Inactivation of SREBP-1a Phosphorylation Prevents Fatty Liver Disease in Mice: Identification of Related Signaling Pathways by Gene Expression Profiles in Liver and Proteomes of Peroxisomes

 , ,
, ,
Abstract
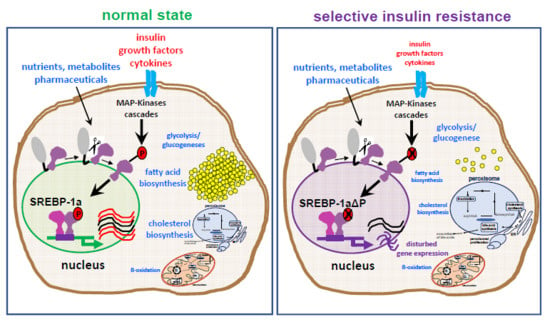
:
1. Introduction
2. Results
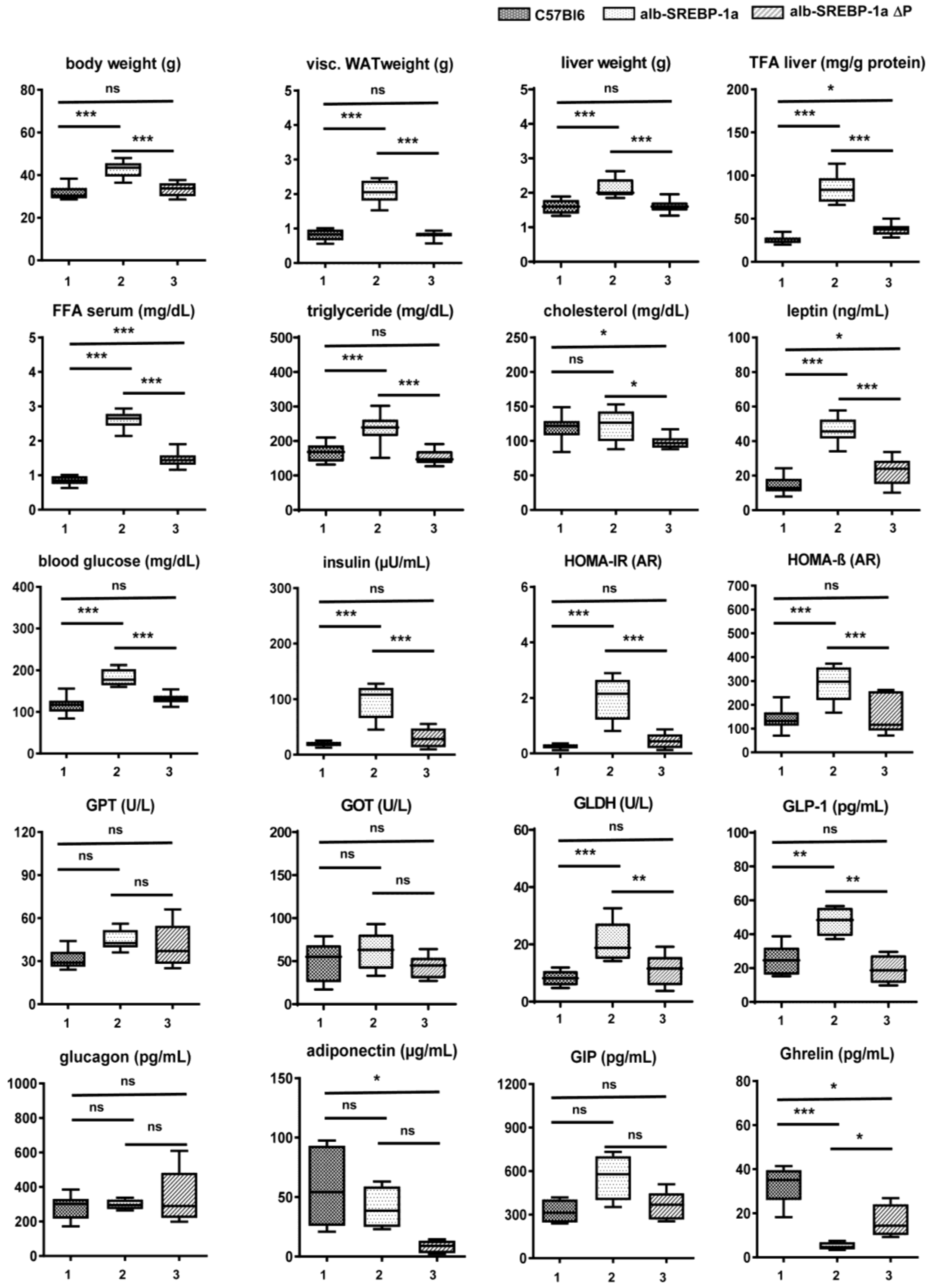
2.1. Influence of SREBP-1a Phosphorylation on the Phenotype
2.2. Role of Functional MAPK-Related Phosphorylation Sites in SREBP-1a for Hepatic Gene Expression
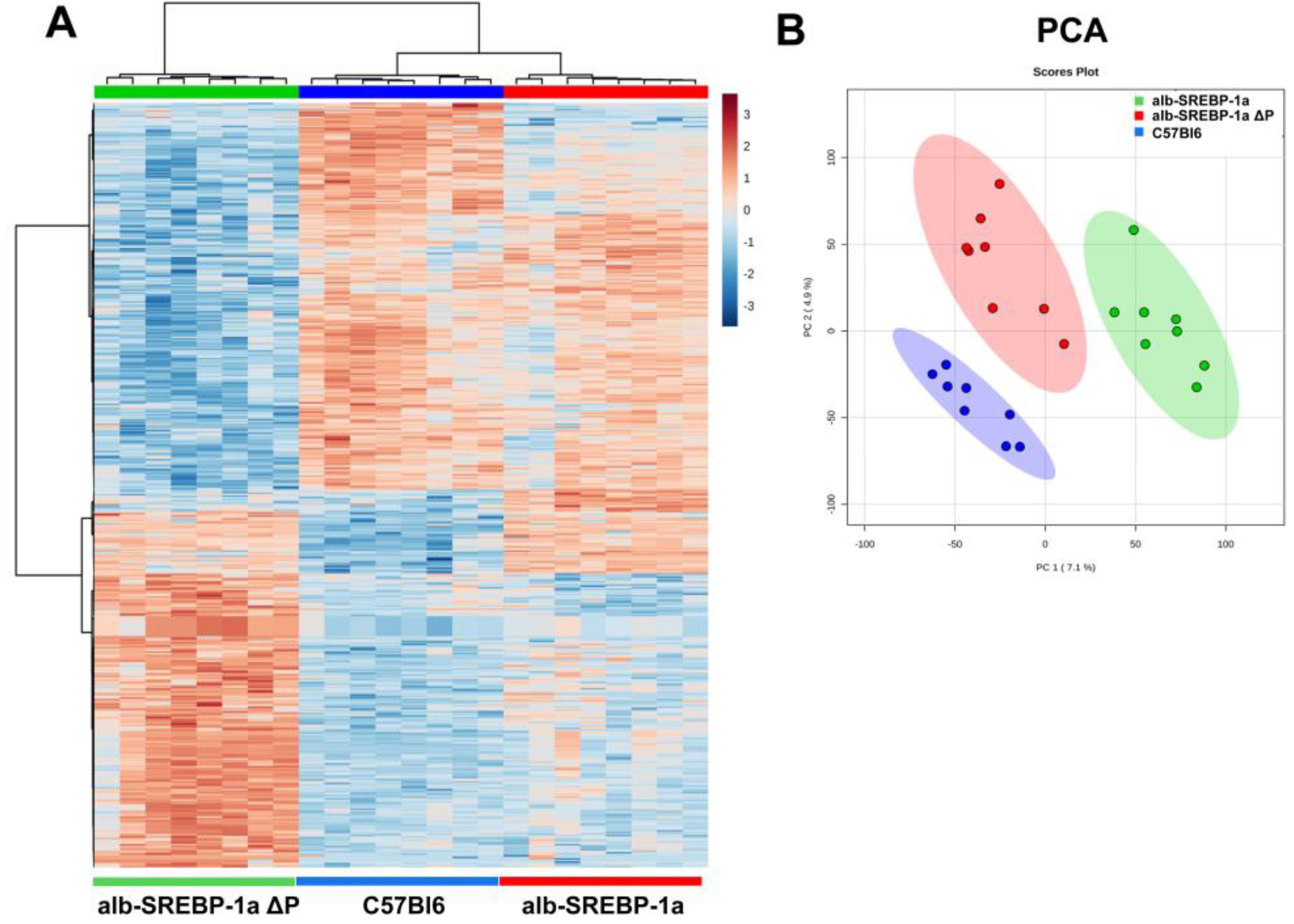
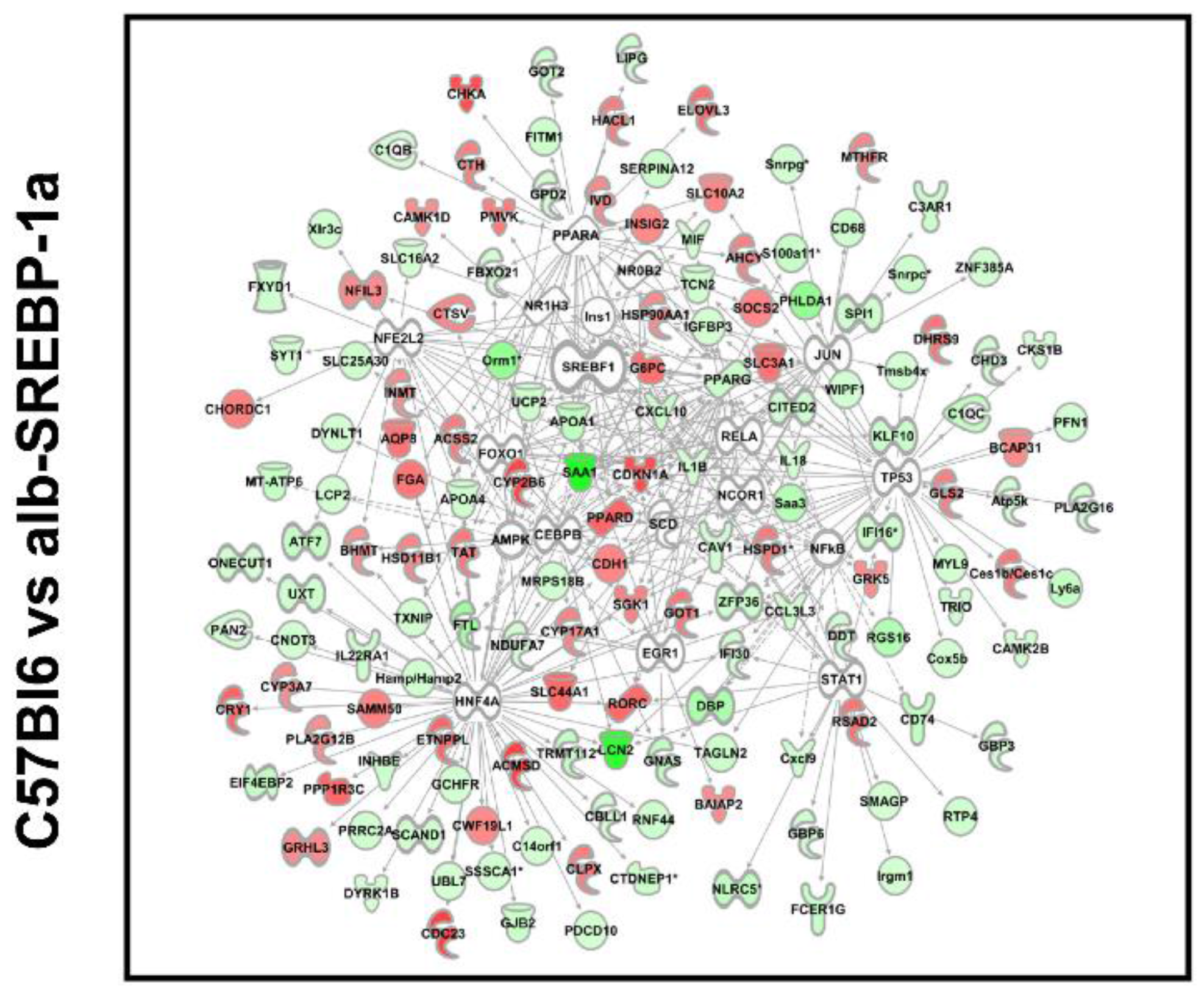
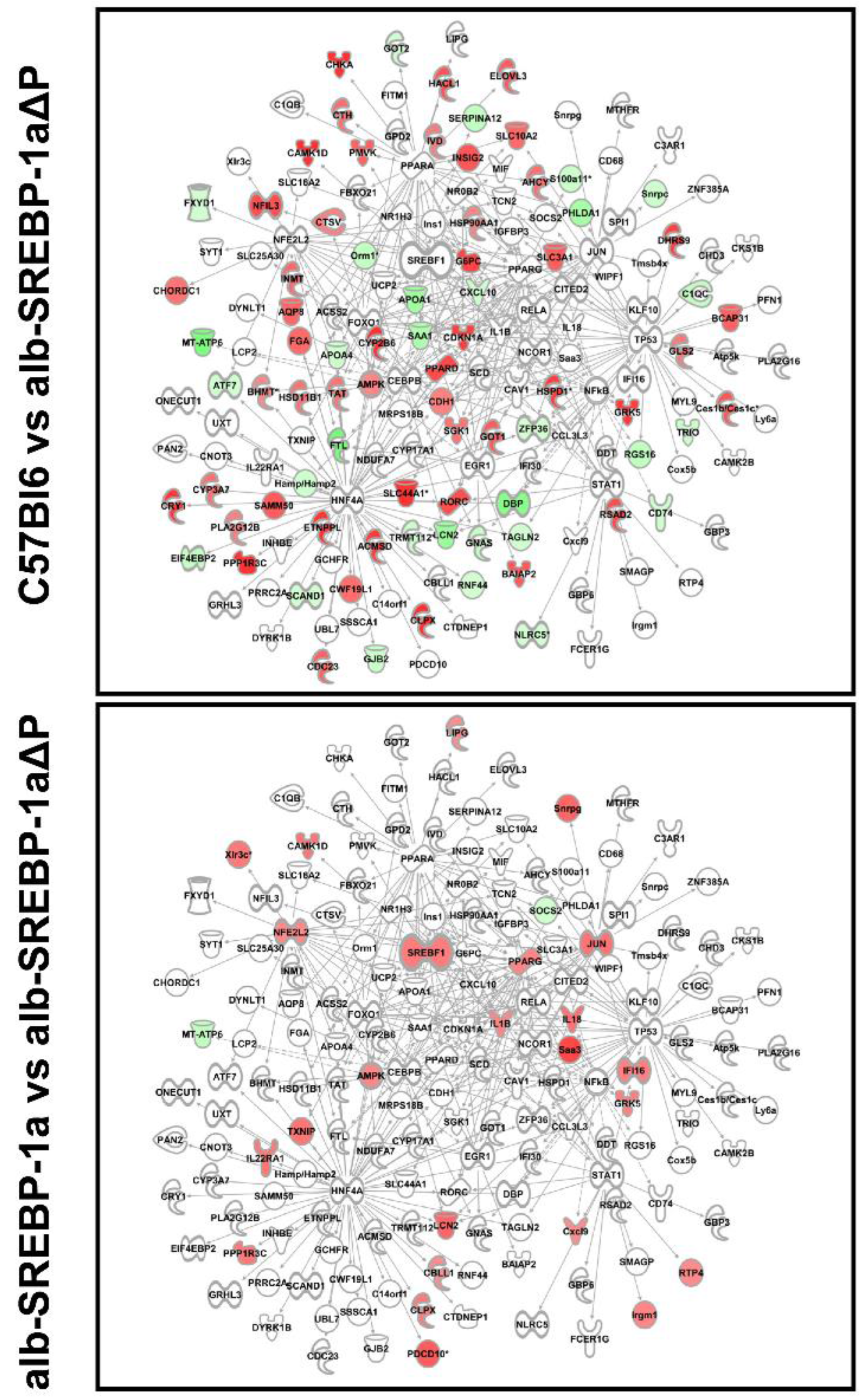
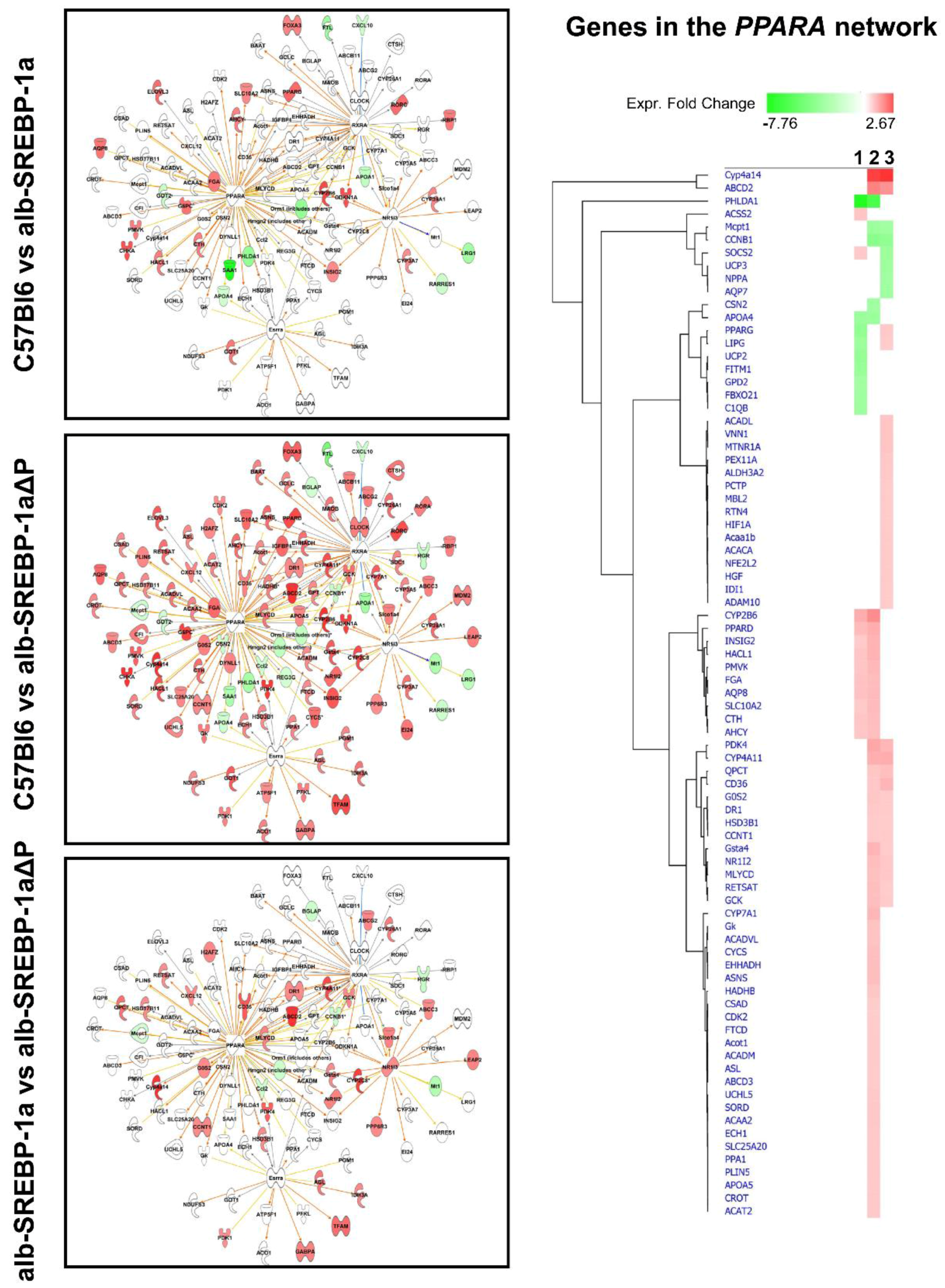
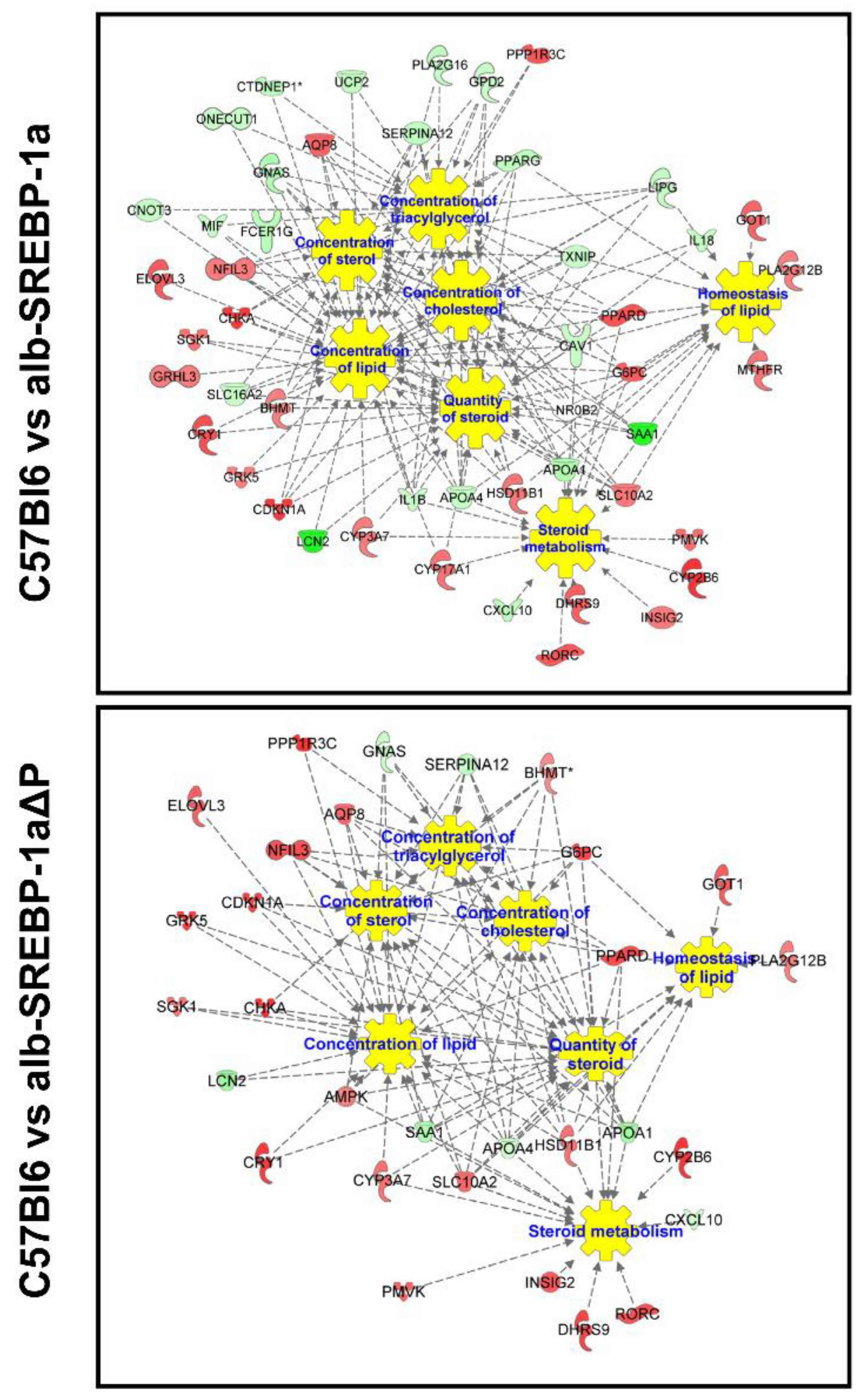
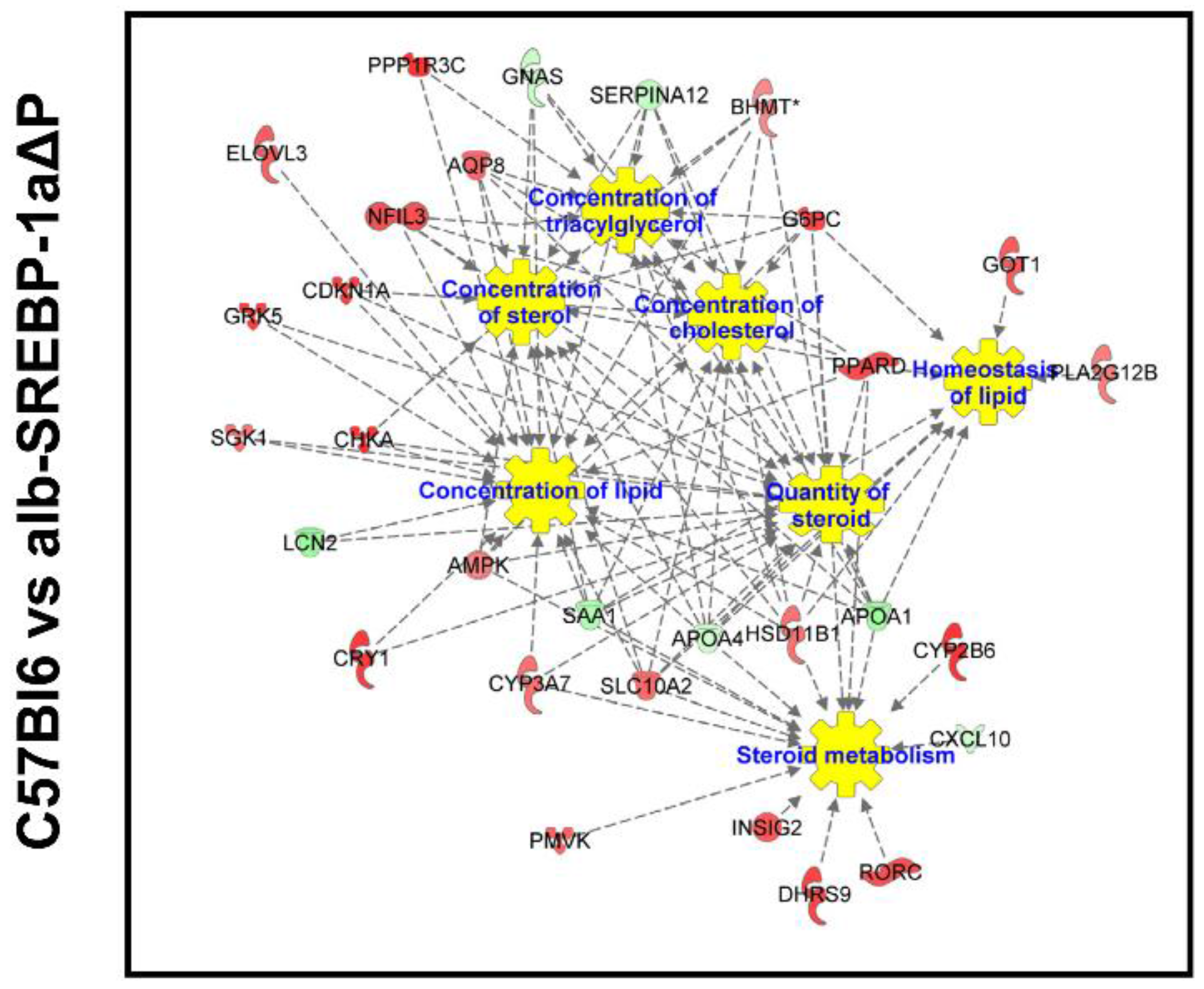
2.3. Overall Hepatic Gene Expression Differences in C57Bl6 vs. alb-SREBP-1a
2.4. Overall Hepatic Gene Expression Differences in C57Bl6 vs. alb-SREBP-1a∆P
2.5. Overall Hepatic Gene Expression Differences in alb-SREBP-1a vs. alb-SREBP-1a∆P
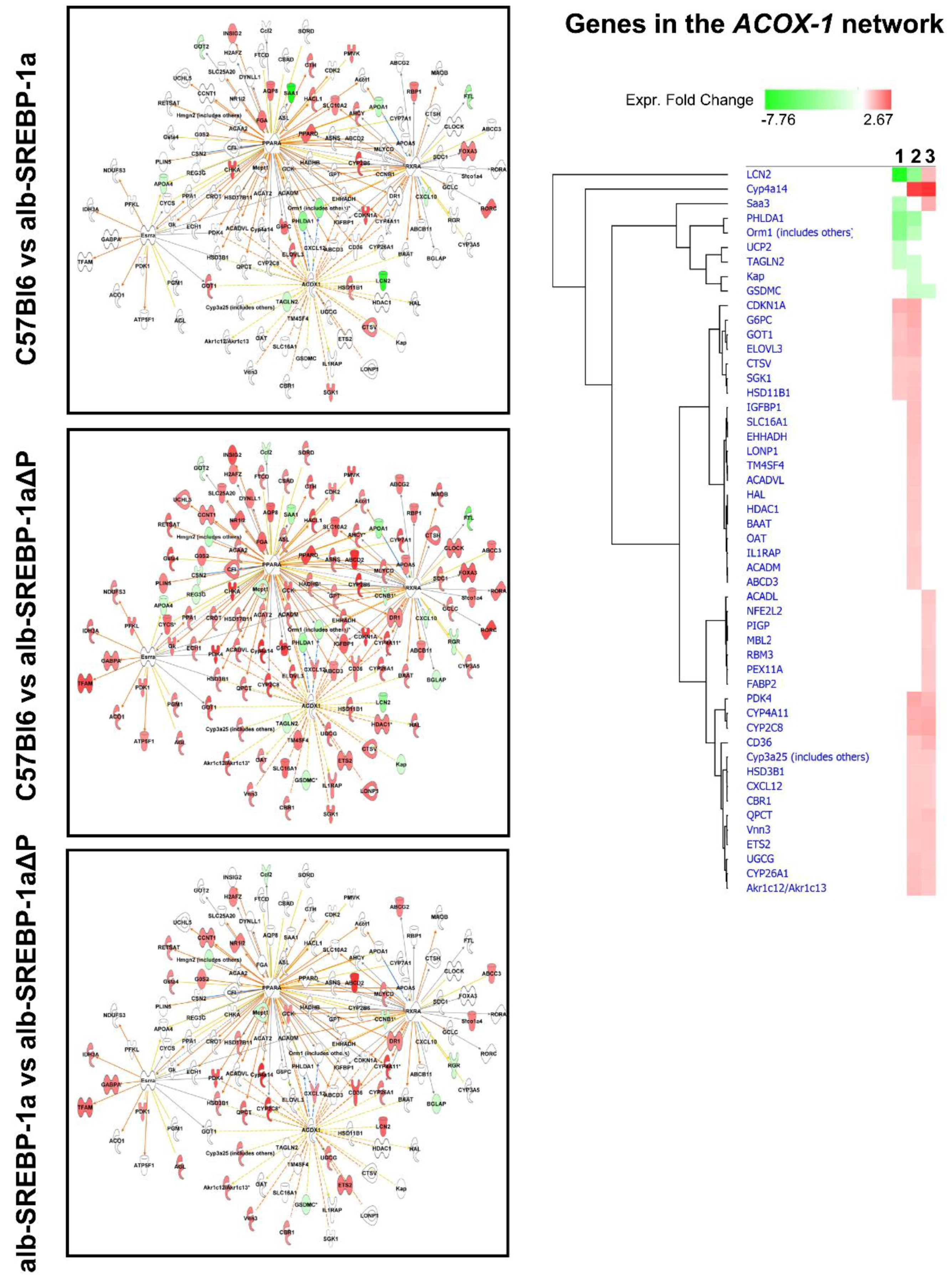
2.6. Systemic Influence of SREBP-1a Phosphorylation on Hepatic Gene Expression
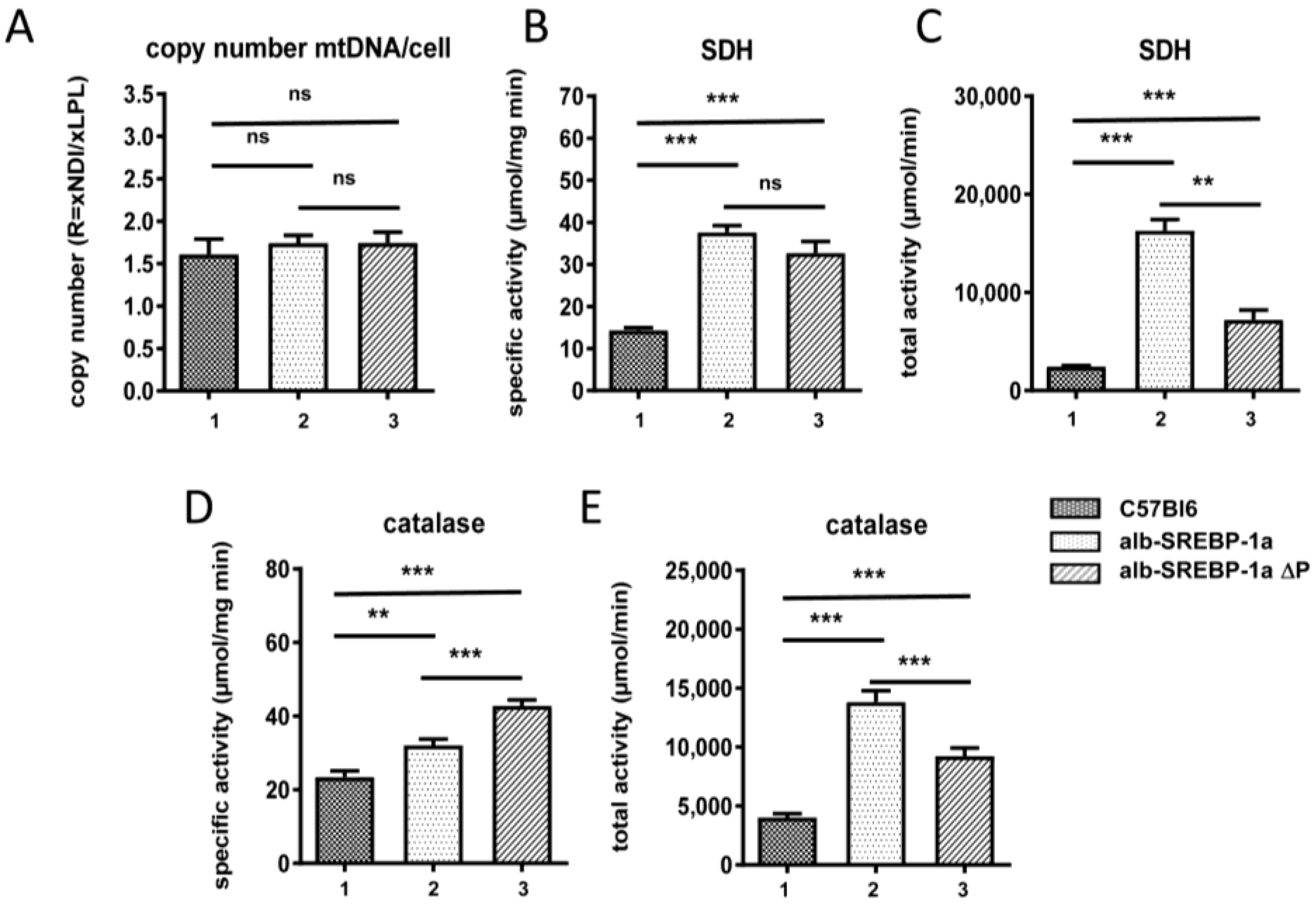
2.7. Effect of Disruption of SREBP-1a Phosphorylation at MAPK Sites on Peroxisomal Proteome Patterns
3. Discussion
4. Materials and Methods
4.1. Animals and Phenotype Characterization
4.2. Gene Expression Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflict of Interest
Abbreviations
| ABCB1 | ATP-binding cassette, sub-family B (MDR/TAP), member 1 |
| ABDH5 | 1-acylglycerol-3-phosphate O-acyltransferase |
| ACOX1 | acyl-Coenzyme A oxidase 1 |
| DUS | desmoplakin |
| ERK | mitogen-activated protein kinase 1 |
| Essra | estrogen receptor releated alpha |
| FABP1 | fatty acid binding protein 1 |
| FBXO32 | F-box protein 32 |
| FBXW | F-box and WD repeat containing |
| FOXO4 | forkhead box O4 |
| FXR | nuclear receptor subfamily 1, group H, member 4 |
| GPD1 | glycerol-3-phosphate dehydrogenase 1 |
| GPD1 | hexose-6-phosphate dehydrogenase |
| GRB14 | growth factor receptor bound protein 14 |
| GSK3 | glycogen synthase kinase 3 beta |
| HNF4A | hepatic nuclear factor 4, alpha |
| INSR | insulin receptor |
| JNK | mitogen-activated protein kinase 8 |
| KLF15 | Kruppel-like factor 15 |
| Lcn2 | lipocalin 2 |
| LDL | low density lipoprotein |
| LEP | leptin |
| LONP1 | lon peptidase 1 |
| LXR | Liver X receptor |
| MAPK | mitogen-activated protein kinase |
| MAPKAP4 | mitogen-activated protein kinase activating protein 4 |
| MYC | myelocytomatosis oncogene |
| NCAM1 | neural cell adhesion molecule 1 |
| NR1D1 | nuclear receptor subfamily 1, group D, member 1 |
| NR2F1 | nuclear receptor subfamily 2, group F, member 1 |
| NR2F1 | nuclear receptor subfamily 2, group F, member 2 |
| ONECUT1 | one cut domain, family member 1 |
| PPARA | peroxisome proliferator activated receptor alpha |
| PPARD | peroxisome proliferator activator receptor delta |
| PPARGC1A | peroxisome proliferative activated receptor, gamma, coactivator 1 alpha |
| RORA | retinoic acid receptor (RAR)-related orphan receptor alpha |
| RORC | retinoic acid receptor (RAR)-related orphan receptor gamma |
| RXR | retinoid x receptor |
| SIRT4 | sirtuin 4 |
| SLC25A13 | solute carrier family 25 member 13 |
| TCA | tricarboxylic acid |
| VLDL | very low density lipoprotein |
References
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochimica et Biophysica Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Parekh, S.; Anania, F.A. Abnormal lipid and glucose metabolism in obesity: Implications for nonalcoholic fatty liver disease. Gastroenterology 2007, 132, 2191–2207. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Ann. Rev. Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Shimano, H.; Horton, J.D.; Goldstein, J.L.; Brown, M.S. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J. Clin. Investig. 1997, 99, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Streicher, R.; Kotzka, J.; Muller-Wieland, D.; Siemeister, G.; Munck, M.; Avci, H.; Krone, W. SREBP-1 mediates activation of the low density lipoprotein receptor promoter by insulin and insulin-like growth factor-I. J. Biol. Chem. 1996, 271, 7128–7133. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.; Kotzka, J.; Kremer, L.; Lehr, S.; Lohaus, C.; Meyer, H.E.; Krone, W.; Muller-Wieland, D. MAP kinases Erk1/2 phosphorylate sterol regulatory element-binding protein [SREBP]-1a at serine 117 in vitro. J. Biol. Chem. 2000, 275, 33302–33307. [Google Scholar] [CrossRef] [PubMed]
- Kotzka, J.; Knebel, B.; Haas, J.; Kremer, L.; Jacob, S.; Hartwig, S.; Nitzgen, U.; Muller-Wieland, D. Preventing phosphorylation of sterol regulatory element-binding protein 1a by MAP-kinases protects mice from fatty liver and visceral obesity. PLoS ONE 2012, 7, e32609. [Google Scholar] [CrossRef] [PubMed]
- Kotzka, J.; Knebel, B.; Avci, H.; Jacob, S.; Nitzgen, U.; Jockenhovel, F.; Heeren, J.; Haas, J.; Muller-Wieland, D. Phosphorylation of sterol regulatory element-binding protein [SREBP]-1a links growth hormone action to lipid metabolism in hepatocytes. Atherosclerosis 2010, 213, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Kotzka, J.; Lehr, S.; Roth, G.; Avci, H.; Knebel, B.; Muller-Wieland, D. Insulin-activated Erk-mitogen-activated protein kinases phosphorylate sterol regulatory element-binding Protein-2 at serine residues 432 and 455 in vivo. J. Biol. Chem. 2004, 279, 22404–22411. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea-Alonso, M.T.; Ericsson, J. The phosphorylation-dependent regulation of nuclear SREBP1 during mitosis links lipid metabolism and cell growth. Cell Cycle 2016, 15, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Hartwig, S.; Haas, J.; Lehr, S.; Goeddeke, S.; Susanto, F.; Bohne, L.; Jacob, S.; Koellmer, C.; Nitzgen, U.; et al. Peroxisomes compensate hepatic lipid overflow in mice with fatty liver. Biochimica et Biophysica Acta 2015, 1851, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Goddeke, S.; Hartwig, S.; Horbelt, T.; Fahlbusch, P.; Al-Hasani, H.; Jacob, S.; Koellmer, C.; Nitzgen, U.; Schiller, M.; et al. Alteration of Liver Peroxisomal and Mitochondrial Functionality in the NZO Mouse Model of Metabolic Syndrome. Proteom. Clin. Appl. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Han, S.; Kitamura, T.; Accili, D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J. Clin. Investig. 2006, 116, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; White, M.F. Targeting Forkhead box O1 from the concept to metabolic diseases: Lessons from mouse models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Kollmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef] [PubMed]
- Engelking, L.J.; Cantoria, M.J.; Xu, Y.; Liang, G. Developmental and extrahepatic physiological functions of SREBP pathway genes in mice. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Pacini, G.; Musso, G.; Gambino, R.; Mecca, F.; Depetris, N.; Cassader, M.; David, E.; Cavallo-Perin, P.; Rizzetto, M. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: Further evidence for an etiologic association. Hepatology 2002, 35, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tsuchiya, H.; Zhang, Y.; Lee, S.; Liu, C.; Huang, Y.; Vargas, G.M.; Wang, L. REV-ERBα Activates C/EBP Homologous Protein to Control Small Heterodimer Partner-Mediated Oscillation of Alcoholic Fatty Liver. Am. J. Pathol. 2016, 186, 2909–2920. [Google Scholar] [CrossRef] [PubMed]
- Le Martelot, G.; Claudel, T.; Gatfield, D.; Schaad, O.; Kornmann, B.; Lo Sasso, G.; Moschetta, A.; Schibler, U. REV-ERBα participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. 2009, 7, e1000181. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, H.; Zheng, H.; Jiang, Y. Hepatic inflammation scores correlate with common carotid intima-media thickness in rats with NAFLD induced by a high-fat diet. BMC Vet. Res. 2014, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Liu, Y.H.; Fu, Y.C.; Liu, X.M.; Zhou, X.H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- Cheng, J.; Liu, C.; Hu, K.; Greenberg, A.; Wu, D.; Ausman, L.M.; McBurney, M.W.; Wang, X.D. Ablation of systemic SIRT1 activity promotes nonalcoholic fatty liver disease by affecting liver-mesenteric adipose tissue fatty acid mobilization. Biochimica et Biophysica Acta 2017, 1863, 2783–2790. [Google Scholar] [CrossRef] [PubMed]
- Shum, E.Y.; Espinoza, J.L.; Ramaiah, M.; Wilkinson, M.F. Identification of novel post-transcriptional features in olfactory receptor family mRNAs. Nucleic Acids Res. 2015, 43, 9314–9326. [Google Scholar] [CrossRef] [PubMed]
- Munakata, Y.; Yamada, T.; Imai, J.; Takahashi, K.; Tsukita, S.; Shirai, Y.; Kodama, S.; Asai, Y.; Sugisawa, T.; Chiba, Y.; et al. Olfactory receptors are expressed in pancreatic β-cells and promote glucose-stimulated insulin secretion. Sci. Rep. 2018, 8, 1499. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hwang, S.H.; Jia, Y.; Choi, J.; Kim, Y.J.; Choi, D.; Pathiraja, D.; Choi, I.G.; Koo, S.H.; Lee, S.J. Olfactory receptor 544 reduces adiposity by steering fuel preference toward fats. J. Clin. Investig. 2017, 127, 4118–4123. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L. Renal and cardiovascular sensory receptors and blood pressure regulation. Am. J. Physiol. Ren. Physiol. 2013, 305, F439–F444. [Google Scholar] [CrossRef] [PubMed]
- Paz-Filho, G.; Mastronardi, C.A.; Parker, B.J.; Khan, A.; Inserra, A.; Matthaei, K.I.; Ehrhart-Bornstein, M.; Bornstein, S.; Wong, M.L.; Licinio, J. Molecular pathways involved in the improvement of non-alcoholic fatty liver disease. J. Mol. Endocrinol. 2013, 51, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Osborne, T.F. Activation domains from both monomers contribute to transcriptional stimulation by sterol regulatory element-binding protein dimers. J. Biol. Chem. 2005, 280, 3338–3345. [Google Scholar] [CrossRef] [PubMed]
- Rishi, V.; Gal, J.; Krylov, D.; Fridriksson, J.; Boysen, M.S.; Mandrup, S.; Vinson, C. SREBP-1 dimerization specificity maps to both the helix-loop-helix and leucine zipper domains: Use of a dominant negative. J. Biol. Chem. 2004, 279, 11863–11874. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, J.E.; Ko, M.S.; Woo, S.H.; Kim, B.J.; Kim, H.S.; Park, I.S.; Koh, E.H.; Lee, K.U. Statins Increase Mitochondrial and Peroxisomal Fatty Acid Oxidation in the Liver and Prevent Non-Alcoholic Steatohepatitis in Mice. Diabetes Metab. J. 2016, 40, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Chamouton, J.; Latruffe, N. PPARα/HNF4α interplay on diversified responsive elements. Relevance in the regulation of liver peroxisomal fatty acid catabolism. Curr. Drug Metab. 2012, 13, 1436–1453. [Google Scholar] [CrossRef] [PubMed]
- Chamouton, J.; Hansmannel, F.; Bonzo, J.A.; Clemencet, M.C.; Chevillard, G.; Battle, M.; Martin, P.; Pineau, T.; Duncan, S.; Gonzalez, F.J.; et al. The Peroxisomal 3-keto-acyl-CoA thiolase B Gene Expression Is under the Dual Control of PPARα and HNF4α in the Liver. PPAR Res. 2010, 2010, 352957. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Choi, J.; Kwon, G.; Ha, H. Endogenous catalase delays high-fat diet-induced liver injury in mice. Korean J. Physiol. Pharmacol. 2017, 21, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, A.; Kitamoto, T.; Nakamura, T.; Ogawa, Y.; Yoneda, M.; Hyogo, H.; Ochi, H.; Mizusawa, S.; Ueno, T.; Nakao, K.; et al. Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits. Endocr. J. 2014, 61, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, M.F.; Wojczynski, M.K.; North, K.E.; Zhang, Q.; Province, M.A.; Carr, J.J.; Borecki, I.B. The ERLIN1-CHUK-CWF19L1 gene cluster influences liver fat deposition and hepatic inflammation in the NHLBI Family Heart Study. Atherosclerosis 2013, 228, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. Second AKT: The rise of SGK in cancer signalling. Growth Factors 2010, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Lauro, D.; Pastore, D.; Capuani, B.; Pacifici, F.; Palmirotta, R.; Abete, P.; Roselli, M.; Bellia, A.; Federici, M.; Di Daniele, N.; et al. Role of Serum and Glucocorticoid-Inducible Kinase [SGK]-1 in Senescence: A Novel Molecular Target Against Age-Related Diseases. Curr. Med. Chem. 2015, 22, 3765–3788. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulou, A.; Fulop, A.; Borkham-Kamphorst, E.; de Leur, E.V.; Gassler, N.; Berger, T.; Beine, B.; Meyer, H.E.; Mak, T.W.; Hopf, C.; et al. Altered mitochondrial and peroxisomal integrity in lipocalin-2-deficient mice with hepatic steatosis. Biochimica et Biophysica Acta 2017, 1863, 2093–2110. [Google Scholar] [CrossRef] [PubMed]
- Kotzka, J.; Knebel, B.; Janssen, O.E.; Schaefer, J.R.; Soufi, M.; Jacob, S.; Nitzgen, U.; Muller-Wieland, D. Identification of a gene variant in the master regulator of lipid metabolism SREBP-1 in a family with a novel form of severe combined hypolipidemia. Atherosclerosis 2011, 218, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Aleman, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008, 7, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, S.; Knebel, B.; Goeddeke, S.; Koellmer, C.; Jacob, S.; Nitzgen, U.; Passlack, W.; Schiller, M.; Dicken, H.D.; Haas, J.; et al. So close and yet so far: Mitochondria and peroxisomes are one but with specific talents. Arch. Physiol. Biochem. 2013, 119, 126–135. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57Bl6 vs. alb-SREBP-1a | |||||
| All (n) | Unique Transcripts (n) | More Abundant in C57Bl6 (n) | More Abundant in alb-SREBP-1a (n) | Unknown Function (n) | Not Annotated (n) |
| 794 | 586 | 169 | 437 | 250 | 63 |
| Fold Change (Linear) | ANOVA p-Value | FDR p-Value | Gene Symbol | Main Description | |
| −9.35 | 1.56 × 10−12 | 4.52 × 10−8 | Nr1d1 | nuclear receptor subfamily 1, group D, member 1 | |
| −2.34 | 8.09 × 10−11 | 1.00 × 10−6 | Mtmr9 | myotubularin-related protein 9 | |
| −1.92 | 3.05 × 10−9 | 9.00 × 10−6 | Cspp1 | centrosome and spindle pole associated protein 1 | |
| −2.05 | 3.55 × 10−9 | 9.00 × 10−6 | Eif4ebp2 | eukaryotic translation initiation factor 4E binding protein 2 | |
| −2.8 | 1.24 × 10−8 | 1.40 × 10−5 | Arrdc3 | arrestin domain containing 3 | |
| −1.96 | 1.32 × 10−8 | 1.40 × 10−5 | Fcer1g | Fc receptor, IgE, high affinity I, gamma polypeptide | |
| −126.7 | 1.46 × 10−8 | 1.40 × 10−5 | mt-Ts2 | mitochondrially encoded tRNA serine 2 | |
| −1.84 | 1.48 × 10−8 | 1.40 × 10−5 | Hcfc1r1 | host cell factor C1 regulator 1 (XPO1-dependent) | |
| −1.92 | 1.85 × 10−8 | 1.70 × 10−5 | Krtcap2 | keratinocyte associated protein 2 | |
| −114.04 | 2.19 × 10−8 | 1.70 × 10−5 | mt-Ty | mitochondrially encoded tRNA tyrosine | |
| −1.88 | 2.66 × 10−8 | 1.90 × 10−5 | Laptm5 | lysosomal-associated protein transmembrane 5 | |
| −1.93 | 4.28 × 10−8 | 2.20 × 10−5 | Cd52 | CD52 antigen | |
| −1.9 | 4.49 × 10−8 | 2.20 × 10−5 | Ear10 | eosinophil-associated, ribonuclease A family, member 10 | |
| −2.14 | 4.92 × 10−8 | 2.30 × 10−5 | C1qc | complement component 1, q subcomponent, C chain | |
| −1.92 | 6.31 × 10−8 | 2.70 × 10−5 | Mn1 | meningioma 1 | |
| 1.68 | 6.87 × 10−9 | 1.00 × 10−5 | Ahcy | S-adenosylhomocysteine hydrolase | |
| 2.02 | 9.74 × 10−9 | 1.30 × 10−5 | Ppard | peroxisome proliferator activator receptor delta | |
| 1.59 | 2.82 × 10−8 | 1.90 × 10−5 | Sfxn1 | sideroflexin 1 | |
| 1.59 | 4.53 × 10−8 | 2.20 × 10−5 | Ivd | isovaleryl coenzyme A dehydrogenase | |
| 1.55 | 6.83 × 10−8 | 2.80 × 10−5 | Nfil3 | nuclear factor, interleukin 3, regulated | |
| 1.55 | 1.03 × 10−7 | 3.60 × 10−5 | Pttg1ip | pituitary tumor-transforming 1 interacting protein | |
| 1.55 | 1.03 × 10−7 | 3.60 × 10−5 | Hspd1 | heat shock protein 1 (chaperonin); predicted gene 12141 | |
| 1.97 | 1.69 × 10−7 | 5.00 × 10−5 | Acnat1 | acyl-coenzyme A amino acid N-acyltransferase 1 | |
| 2.75 | 2.20 × 10−7 | 5.80 × 10−5 | Snora69 | small nucleolar RNA, H/ACA box 69 | |
| 1.75 | 2.93 × 10−7 | 7.00 × 10−5 | Hacl1 | 2-hydroxyacyl-CoA lyase 1 | |
| 1.53 | 3.60 × 10−7 | 8.00 × 10−5 | Bcap31 | B cell receptor associated protein 31 | |
| 1.92 | 4.28 × 10−7 | 8.90 × 10−5 | Tmem254b | transmembrane protein 254b | |
| 1.8 | 4.64 × 10−7 | 9.40 × 10−5 | Tat | tyrosine aminotransferase | |
| 1.81 | 5.69 × 10−7 | 1.11 × 10−4 | Fga | fibrinogen alpha chain | |
| 1.58 | 5.96 × 10−7 | 1.13 × 10−4 | Spop | speckle-type POZ protein | |
| C57Bl6 vs. alb-SREBP-1a ΔP | |||||
| All (n) | Unique Transcripts (n) | More Abundant in C57Bl6 (n) | more Abundant in alb-SREBP-1a (n) | Unknown Function (n) | not Annotated (n) |
| 4581 | 3723 | 1732 | 1991 | 950 | 370 |
| Fold Change (Linear) | ANOVA p-Value | FDR p-Value | Gene Symbol | Main Description | |
| −108.6 | 1.92 × 10−14 | 2.78 × 10−10 | mt-Ts2 | mitochondrially encoded tRNA serine 2 | |
| −11.38 | 1.09 × 10−14 | 2.78 × 10−10 | Nr1d1 | nuclear receptor subfamily 1, group D, member 1 | |
| −187.66 | 6.92 × 10−14 | 6.67 × 10−10 | mt-Tt | mitochondrially encoded tRNA threonine | |
| −96.52 | 1.31 × 10−13 | 9.45 × 10−10 | mt-Ty | mitochondrially encoded tRNA tyrosine | |
| −15.04 | 2.01 × 10−12 | 1.16 × 10−8 | mt-Tp | mitochondrially encoded tRNA proline | |
| −2.32 | 4.57 × 10−12 | 2.21 × 10−8 | Rnasek | ribonuclease, RNase K | |
| −3.02 | 2.57 × 10−11 | 5.32 × 10−8 | Snord57 | small nucleolar RNA, C/D box 57; NOP56 ribonucleoprotein | |
| −20.09 | 4.35 × 10−11 | 7.86 × 10−8 | mt-Tk | mitochondrially encoded tRNA lysine | |
| −13.23 | 7.61 × 10−11 | 1.10 × 10−7 | mt-Tn | mitochondrially encoded tRNA asparagine | |
| −1.81 | 8.65 × 10−11 | 1.19 × 10−7 | Vmn1r170 | vomeronasal 1 receptor 170 | |
| −1.75 | 3.34 × 10−10 | 1.91 × 10−7 | Gsdmc2 | gasdermin C2; gasdermin C3; gasdermin C4 | |
| −3.31 | 2.94 × 10−10 | 1.91 × 10−7 | Mir692-1 | microRNA 692-1 | |
| −4.02 | 6.36 × 10−10 | 2.25 × 10−7 | Dbp | D site albumin promoter binding protein | |
| −3.18 | 6.82 × 10−10 | 2.32 × 10−7 | mt-Te | mitochondrially encoded tRNA glutamic acid | |
| −8.36 | 1.41 × 10−9 | 3.90 × 10−7 | mt-Tc | mitochondrially encoded tRNA cysteine | |
| 1.58 | 7.82 × 10−12 | 2.52 × 10−8 | Timm23 | translocase of inner mitochondrial membrane 23 | |
| 1.78 | 5.46 × 10−10 | 2.00 × 10−7 | Adck3 | aarF domain containing kinase 3 | |
| 1.86 | 4.19 × 10−10 | 1.91 × 10−7 | Mrpl36 | mitochondrial ribosomal protein L36 | |
| 1.9 | 7.46 × 10−11 | 1.10 × 10−7 | Hsd11b1 | hydroxysteroid 11-beta dehydrogenase 1 | |
| 1.93 | 2.84 × 10−10 | 1.91 × 10−7 | Sgpl1 | sphingosine phosphate lyase 1 | |
| 1.98 | 1.93 × 10−10 | 1.91 × 10−7 | Spop | speckle-type POZ protein | |
| 2.11 | 1.26 × 10−10 | 1.66 × 10−7 | Bcap31 | B cell receptor associated protein 31 | |
| 2.4 | 4.19 × 10−10 | 1.91 × 10−7 | Tubb2a | tubulin, beta 2A class IIA | |
| 2.43 | 2.23 × 10−11 | 4.96 × 10−8 | Ppard | peroxisome proliferator activator receptor delta | |
| 2.57 | 2.09 × 10−10 | 1.91 × 10−7 | Snord104 | small nucleolar RNA, C/D box 104 | |
| 3.56 | 1.96 × 10−10 | 1.91 × 10−7 | Npas2 | neuronal PAS domain protein 2 | |
| 1.51 | 1.40 × 10−9 | 3.90 × 10−7 | G3bp1 | GTPase activating protein (SH3 domain) binding protein 1 | |
| 1.53 | 2.81 × 10−9 | 6.41 × 10−7 | Rbmxl1 | RNA binding motif protein | |
| 1.55 | 3.46 × 10−9 | 7.10 × 10−7 | Nubp2 | nucleotide binding protein 2 | |
| 1.57 | 3.01 × 10−9 | 6.74 × 10−7 | Sumo2 | small ubiquitin-related modifier 2-like | |
| alb-SREBP-1a vs. alb-SREBP-1aΔP | |||||
| All (n) | Unique Transcripts (n) | More Abundant in C57Bl6 (n) | More Abundant in alb-SREBP-1a (n) | Unknown Function (n) | Not Annotated (n) |
| 3385 | 2544 | 1143 | 1401 | 276 | 930 |
| Fold Change (Linear)) | ANOVA p-Value | FDR p-Value | Gene Symbol | Main Description | |
| −1.98 | 6.42 × 10−9 | 7.10 × 10−5 | Xlr5a | X-linked lymphocyte-regulated 5A, pseudogene | |
| −1.66 | 7.17 × 10−8 | 8.20 × 10−5 | Olfr1410 | olfactory receptor 1410 | |
| −1.91 | 8.85 × 10−8 | 8.20 × 10−5 | Olfr796 | olfactory receptor 796 | |
| −2.04 | 2.14 × 10−7 | 9.50 × 10−5 | Olfr154 | olfactory receptor 154 | |
| −1.84 | 2.37 × 10−7 | 9.50 × 10−5 | Olfr169 | olfactory receptor 169 | |
| −2.06 | 2.52 × 10−7 | 9.50 × 10−5 | Olfr644 | olfactory receptor 644 | |
| −1.54 | 2.97 × 10−7 | 9.80 × 10−5 | Slc12a8 | solute carrier family 12, member 8 | |
| −2 | 3.24 × 10−7 | 1.02 × 10−4 | Vmn1r193 | vomeronasal 1 receptor 193 | |
| −2.52 | 3.40 × 10−7 | 1.04 × 10−4 | Vmn1r37 | vomeronasal 1 receptor 37pseudogene 21 | |
| −1.7 | 5.92 × 10−7 | 1.24 × 10−4 | Olfr1269 | olfactory receptor 1269 | |
| −2.15 | 6.13 × 10−7 | 1.24 × 10−4 | Krtap10-10 | keratin associated protein 10-10 | |
| −1.84 | 8.08 × 10−7 | 1.30 × 10−4 | Clps | colipase, pancreatic | |
| −2.49 | 8.93 × 10−7 | 1.37 × 10−4 | Mir301b | microRNA 301b | |
| −2.34 | 9.64 × 10−7 | 1.42 × 10−4 | Olfr488 | olfactory receptor 488 | |
| −1.57 | 1.00 × 10−6 | 1.56 × 10−4 | Adamts17 | a disintegrin-like and metallopeptidase thrombospondin type 1 motif, 17 | |
| 1.6 | 1.59 × 10−8 | 7.10 × 10−5 | Pde8a | phosphodiesterase 8A | |
| 2.3 | 3.24 × 10−8 | 7.10 × 10−5 | Ppp2r5c | protein phosphatase 2, regulatory subunit B, gamma | |
| 1.9 | 3.46 × 10−8 | 7.10 × 10−5 | Qpct | glutaminyl-peptide cyclotransferase | |
| 1.86 | 3.60 × 10−8 | 7.10 × 10−5 | Slirp | SRA stem-loop interacting RNA binding protein | |
| 1.77 | 3.66 × 10−8 | 7.10 × 10−5 | Asf1a | anti-silencing function 1A histone chaperone | |
| 1.65 | 4.60 × 10−8 | 7.60 × 10−5 | Mrpl36 | mitochondrial ribosomal protein L36 | |
| 1.66 | 6.08 × 10−8 | 8.20 × 10−5 | Psma5 | proteasome (prosome, macropain) subunit, alpha type 5 | |
| 1.61 | 7.01 × 10−8 | 8.20 × 10−5 | Syne2 | spectrin repeat containing, nuclear envelope 2 | |
| 1.62 | 7.94 × 10−8 | 8.20 × 10−5 | Usp40 | ubiquitin specific peptidase 40 | |
| 1.75 | 8.06 × 10−8 | 8.20 × 10−5 | Gtf2a2 | general transcription factor II A, 2 | |
| 1.64 | 9.07 × 10−8 | 8.20 × 10−5 | Nt5c2 | 5-nucleotidase, cytosolic II | |
| 1.54 | 1.01 × 10−7 | 8.60 × 10−5 | Pccb | propionyl Coenzyme A carboxylase, beta polypeptide | |
| 1.62 | 1.20 × 10−7 | 8.60 × 10−5 | Pdzd11 | PDZ domain containing 11 | |
| 1.62 | 1.22 × 10−7 | 8.60 × 10−5 | Lig3 | ligase III, DNA, ATP-dependent | |
| 1.75 | 1.23 × 10−7 | 8.60 × 10−5 | Acadl | acyl-Coenzyme A dehydrogenase, long-chain | |
| C57Bl6 vs. alb-SREBP-1a | |||||||
| Canonical Pathways | p-Value | Upstream Activator | p-Value | Disease or Tox Function | p-Value | Interaction Network | p-Value |
| FXR/RXR | 6.76 × 10−6 | RORA | 2.67 × 10−18 | hepatic steatosis associated pathways. | 1.60 × 10−7 | ACOX1 | 1.76 × 10−12 |
| PPAR | 1.58 × 10−4 | RORC | 1.98 × 10−13 | liver cholestasis | 1.30 × 10−3 | MAPK7 | 2.93 × 10−12 |
| sirtuin signaling | 2.63 × 10−4 | PPARA | 1.22 × 10−11 | liver hyperplasia/hyperproliferation | 2.76 × 10−3 | ONECUT1 | 3.16 × 10−11 |
| PPARD | 1.16 × 10−8 | liver proliferation | 1.80 × 10−3 | PPARA | 3.33 × 10−9 | ||
| GPD1 | 5.31 × 10−11 | renal damage | 6.15 × 10−4 | ||||
| SLC25A13 | 7.24 × 10−11 | nonalcoholic fatty liver disease | 8.13 × 10−3 | ||||
| HNF4A | 4.59 × 10−6 | ||||||
| C57Bl6 vs. alb-SREBP-1a ΔP | |||||||
| Canonical Pathways | p-Value | Upstream Activator | p-Value | Disease or Tox Function | p-Value | Interaction Network | p-Value |
| methionine degradation | 3.24 × 10−6 | HNF4A | 9.23 × 10−29 | olfactory response | 2.32 × 10−144 | ACOX1 | 2.51 × 10−9 |
| isoleucinedegradation | 3.89 × 10−4 | RORC | 1.56 × 10−9 | olfaction | 1.68 × 10−139 | GRB14 | 6.14 × 10−9 |
| valine degradation | 2.40 × 10−3 | RORA | 7.56 × 10−9 | signal transduction | 4.91 × 10−60 | PPARGC1A | 2.10 × 10−6 |
| cysteine biosynthesis | 9.33 × 10−5 | ONECUT1 | 2.85 × 10−8 | Cell communication | 1.30 × 10−55 | ||
| autophagy | 1.12 × 10−3 | MYC | 4.47 × 10−7 | RNA post-transcriptional modification | 2.08 × 10−6 | ||
| protein ubiquitination | 1.12 × 10−3 | NR1D1 | 1.76 × 10−6 | synthesis of ribonucleoside monophosphate | 7.74 × 10−6 | ||
| sirtuin signaling | 8.51 × 10−3 | ACOX1 | 4.78 × 10−10 | amino acids metabolism | 6.02 × 10−10 | ||
| ceramide biosynthesis | 4.27 × 10−3 | PPARA | 6.16 × 10−6 | oxidation of lipid | 3.08 × 10−6 | ||
| fatty acid β-oxidation | 2.29 × 10−2 | RXRA | 8.74 × 10−4 | oxidation of fatty acids | 8.63 × 10−6 | ||
| fatty acid metabolism | 9.55 × 10−6 | ||||||
| bleeding of liver | 1.55 × 10−3 | ||||||
| compensated cirrhosis | 7.29 × 10−3 | ||||||
| liver degeneration | 7.31 × 10−3 | ||||||
| cholestasis | 8.51 × 10−3 | ||||||
| alb-SREBP-1a vs. alb-SREBP-1aΔP | |||||||
| Canonical Pathways | p-Value | Upstream Activator | p-Value | Disease or Tox Function | p-Value | Interaction Network | p-Value |
| RNA polymerase II complex assembly | 9.33 × 10−5 | HNF4A | 4.34 × 10−13 | olfactory response | 7.51 × 10−123 | NCAM1 | 2.17 × 10−5 |
| protein ubiquitination | 9.12 × 10−4 | RORC | 6.11 × 10−5 | olfaction | 2.92 × 10−120 | ACOX1 | 4.32 × 10−5 |
| sirtuin signaling | 1.02 × 10−2 | RORA | 6.26 × 10−5 | signal transduction | 7.55 × 10−61 | SIRT4 | 1.56 × 10−4 |
| sumoylation | 3.72 × 10−2 | PPARA | 2.13 × 10−4 | cell communication | 1.49 × 10−57 | NR2F1 | 2.55 × 10−4 |
| ACOX1 | 1.71 × 10−5 | RNA post-transcriptional processing | 2.69 × 10−7 | FOXO4 | 4.82 × 10−4 | ||
| RNA transport | 2.66 × 10−6 | FABP1 | 5.98 × 10−4 | ||||
| differentiation processes | 1.78 × 10−5 | ABCB1 | 9.88 × 10−4 | ||||
| clearance of bilirubin | 6.17 × 10−3 | PPARA | 1.17 × 10−3 | ||||
| proliferation of hepatic stellate cells | 4.42 × 10−2 | ||||||
| hepatomegaly | 6.43 × 10−2 | ||||||
| C57Bl6 vs. alb-SREBP-1a | |||||||
| Canonical Pathways | p-Value | Upstream Activators | p-Value | Disease, Function | p-Value | Interaction Networks | p-Value |
| INSR | 9.95 × 10−7 | DSP | 1.23 × 10−8 | ||||
| MYC | 1.00 × 10−5 | mediator | 4.45 × 10−8 | ||||
| PPARA | 7.24 × 10−5 | FBXO32 | 3.95 × 10−7 | ||||
| FBXW | 4.0 × 10−7 | ||||||
| HNF4A | 5.46 × 10−7 | ||||||
| C57Bl6 vs. alb-SREBP-1a ΔP | |||||||
| Canonical Pathways | p-Value | Upstream Activators | p-Value | Disease, Function | p-Value | Interaction Networks | p-Value |
| fatty acid β-oxidation | 3.98 × 10−16 | PPARA | 2.67 × 10−34 | microvesicular hepatic steatosis | 1.21 × 10−17 | INSR | 9.63 × 10−30 |
| mitochondrial dysfunction | 1.585 × 10−13 | PPARG | 1.64 × 10−12 | hepatic steatosis | 1.2 × 10−16 | ACOX1 | 1.64 × 10−14 |
| TCA Cycle II | 1.585 × 10−11 | PPARGC1A | 2.05 × 10−11 | liver hyperplasia/hyperproliferation | 5.33 × 10−4 | MAP4K4 | 7.86 × 10−13 |
| valine degradation | 2.45 × 10−10 | HNF4A | 3.67 × 10−13 | nonalcoholic fatty liver disease | 0.000106 | LONP1 | 4.36 × 10−11 |
| Methionine degradation | 9.55 × 10−7 | fatty acid metabolism | 7.94 × 10−24 | ||||
| arginine biosynthesis | 8.91 × 10−9 | mitochondrial dysfunction | 1.99 × 10−13 | ||||
| sirtuin signaling | 1.99 × 10−9 | inhibition of RXR function | 4.89 × 10−7 | ||||
| oxidative phosphorylation | 3.72 × 10−9 | PPARα/RXRα activation | 3.31 × 10−5 | ||||
| urea cycle | 2.4 × 10−6 | ||||||
| alb-SREBP-1a vs. alb-SREBP-1aΔP | |||||||
| Canonical Pathways | p-Value | Upstream Activators | p-Value | Disease, Function | p-Value | Interaction Networks | p-Value |
| mitochondrial dysfunction | 2.51 × 10−26 | PPARA | 8.36 × 10−34 | microvascular hepatic steatosis | 9.98 × 10−15 | INSR | 3.25 × 10−38 |
| oxidative phosphorylation | 1.26 × 10−19 | PPARGC1A | 1.38 × 10−18 | liver growth regulation | 1.24 × 10−6 | ABHD5 | 7.00 × 10−35 |
| sirtuin signaling | 1.58 × 10−19 | HNF4A | 9.90 × 10−14 | nonalcoholic fatty liver disease | 1.16 × 10−5 | ACOX1 | 2.57 × 10−31 |
| fatty acid β-oxidation | 2.51 × 10−15 | PPARG | 5.48 × 10−13 | TCA Cycle | 6.31 × 10−13 | PPARA | 3.60 × 10−31 |
| branched-chain α-keto acid dehydrogenase complex | 8.71 × 10−7 | KLF15 | 4.72 × 10−12 | valine degradation | 7.94 × 10−12 | ||
| 2-oxobutanoate degradation | 2.14 × 10−6 | Esrra | 1.26 × 10−11 | methionine degradation | 1.82 × 10−9 | ||
| acetyl-CoA oxidation | 7.41 × 10−6 | INSR | 2.07 × 10−28 | citrulline degradation | 2.14 × 10−8 | ||
| fatty acid α-oxidation | 2.29 × 10−4 | MAP4K4 | 5.20 × 10−19 | Isoleucine biosynthesis | 1.23 × 10−6 | ||
| LONP1 | 1.17 × 10−16 | arginine biosynthesis | 1.91 × 10−8 | ||||
| LEP | 1.37 × 10−11 | urea cycle | 1.91 × 10−8 | ||||
| growth hormone | 5.60 × 10−11 | ethanol degradation | 2.34 × 10−7 | ||||
| glycine betaine degradation | 2.63 × 10−7 | ||||||
| xenobiotic metabolism signaling | 1.38 × 10−6 | ||||||
| PPARα/RXRα activation | 1.00 × 10−4 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knebel, B.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Passlack, W.; Koellmer, C.; Lehr, S.; Müller-Wieland, D.; Kotzka, J. Inactivation of SREBP-1a Phosphorylation Prevents Fatty Liver Disease in Mice: Identification of Related Signaling Pathways by Gene Expression Profiles in Liver and Proteomes of Peroxisomes. Int. J. Mol. Sci. 2018, 19, 980. https://doi.org/10.3390/ijms19040980
Knebel B, Hartwig S, Jacob S, Kettel U, Schiller M, Passlack W, Koellmer C, Lehr S, Müller-Wieland D, Kotzka J. Inactivation of SREBP-1a Phosphorylation Prevents Fatty Liver Disease in Mice: Identification of Related Signaling Pathways by Gene Expression Profiles in Liver and Proteomes of Peroxisomes. International Journal of Molecular Sciences. 2018; 19(4):980. https://doi.org/10.3390/ijms19040980
Chicago/Turabian StyleKnebel, Birgit, Sonja Hartwig, Sylvia Jacob, Ulrike Kettel, Martina Schiller, Waltraud Passlack, Cornelia Koellmer, Stefan Lehr, Dirk Müller-Wieland, and Jorg Kotzka. 2018. "Inactivation of SREBP-1a Phosphorylation Prevents Fatty Liver Disease in Mice: Identification of Related Signaling Pathways by Gene Expression Profiles in Liver and Proteomes of Peroxisomes" International Journal of Molecular Sciences 19, no. 4: 980. https://doi.org/10.3390/ijms19040980