22915619 | SPAG6 | Enhanced | Ciliated cells | Cytoplasm and brush border |
| GWAS_EOC_8 | chr11:7404500-9404501 | STK33 | Uncertain | Ciliated cells (not all) | Cytoplasm |
| GWAS_EOC_34 | chr3:155397748-157435952 | TIPARP | Uncertain | Ambiguous | NA |
| GWAS_EOC_11 | chr11:35386754-37386755 | TNXB | NA | Not available | NA |
| GWAS_EOC_38 | chr5:279789-2279790 | TPPP | Enhanced | Ciliated cells (not all) | Cytoplasm and brush border |
| GWAS_EOC_1 | chr1:21415409-23490724 | WNT4 | Uncertain | Serous | NA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coan, M.; Rampioni Vinciguerra, G.L.; Cesaratto, L.; Gardenal, E.; Bianchet, R.; Dassi, E.; Vecchione, A.; Baldassarre, G.; Spizzo, R.; Nicoloso, M.S. Exploring the Role of Fallopian Ciliated Cells in the Pathogenesis of High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 2512. https://doi.org/10.3390/ijms19092512
Coan M, Rampioni Vinciguerra GL, Cesaratto L, Gardenal E, Bianchet R, Dassi E, Vecchione A, Baldassarre G, Spizzo R, Nicoloso MS. Exploring the Role of Fallopian Ciliated Cells in the Pathogenesis of High-Grade Serous Ovarian Cancer. International Journal of Molecular Sciences. 2018; 19(9):2512. https://doi.org/10.3390/ijms19092512
Chicago/Turabian StyleCoan, Michela, Gian Luca Rampioni Vinciguerra, Laura Cesaratto, Emanuela Gardenal, Riccardo Bianchet, Erik Dassi, Andrea Vecchione, Gustavo Baldassarre, Riccardo Spizzo, and Milena Sabrina Nicoloso. 2018. "Exploring the Role of Fallopian Ciliated Cells in the Pathogenesis of High-Grade Serous Ovarian Cancer" International Journal of Molecular Sciences 19, no. 9: 2512. https://doi.org/10.3390/ijms19092512