4-Hydroxypiperidines and Their Flexible 3-(Amino)propyloxy Analogues as Non-Imidazole Histamine H3 Receptor Antagonist: Further Structure–Activity Relationship Exploration and In Vitro and In Vivo Pharmacological Evaluation

Abstract
:1. Introduction
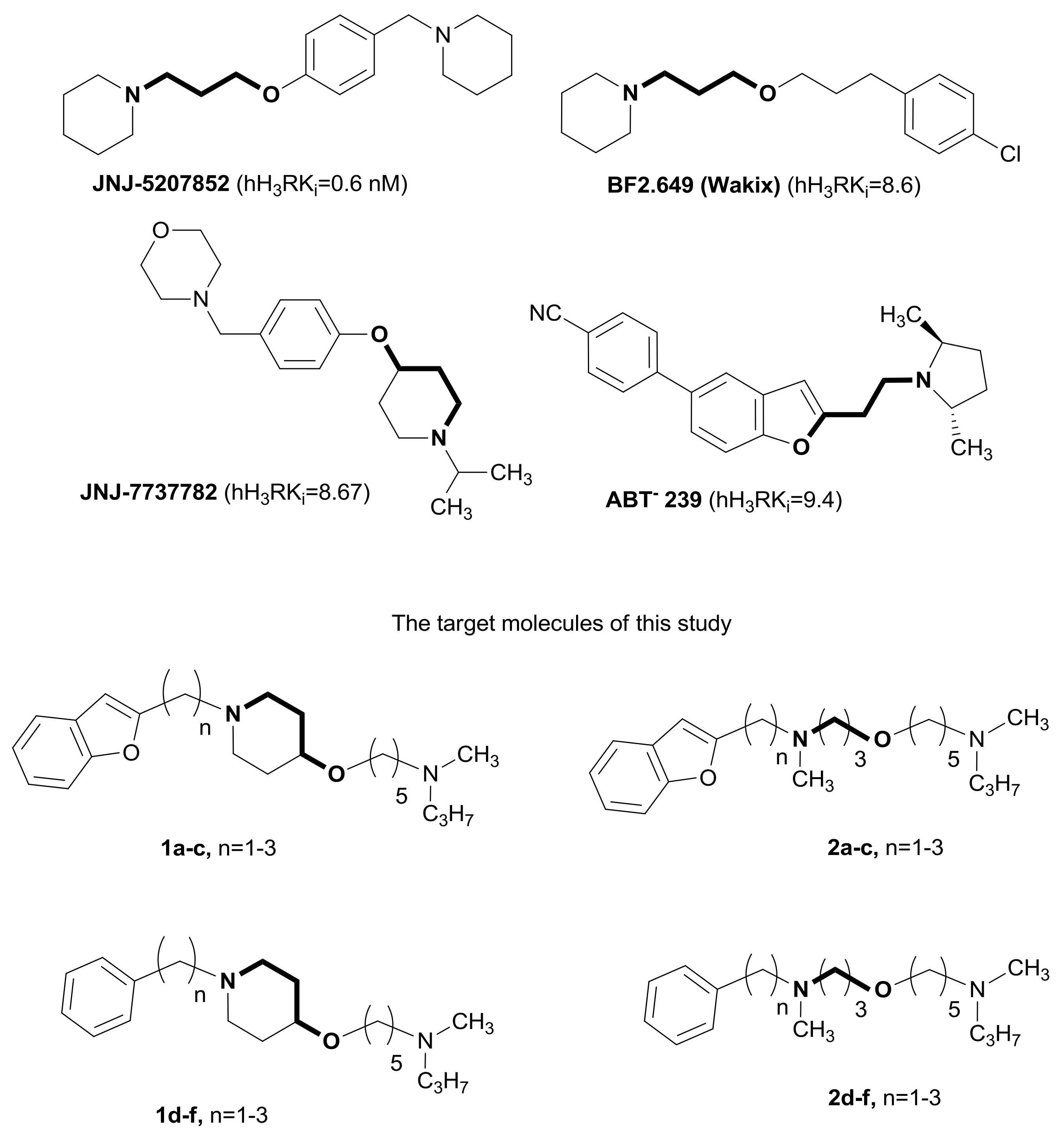
2. Results and Discussion
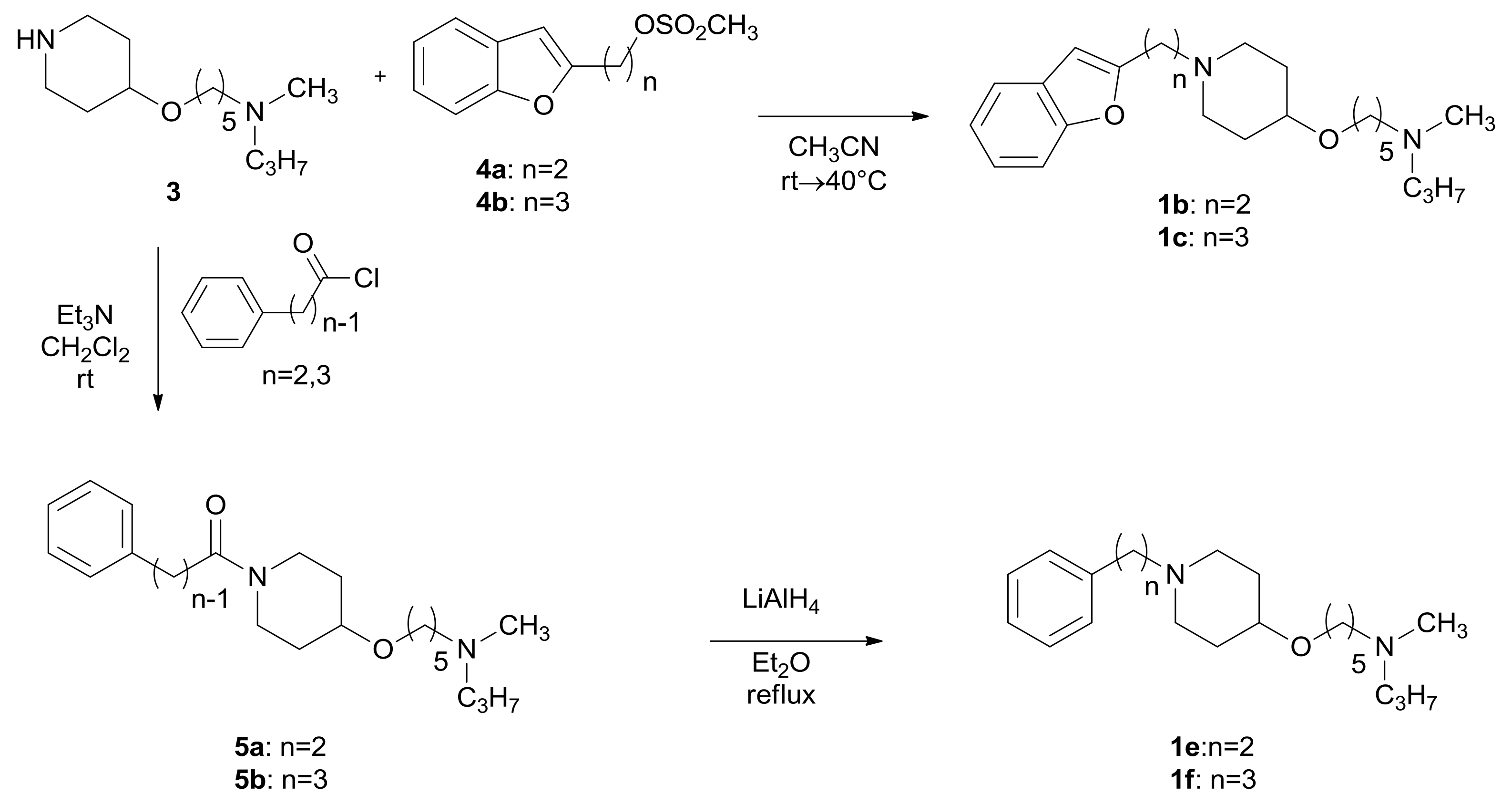
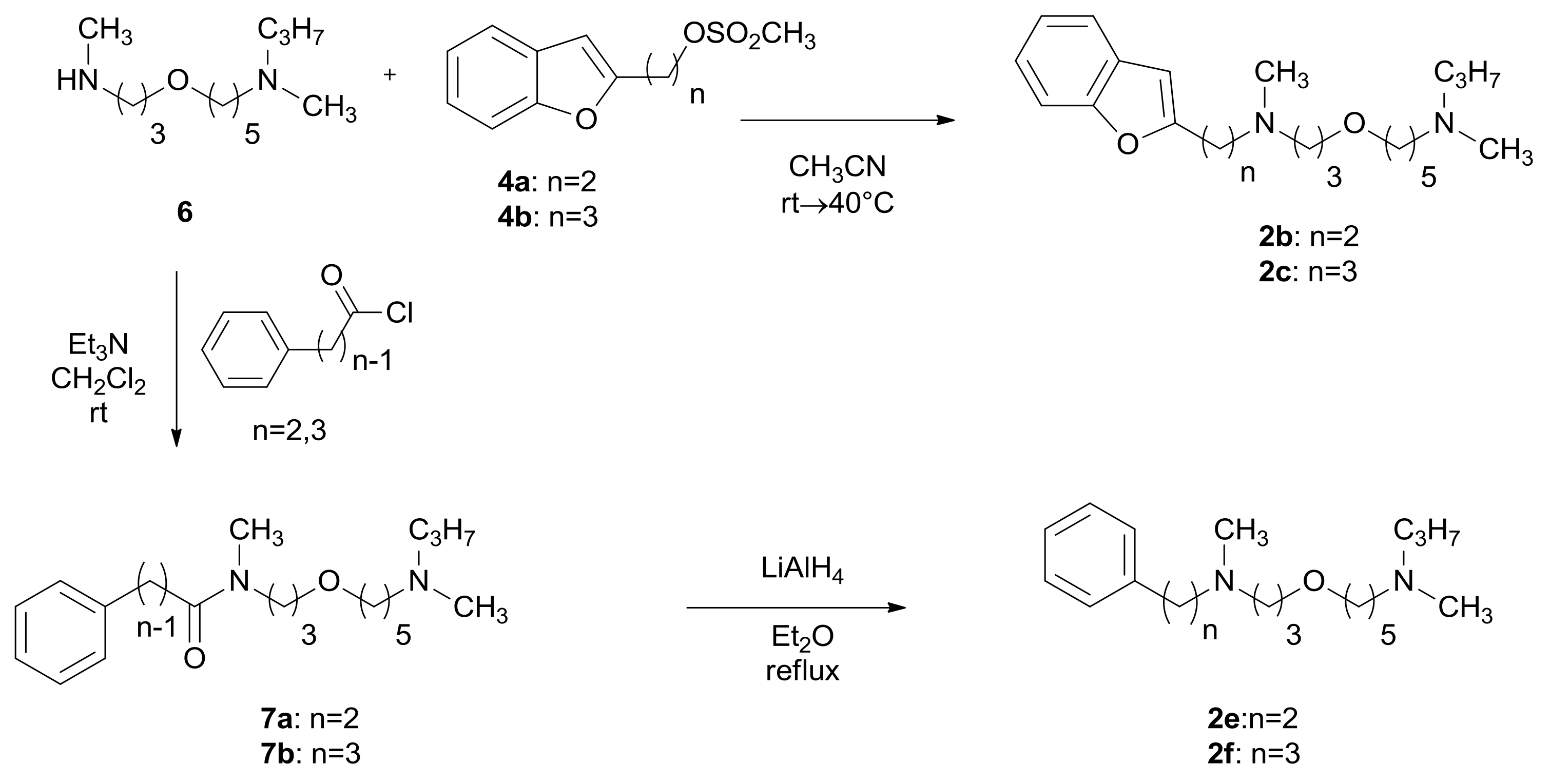
2.1. Chemistry
2.2. Pharmacology
2.2.1. In Vitro Pharmacological Studies
H3 Antagonistic Activity for Compounds 1a–f, and 2a–f
H1 Antagonistic Activity for Compounds 1b and 1f
2.2.2. Histamine H3 Receptor Affinity
Saturation of Rat and Human H3 Receptors
Competition Binding of H3 Receptor Ligands
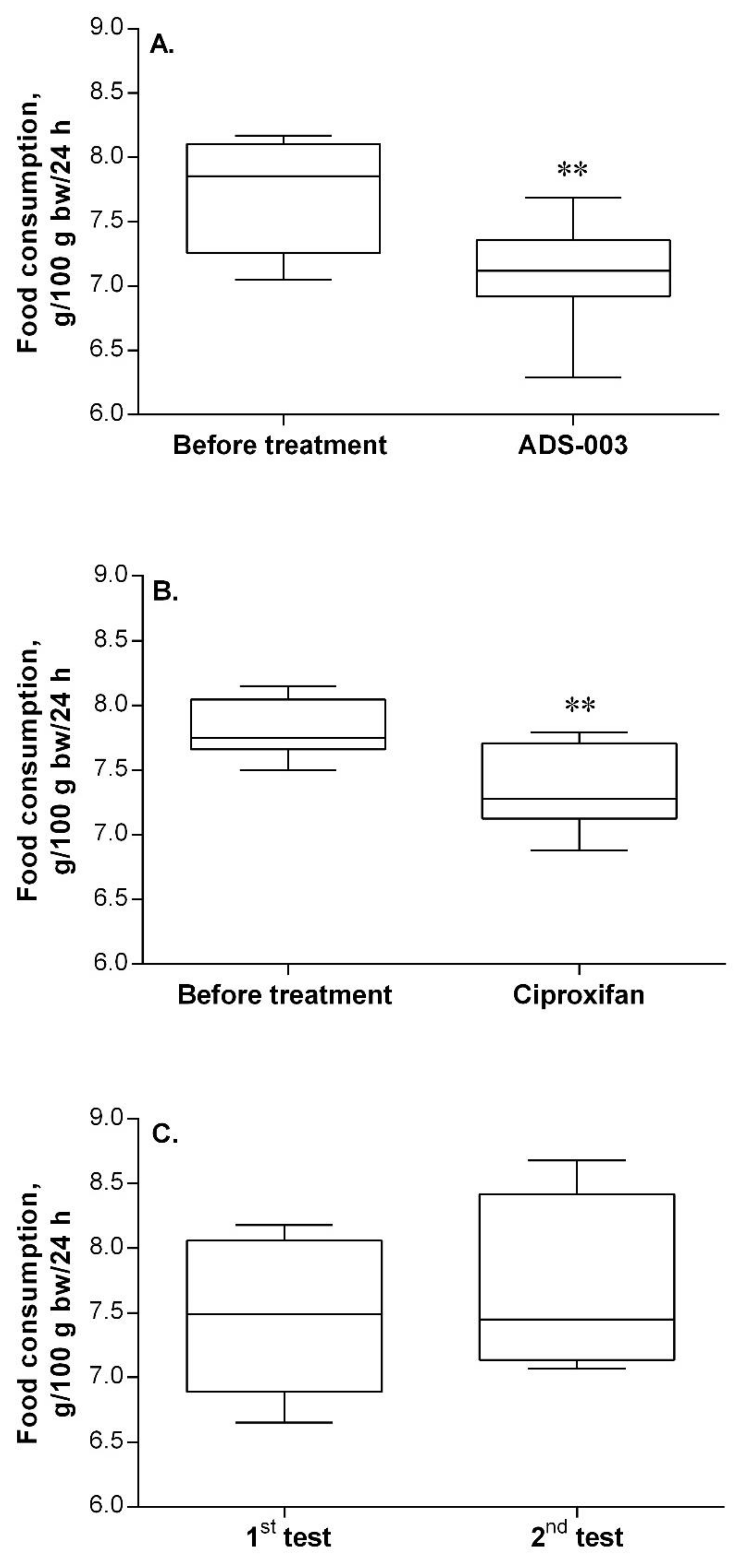
2.2.3. Verification of In Vivo Activity for Compound ADS-003 (1a)
- -
- The effects of the compound on the feeding behavior of rats after its repeated peripheral administration. Given that the compound enters the CNS and blocks the H3R, it should release histamine. Histamine, in turn, acting via H1R, would induce loss of appetite, i.e., the food intake of rats would decrease,
- -
- The influence on the cerebral amine neurotransmitter concentrations, as well as the activity of the monoamine oxidases A and B and histamine N-methyltransferase. The latter was accomplished by postmortem analyses of the brain tissues of the treated rats.
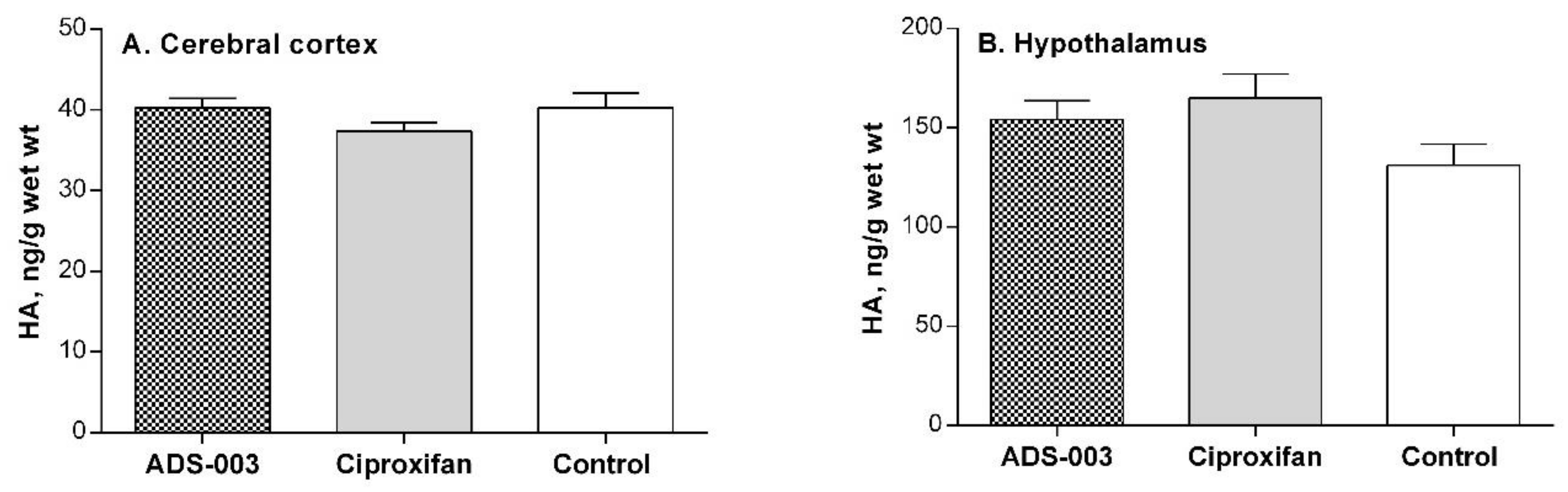
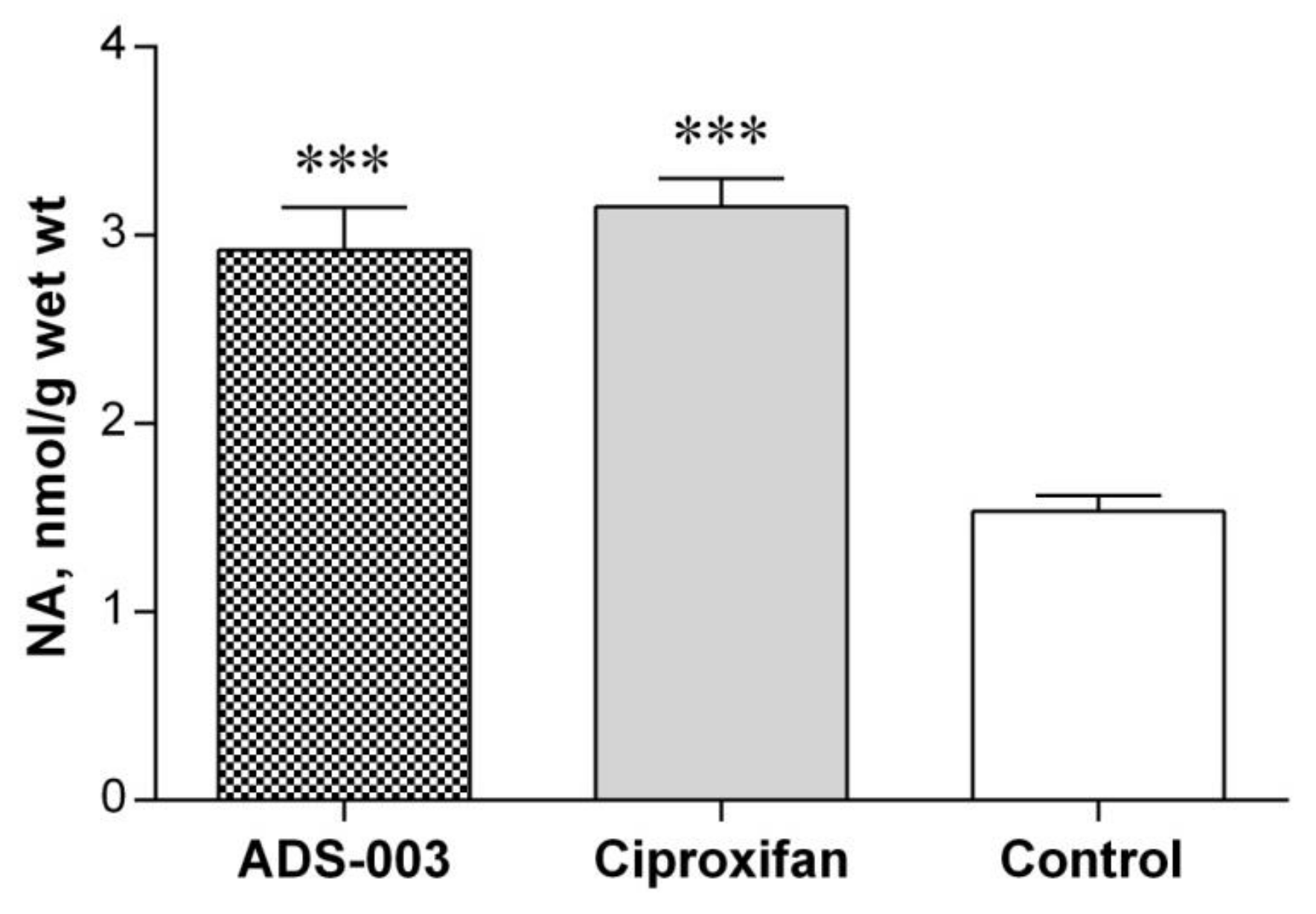
Post-Mortem Biochemical Analysis of the Brain Tissues of ADS-003 (1a)-Treated Rats
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
Chemicals
3.1.2. General Procedure for the Preparation of Compounds 1b,c and 2b,c
3.1.3. General Procedure for the Preparation of Compounds 5a,b and 7a,b
3.1.4. General Procedure for the Preparation of Compounds 1e,f and 2e,f
3.2. In Vitro Pharmacology
3.2.1. H3 Antagonistic Activity for Compounds 1a–f and 2a–f
3.2.2. H1 Antagonistic Activity for Compounds 1b and 1f
Chemicals
3.2.3. Antagonist Binding to Rat rH3R and Human hH3R
Cell Culture and Transfection
Crude Membrane Extracts
[3H]-Nα-Methylhistamine Binding
Chemicals
3.3. Verification of In Vivo Activity of Compound ADS-003 (1a)
3.3.1. Post-Mortem Biochemical Analyses
Chemicals
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arrang, J.-M.; Garbarg, M.; Schwartz, J.-C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 1983, 302, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Arrang, J.-M.; Devaux, B.; Chodkiewicz, J.-P.; Schwartz, J.-C. H3 receptors control release of histamine in human brain. J. Neurochem. 1988, 51, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Lovenberg, T.W.; Roland, B.L.; Wilson, S.J.; Jiang, X.; Pyati, J.; Huvar, A.; Jackson, M.R.; Erlander, M.G. Cloning and functional expression of the human histamine H3 receptor. Mol. Pharmacol. 1999, 55, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ramirez, J.; Ortiz, J.; Blanco, I. Presynaptic H3 autoreceptors modulate histamine synthesis through cAMP pathway. Mol. Pharmacol. 2002, 61, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Blandizzi, C.; Toghetti, M.; Colucci, R.; Del Tacca, M. Histamine H3 receptors mediate inhibition of noradrenaline release from intestinal sympathetic nerves. Br. J. Pharmacol. 2000, 129, 1387–1396. [Google Scholar] [CrossRef]
- Schlicker, E.; Fink, K.; Detzner, M.; Göthert, M.J. Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. Neural Transm. Gen. Sect. 1993, 93, 1–10. [Google Scholar] [CrossRef]
- Schlicker, E.; Betz, R.; Göthert, M. Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1988, 337, 588–590. [Google Scholar] [CrossRef]
- Bergquist, F.; Ruthven, A.; Ludwig, M.; Dutia, M.B. Histaminergic and glycinergic modulation of GABA release in the vestibular nuclei of normal and labyrinth ctomised rat. J. Physiol. 2006, 577, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Garduño-Torres, B.; Treviño, M.; Gutierrez, R.; Arias-Montaño, J.A. Pre-synaptic histamine H3 receptors regulate glutamate, but not GABA release in rat thalamus. Neuropharmacology 2007, 52, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Blandina, P.; Giorgetti, M.; Bartolini, L.; Cecchi, M.; Timmerman, H.; Leurs, R.; Pepeu, G.; Giovannini, M.G. Inhibition of cortical acetylcholine release and cognitive performance by histamine H3 receptor activation in rats. Br. J. Pharmacol. 1996, 119, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Hancock, A.A.; Fox, G.B. Cognitive enhancing effects of drugs that target histamine receptors. In Cognition-Enhancing Drugs Milestones in Drug Therapy; Buccafusco, J.J., Ed.; Birkhäuser Verlag: Basel, Switzerland, 2004; pp. 97–114. [Google Scholar]
- Onodera, K.; Miyazaki, S.; Imaizumi, M.; Stark, H.; Schunack, W. Improvement by FUB 181, a novel histamine H3-receptor antagonist, of learning and memory in the elevated plus-maze test in mice. Naunyn Schmiedeberg’s Arch. Pharmacol. 1998, 357, 508–513. [Google Scholar] [CrossRef]
- Passani, M.B.; Cangioli, I.; Bocciottini, L.; Mannaioni, P.F. Thioperamide, and cimetidine modulate acetylcholine release from the amygdala of freely moving rats. Inflamm. Res. 2000, 49, S43–S44. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, M.; Khanam, R.; Vohora, D. Histamine H3 receptor antagonists in relation to epilepsy and neurodegeneration: A systemic consideration of recent progress and perspectives. Br. J. Pharmacol. 2012, 167, 1398–1414. [Google Scholar] [CrossRef] [PubMed]
- Pillot, C.; Ortiz, J.; Héron, A.; Ridray, S.; Schwartz, J.-C.; Arrang, J.-M. Ciproxifan, a histamine H3-receptor antagonist/inverse agonist, potentiates neurochemical and behavioral effects of haloperidol in the rat. J. Neurosci. 2002, 22, 7272–7280. [Google Scholar] [CrossRef]
- Takahashi, K.; Suwa, H.; Ishikawa, T.; Kotani, H.J. Targeted disruption of H3 receptors results in changes in brain histamine tone leading to an obese phenotype. J. Clin. Investig. 2002, 110, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Van der Goot, H.; Timmerman, H. Selective ligands as tools to study histamine receptors. Eur. J. Med. Chem. 2000, 35, 5–20. [Google Scholar] [CrossRef]
- Stark, H.; Kathmann, M.; Schlicker, E.; Schunack, W.; Schlegel, B.; Sippl, W. Medicinal chemical and pharmacological aspects of imidazole-containing histamine H3 receptor antagonists. Mini-Rev. Med. Chem. 2004, 4, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, S.; Flau, K.; Kathmann, M.; Schlicker, E.; Stark, H.; Meier, G.; Schunack, W. Replacement of imidazole by a piperidine moiety differentially affects the potency of histamine H3-receptor antagonists. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 367, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Hey, J.A.; Aslanian, R.; Rizzo, C.A. Coordination of histamine H3 receptor antagonists with human adrenal cytochrome P450 enzymes. Pharmacology 2002, 66, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, S.; Wijtmans, M.; Lim, H.D.; Leurs, R.; de Esch, I.J.P. Several down, a few to go: Histamine H3 receptor ligands making the final push towards the market? Expert Opin. Investig. Drugs 2011, 20, 1629–1648. [Google Scholar] [CrossRef] [PubMed]
- Pomponi, S.A.; Gullo, V.P.; Horan, A.C.; Patel, M.G. Coval SUS (Harbor Branch Oceanographic Institution). U.S. Patent 5352707, 4 October 1994. [Google Scholar]
- Swanson, D.M.; Wilson, S.J.; Boggs, J.D.; Xiao, W.; Apodaca, R.; Barbier, A.J.; Lovenberg, T.W.; Carruthers, N.I. Aplysamine-1 and related analogs as histamine H3 receptor antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Apodaca, R.; Dvorak, C.A.; Xiao, W.; Barbier, A.J.; Boggs, J.D.; Wilson, S.J.; Lovenberg, T.W.; Carruthers, N.I. A new class of diamine-based human histamine H3 receptor antagonists: 4-(Aminoalkoxy)benzylamines. J. Med. Chem. 2003, 46, 3938–3944. [Google Scholar] [CrossRef] [PubMed]
- Ligneau, X.; Perrin, D.; Landais, L.; Camelin, J.C.; Calmels, T.P.; Berrebi-Bertrand, I.; Lecomte, J.M.; Parmentrier, R.; Anaclet, C.; Lin, J.S.; et al. BF2.649 [1-{3-[3-(4 Chlorophenyl)propoxy]propyl}piperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor. J. Pharmacol. Exp. Ther. 2007, 320, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, C.A.; Apodaca, R.; Barbier, A.J.; Berridge, C.W.; Wilson, S.J.; Boggs, J.D.; Xiao, W.; Lovenberg, T.W.; Carruthers, N.I. 4-phenoxypiperidines: Potent, conformationally restricted, non-imidazolehistamine H3 antagonists. J. Med. Chem. 2005, 48, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Cowart, M.D.; Bennani, Y.L.; Faghih, R.; Gfesser, G.A.; Black, L.A. Abbott Laboratories, USA. U.S. Patent 20020183309, 12 May 2002. [Google Scholar]
- Staszewski, M.; Walczyński, K. Synthesis and preliminary pharmacological investigation of new N-substituted-N-[ω-(ω-phenoxy-alkylpiperazin-1-yl)alkyl]guanidines as non-imidazole histamine H3 antagonists. Arch. Pharm. Chem. Life Sci. 2012, 345, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Staszewski, M.; Walczyński, K. 1-Phenoxyalkyl-4-[(N,N-disubstitutedamino)alkyl]-piperazine derivatives as non-imidazole histamine H3-antagonists. Med. Chem. Res. 2013, 22, 1287–1304. [Google Scholar] [CrossRef]
- Masłowska-Lipowicz, I.; Figlus, M.; Zuiderveld, O.P.; Walczyński, K. New 1-benzyl-4-hydroxypiperidine derivatives as non-imidazole histamine H3 receptor antagonists. Arch. Pharm. 2008, 341, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Masłowska-Lipowicz, I.; Walczyński, K. Structure-activity relationships of new 1-substitutedmethyl-4-[5-(N-methyl-N-propylamino)pentyloxy]piperidines and selected 1-[(N-substituted-N-methyl)-3-propyloxy]-5-(N-methy-l-N-propyl)-pentanediamines as H3-antagonists. Chem. Biol. Drug Des. 2014, 83, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Vollinga, R.C.; Zuiderveld, O.P.; Scheerens, H.; Bast, A.; Timmerman, H. A simple and rapid in vitro test system for the screening of histamine H3 ligands. Methods Find. Exp. Clin. Pharmacol. 1992, 105, 747. [Google Scholar]
- Sibasish, P.; Sankha, P.; Surajit, S. Synthesis of new series of iboga analogues. Tetrahedron Lett. 2011, 52, 6166–6169. [Google Scholar] [CrossRef]
- Arunlakshana, O.; Schild, H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1959, 14, 48–55. [Google Scholar] [CrossRef]
- Bongers, G.; Sallmen, T.; Passani, M.B.; Mariottini, C.; Wendelin, D.; Lozada, A.; van Marle, A.; Navis, M.; Blandina, P.; Bakker, R.A.; et al. The Akt/GSK-3b axis as a new signaling pathway of the histamine H3 receptor. J. Neurochem. 2007, 103, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Guryn, R.; Staszewski, M.; Stasiak, A.; McNaught Flores, D.; Fogel, W.A.; Leurs, R.; Walczyński, K. Non-imidazole histamine H3 ligands. Part VII. Synthesis, in vitro and in vivo characterization of 5-substituted-2-thiazol-4-n-propylpiperazines. Molecules 2018, 23, 326. [Google Scholar] [CrossRef] [PubMed]
- Glowinski, J.; Iversen, L.L. Regional studies of catecholamines in the rat Brain-I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various region of the brain. J. Neurochem. 1966, 13, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Clineschmidt, B.V.; Lotti, V.J. Histamine: Intraventricular injection suppresses ingestive behavior of the cat. Arch. Int. Pharmacodyn. Ther. 1973, 206, 288–298. [Google Scholar] [PubMed]
- Masaki, T.; Chiba, S.; Yasuda, T.; Noguchi, H.; Kakuma, T.; Watanabe, T.; Sakata, T.; Yoshimatsu, H. Involvement of hypothalamic histamine H1 receptor in the regulation of feeding rhythm and obesity. Diabetes 2004, 53, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Provensi, G.; Coccurello, R.; Umehara, H.; Munari, L.; Giacovazzo, G.; Galeotti, N.; Nosi, D.; Gaetani, S.; Romano, A.; Moles, A.; et al. Satiety factor oleoylethanolamide recruits the brain histaminergic system to inhibit food intake. Proc. Natl. Acad. Sci. USA 2014, 111, 11527–11532. [Google Scholar] [CrossRef] [PubMed]
- Provensi, G.; Blandina, P.; Passani, M.B. The histaminergic system as a target for the prevention of obesity and metabolic syndrome. Neuropharmacology 2016, 106, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ligneau, X.; Lin, J.-S.; Vanni-Mercier, G.; Jouvet, M.; Muir, J.L.; Ganellin, C.R.; Stark, H.; Elz, S.; Schunack, W.; Schwartz, J.-C. Neurochemical and behavioral effects of ciproxifan, a potent histamine H3-receptor antagonist. JPET 1998, 297, 658–666. [Google Scholar]
- Schlicker, E.; Werthwein, S.; Zentner, J. Histamine H3 receptor-mediated inhibition of noradrenaline release in the human brain. Fundam. Clin. Pharmacol. 1999, 13, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Flik, G.; Folgering, J.H.; Cremers, T.I.; Westerink, B.H.; Dremencov, E. Interaction between brain histamine and serotonin, norepinephrine, and dopamine systems: In vivo microdialysis and electrophysiology study. J. Mol. Neurosci. 2015, 56, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Snyder, S.H. Isotopic microassay of histamine, histidine, histidine decarboxylase and histamine methyltransferase in brain tissue. J. Neurochem. 1972, 19, 1343–1358. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J.; Tipton, K.F. Deamination of 5-hydroxytryptamine by both forms of monoamine oxidase in the rat brain. J. Neurochem. 1982, 38, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Code Cpd. | Structure | pA2 (sem) a | N (caviae) |
|---|---|---|---|---|
| 1a [31] | ADS-003 |  | 8.27 (0.05) (8.47) * | 12 (4) |
| 1b | ADS-013 |  | 7.26 (0.25) | 12 (4) |
| 1c | ADS-014 |  | 7.11 (0.12) | 12 (4) |
| 1d [30] | ADS-009 |  | 7.79 (0.06) | 12 (4) |
| 1e | ADS-015 |  | 6.67 (0.12) | 9 (4) |
| 1f | ADS-016 |  | 7.51 (0.03) | 8 (4) |
| 2a [31] | ADS-012 |  | 6.23 (0.12) | 12 (4) |
| 2b | ADS-017 |  | 6.32 (0.52) | 5 (4) |
| 2c | ADS-018 |  | 6.37 (0.01) | 6 (4) |
| 2d [31] | ADS-011 |  | 8.06 (0.05) | 12 (4) |
| 2e | ADS-019 |  | 6.72 (0.06) | 6 (4) |
| 2f | ADS-020 |  | 6.79 (0.01) | 6 (4) |
| Thioperamide | 8.44 (0.26) | 36 (12) |
| Group | MAO-A | MAO-B | HMNT | |
|---|---|---|---|---|
| pmol/min/mg Protein | pmol/min/mg Protein | |||
| CTX | CTX | CTX | HTH | |
| ADS-003 | 1536 ± 50 | 1065 ± 48 | 44.41 ± 1.06 | 41.09 ± 0.80 |
| Ciproxifan | 1654 ± 31 | 1049 ± 56 | 46.90 ± 3.59 | 51.76 ± 5.67 |
| Control | 1506 ± 29 | 1054 ± 19 | 45.97 ± 1.28 | 39.81 ± 1.94 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olszewska, B.; Stasiak, A.; McNaught Flores, D.; Fogel, W.A.; Leurs, R.; Walczyński, K. 4-Hydroxypiperidines and Their Flexible 3-(Amino)propyloxy Analogues as Non-Imidazole Histamine H3 Receptor Antagonist: Further Structure–Activity Relationship Exploration and In Vitro and In Vivo Pharmacological Evaluation. Int. J. Mol. Sci. 2018, 19, 1243. https://doi.org/10.3390/ijms19041243
Olszewska B, Stasiak A, McNaught Flores D, Fogel WA, Leurs R, Walczyński K. 4-Hydroxypiperidines and Their Flexible 3-(Amino)propyloxy Analogues as Non-Imidazole Histamine H3 Receptor Antagonist: Further Structure–Activity Relationship Exploration and In Vitro and In Vivo Pharmacological Evaluation. International Journal of Molecular Sciences. 2018; 19(4):1243. https://doi.org/10.3390/ijms19041243
Chicago/Turabian StyleOlszewska, Beata, Anna Stasiak, Daniel McNaught Flores, Wiesława Agnieszka Fogel, Rob Leurs, and Krzysztof Walczyński. 2018. "4-Hydroxypiperidines and Their Flexible 3-(Amino)propyloxy Analogues as Non-Imidazole Histamine H3 Receptor Antagonist: Further Structure–Activity Relationship Exploration and In Vitro and In Vivo Pharmacological Evaluation" International Journal of Molecular Sciences 19, no. 4: 1243. https://doi.org/10.3390/ijms19041243