Sinomenine Hydrochloride Inhibits the Metastasis of Human Glioblastoma Cells by Suppressing the Expression of Matrix Metalloproteinase-2/-9 and Reversing the Endogenous and Exogenous Epithelial-Mesenchymal Transition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
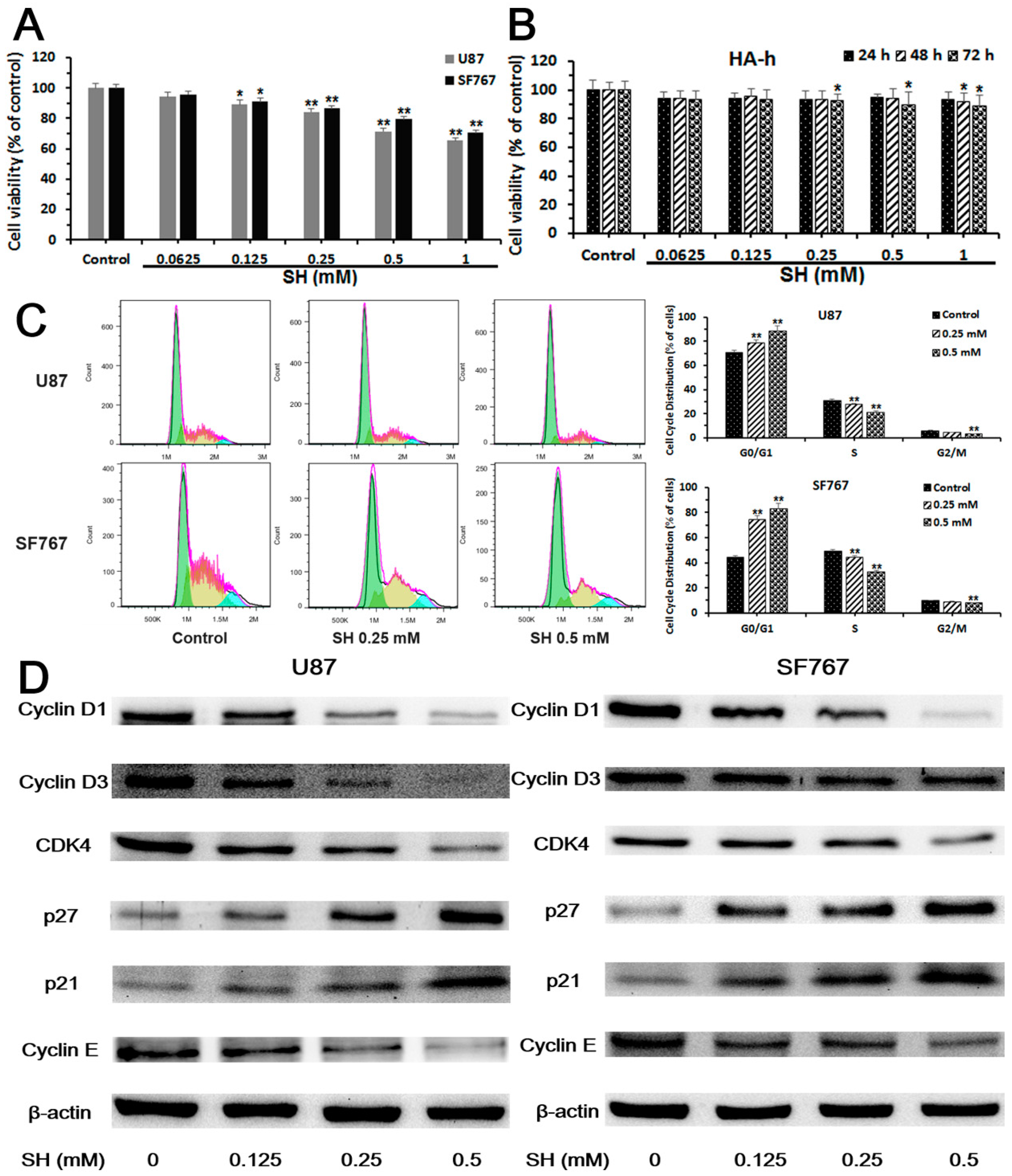
2.1. Sinomenine Hydrochloride (SH) Selectively Kills Human Glioblastoma Cells, But Not Normal Glial Cells, and Induces Human Glioblastoma Cell Cycle Arrest
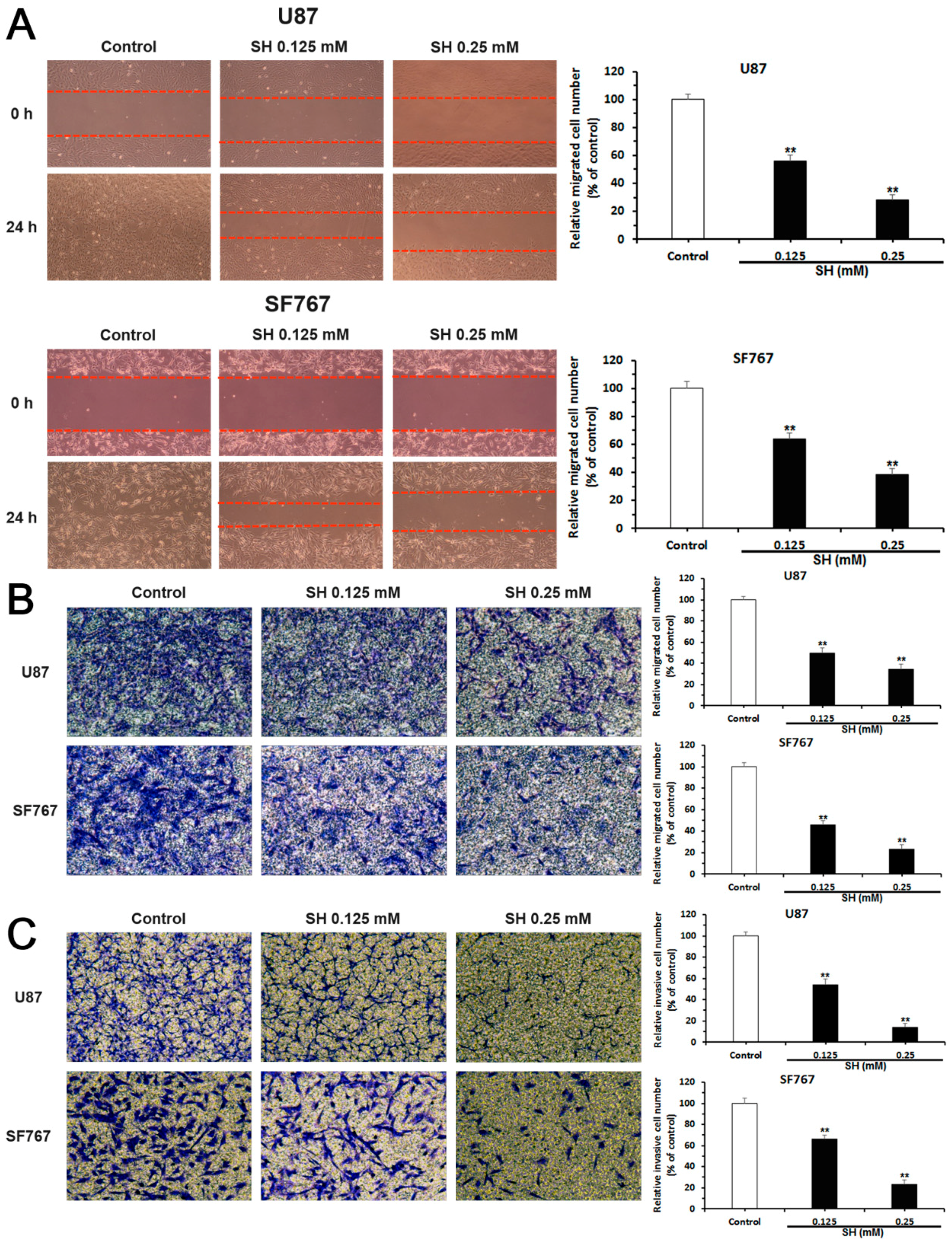
2.2. SH Inhibits the Migration and Invasion of U87 and SF767 Cells
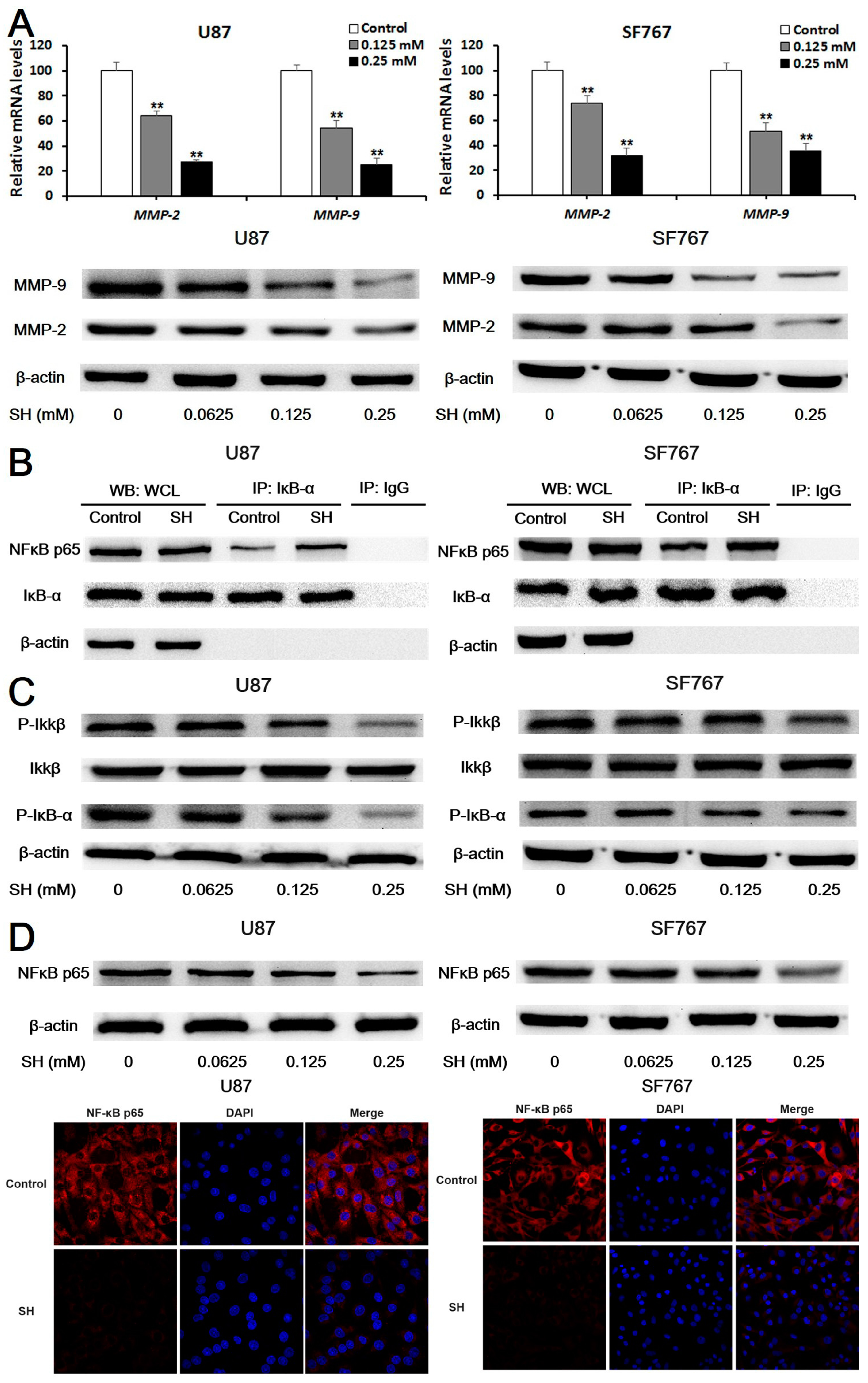
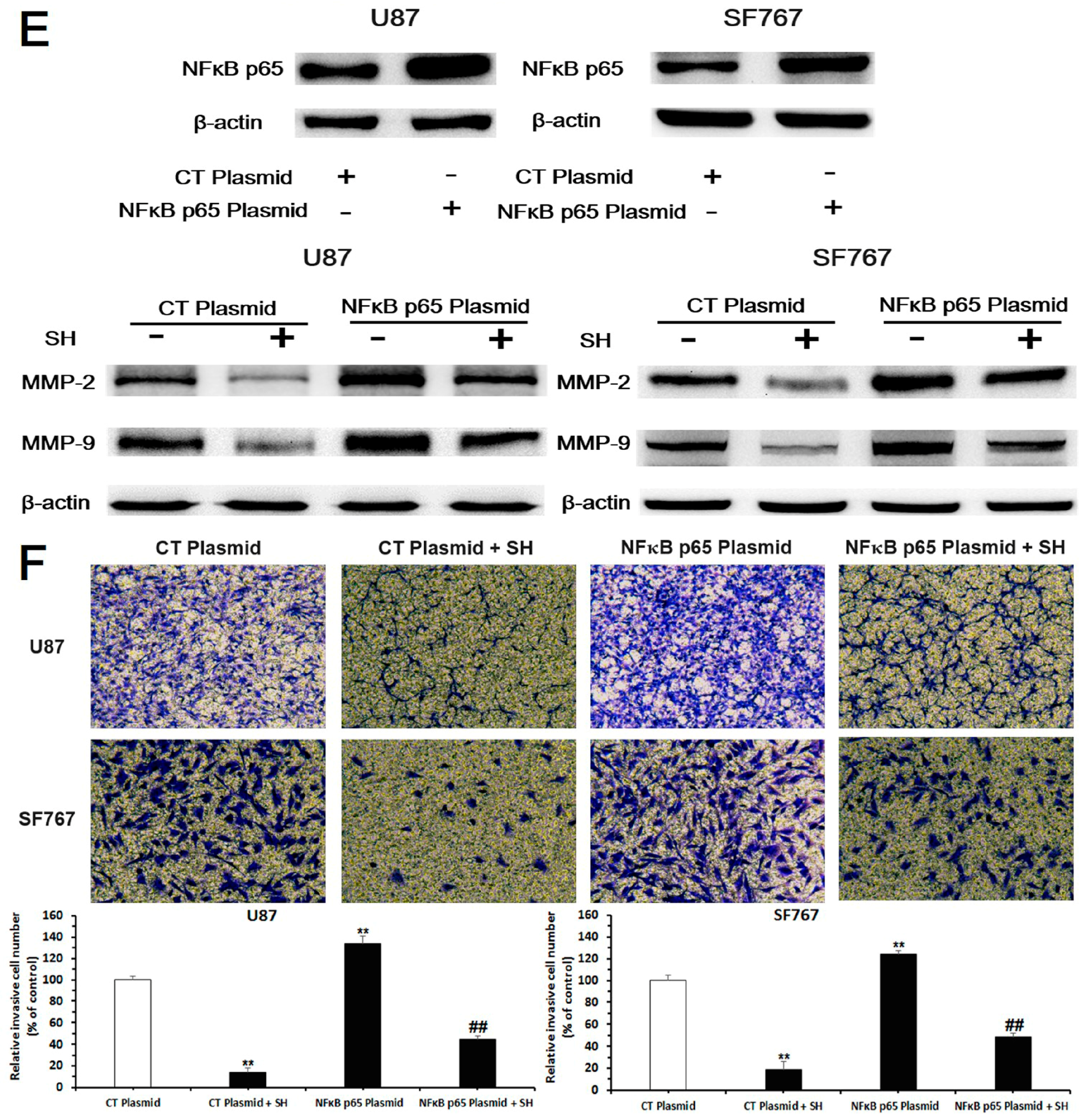
2.3. SH Decreases MMP-2/-9 Expression by Suppressing NFκB Activation
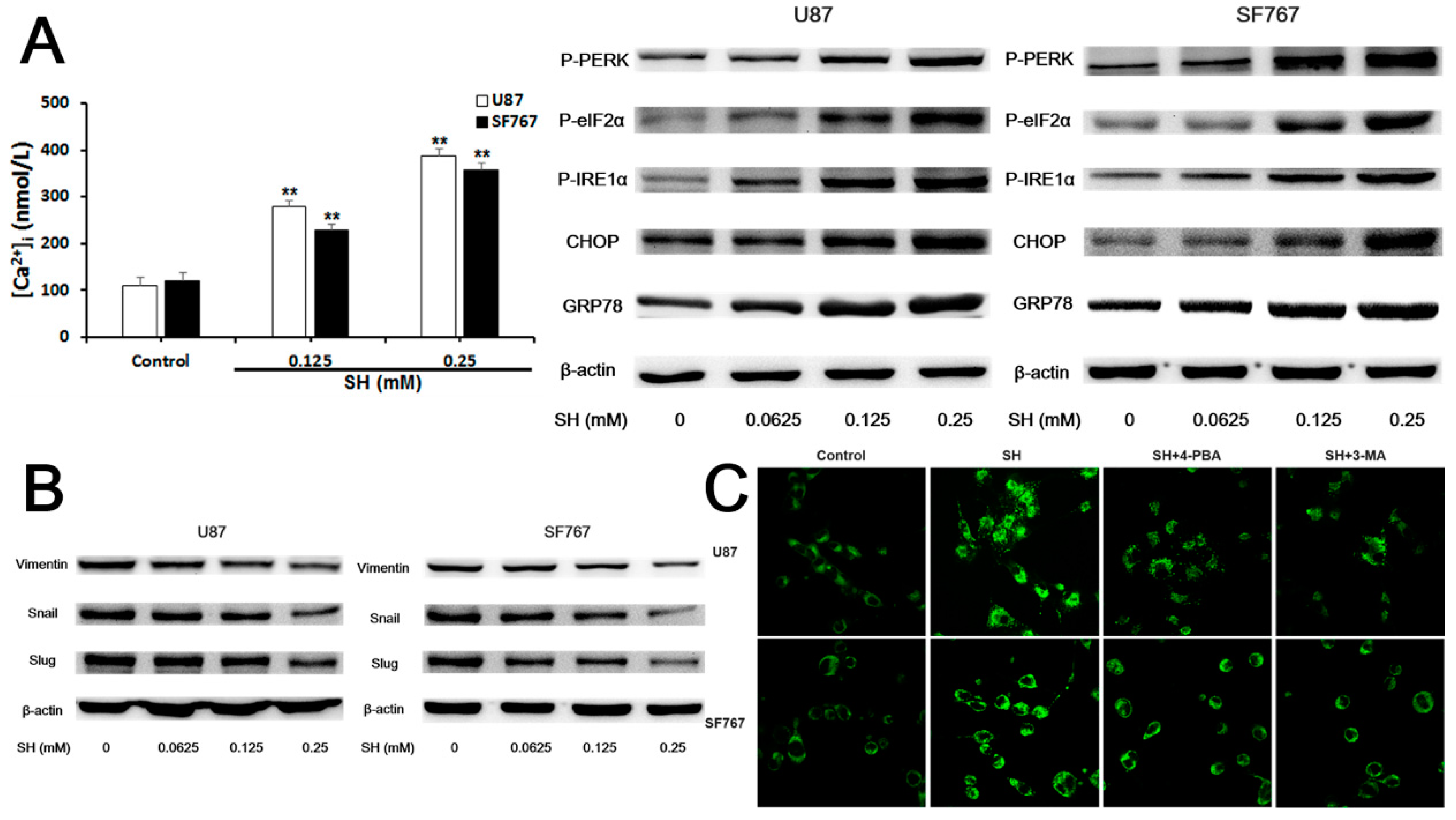
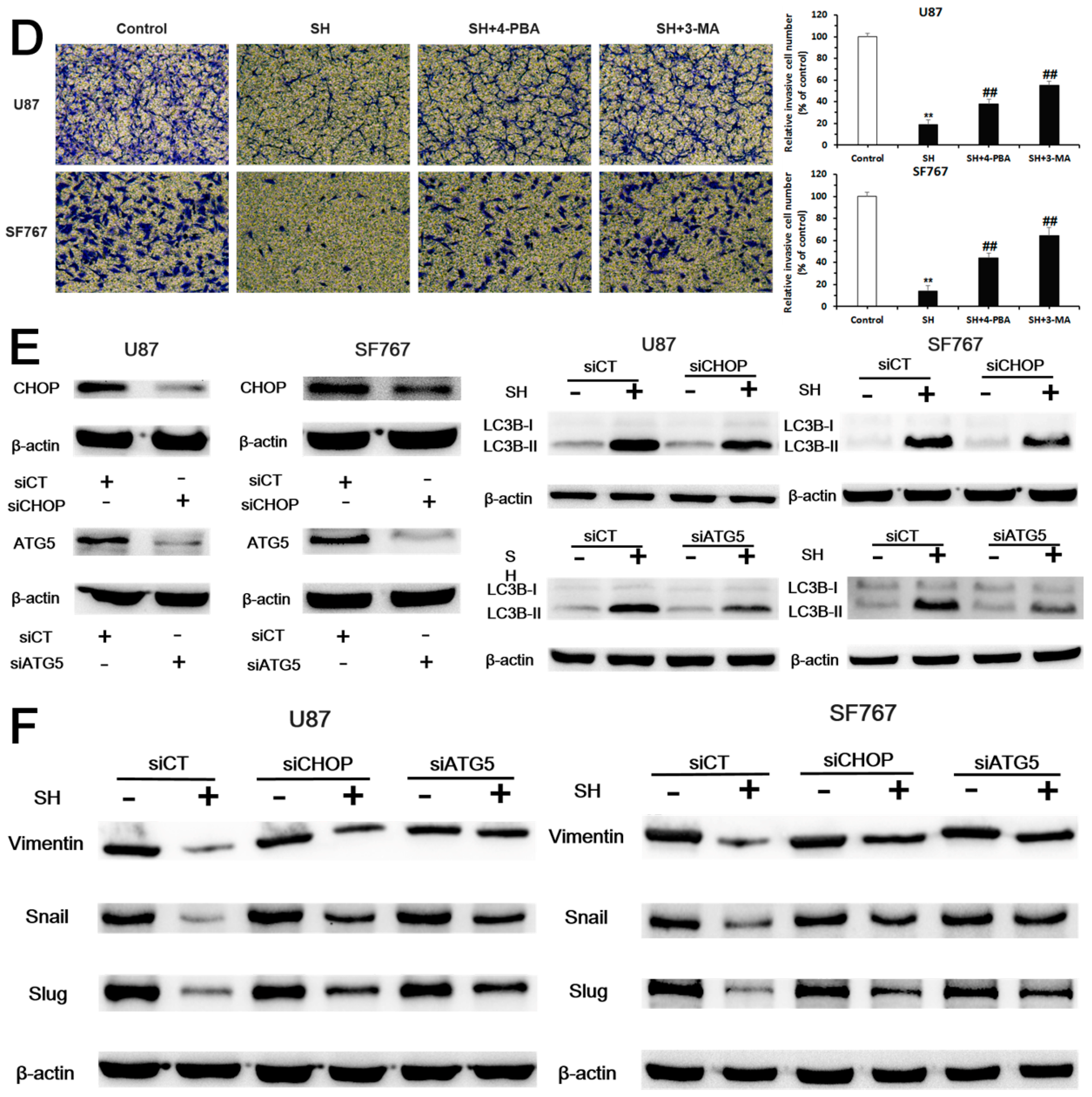
2.4. SH Impairs the Invasiveness of Human Glioblastoma Cells by Reversing the Endogenous EMT through ER Stress-Mediated Autophagy
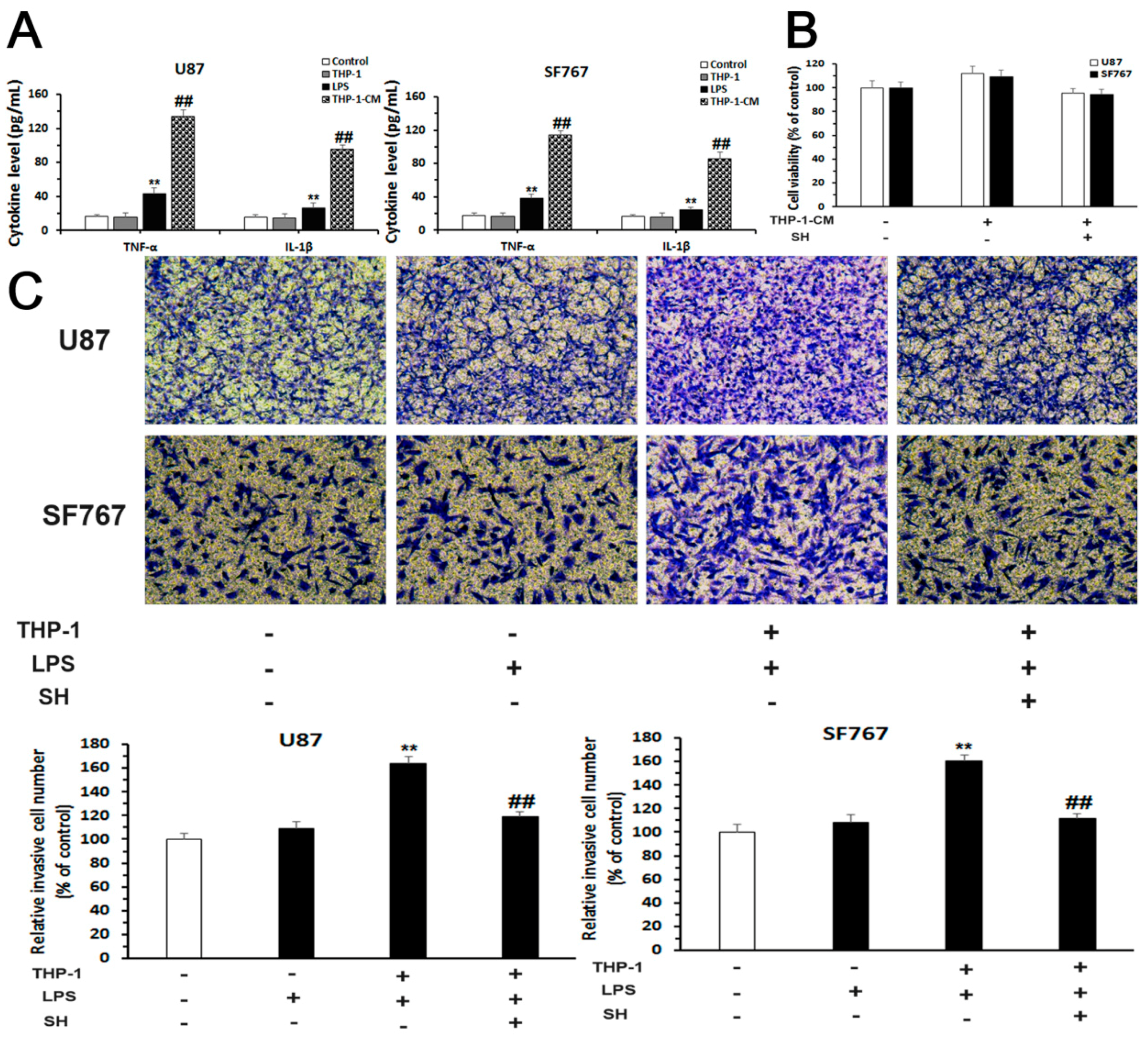
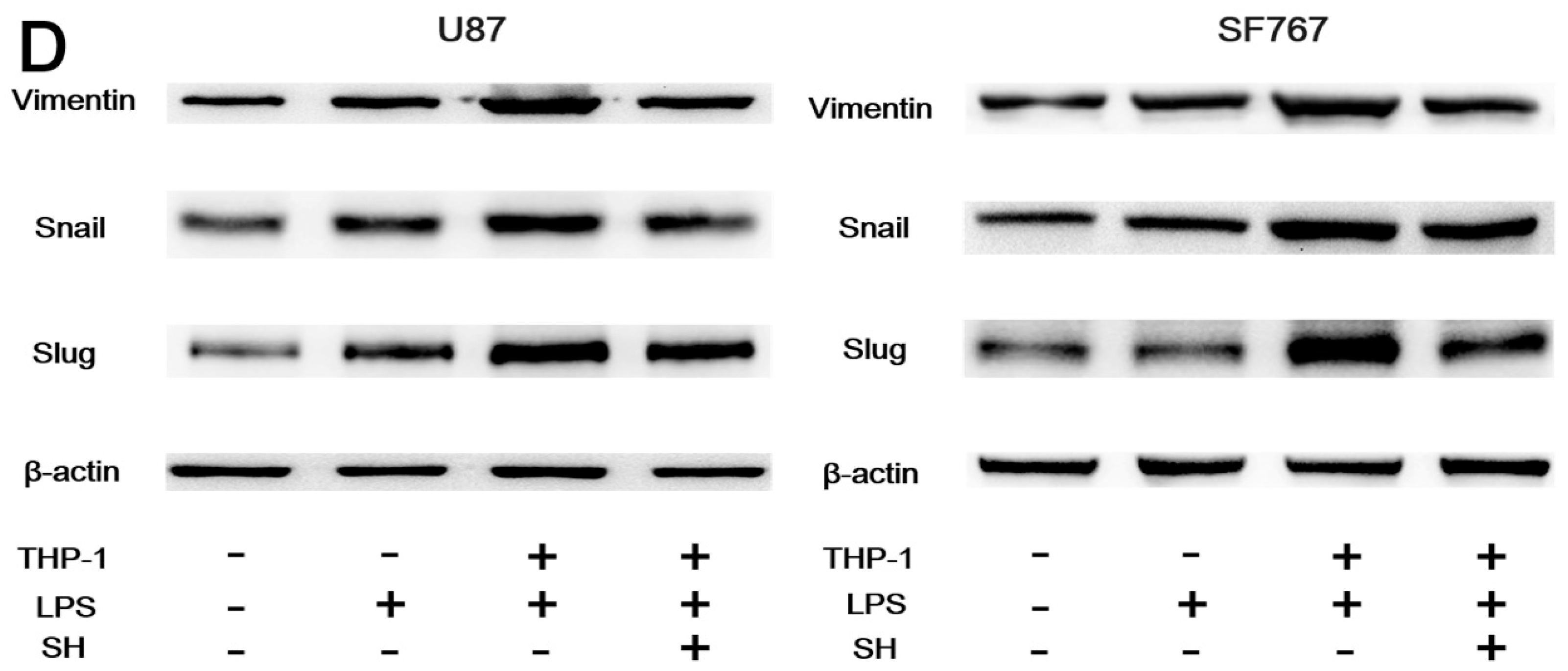
2.5. SH Inhibits the EMT and Invasion of Human Glioblastoma Cells in the Inflammatory Microenvironment
2.5.1. Establishment of an Inflammatory Microenvironment System
2.5.2. SH Abolishes THP-1-CM-Stimulated Exogenous EMT and Invasion in U87 and SF767 Cells
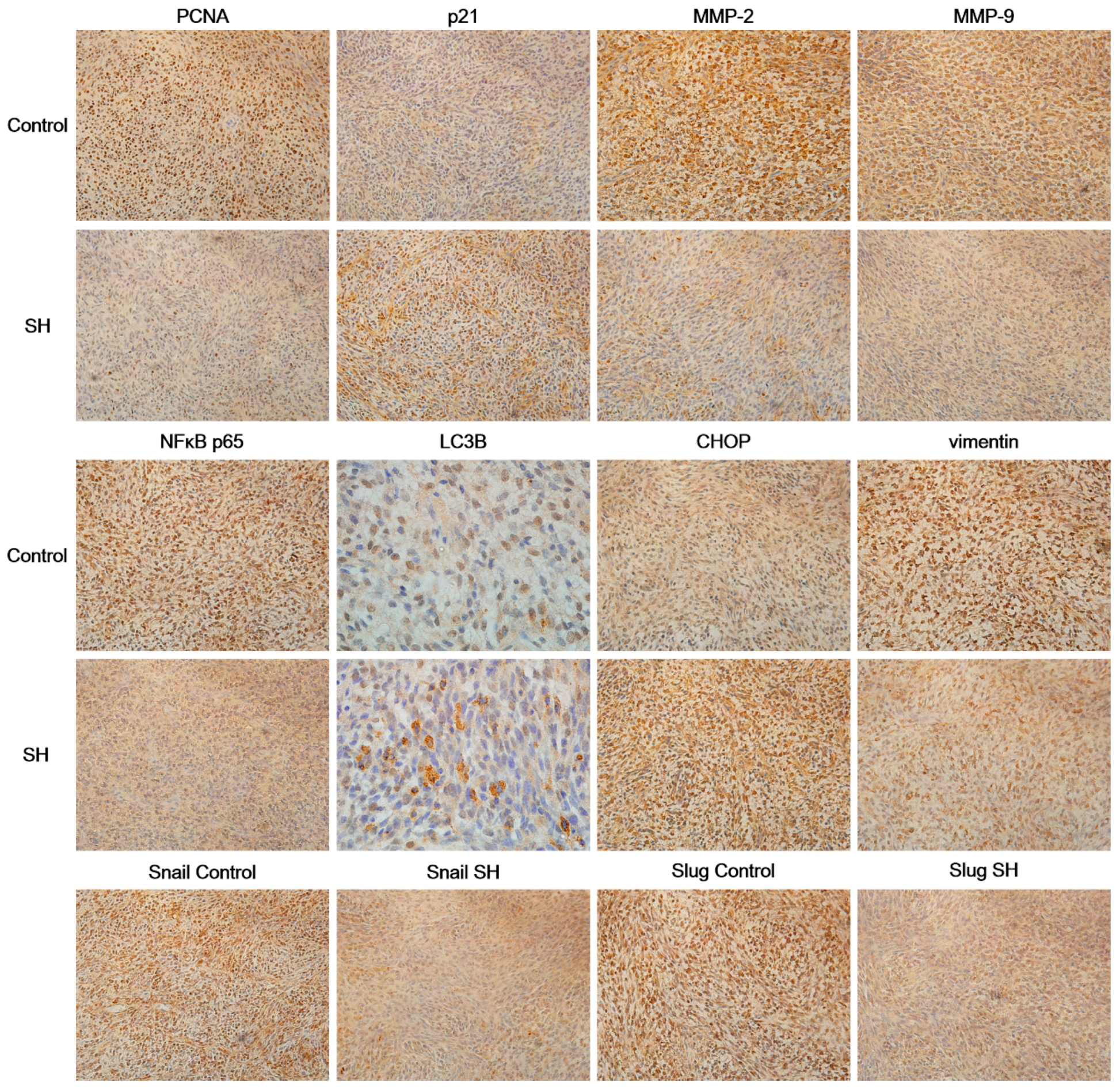
2.6. SH Regulates the Levels of Proliferation and Metastasis-Related Proteins in Glioblastoma Tumors In Vivo
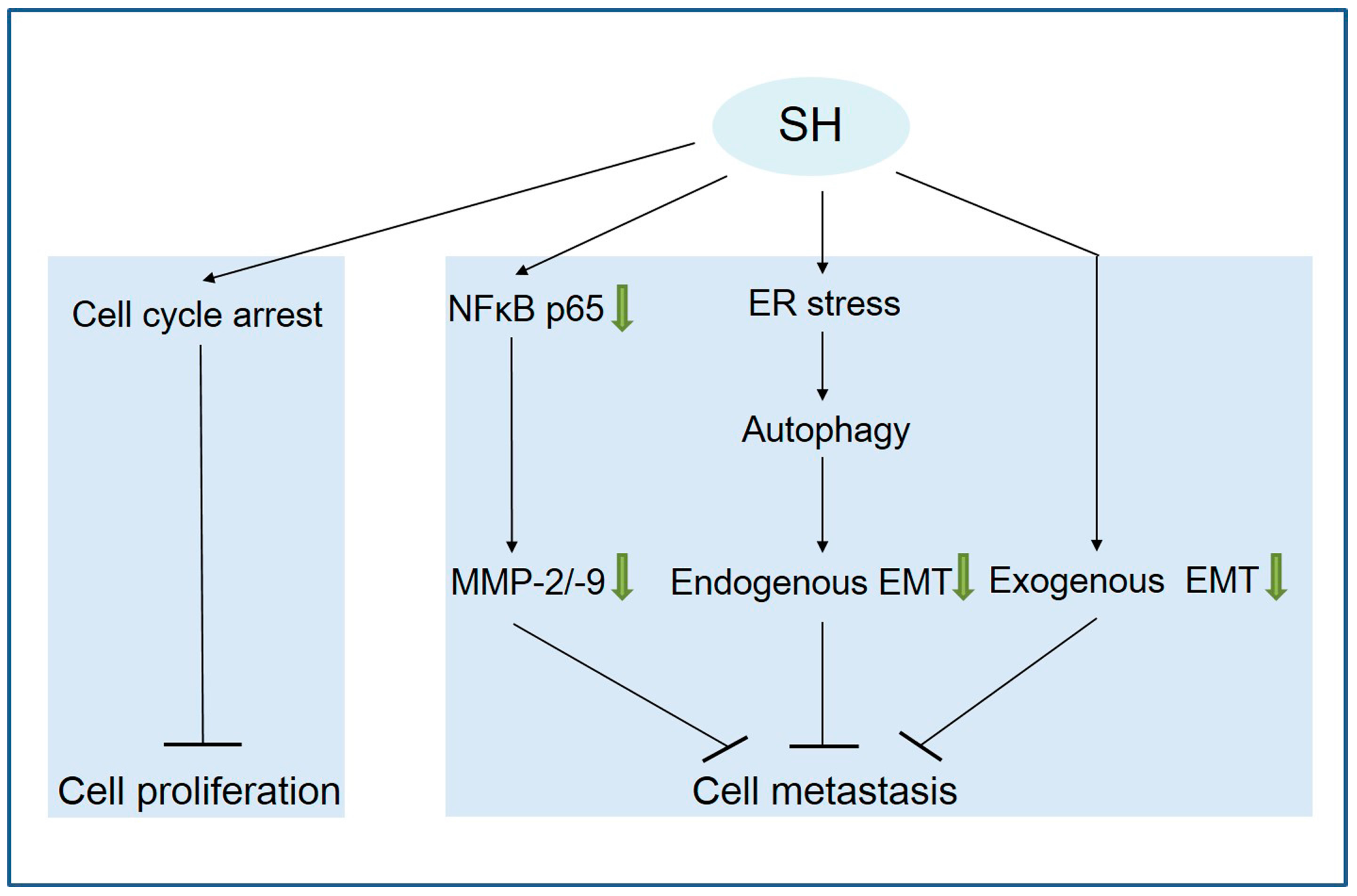
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Treatment
4.3. Cell Viability Measurement
4.4. Cell Cycle Analysis
4.5. In Vitro Migration and Invasion Assays
4.6. Quantitative Real-Time PCR
4.7. Co-IP Analysis
4.8. Confocal Microscopy
4.9. Plasmids and siRNA Transfection
4.10. Measurement of Intracellular Ca2+ Concentrations
4.11. ELISA
4.12. Western Blot
4.13. Immunohistochemistry
4.14. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SH | Sinomenine hydrochloride |
| LC3B | Microtubule-associated protein light chain 3B |
| 3-MA | 3-methyladenine |
| MDC | Monodansylcadaverine |
| ATG5 | Autophagy-related 5 |
| siRNA | Small interfering RNA |
| MMP | Matrix metalloproteinase |
| ER | Endoplasmic reticulum |
| EMT | Epithelial-mesenchymal transition |
| CHOP | C/EBP homologous protein |
| 4-PBA | 4-phenylbutyric acid |
| ECM | Extracellular matrix |
| UPR | Unfolded protein response |
| TNF-α | Tumor necrosis factor-α |
| IL-1β | Interleukin-1β |
| PERK | Eukaryotic translation initiation factor 2α kinase 3 |
| GRP78 | ER intraluminal 78 kDa glucose-regulated protein |
| eIF2α | Eukaryotic translation initiation factor 2α |
| AVs | Autophagic vacuoles |
References
- Afshordel, S.; Kern, B.; Clasohm, J.; König, H.; Priester, M.; Weissenberger, J.; Kögel, D.; Eckert, G.P. Lovastatin and perillyl alcohol inhibit glioma cell invasion, migration, and proliferation-impact of Ras-/Rho-prenylation. Pharmacol. Res. 2015, 91, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Anderson, E.M.; Demuth, T.; Nakada, S.; Reavie, L.B.; Drake, K.L.; Hoelzinger, D.B.; Berens, M.E. The phosphorylation of ephrin-B2 ligand promotes glioma cell migration and invasion. Int. J. Cancer 2010, 126, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Fridman, R.; Toth, M.; Peña, D.; Mobashery, S. Activation of progelatinase B (MMP-9) by gelatinase A (MMP-2). Cancer Res. 1995, 55, 2548–2555. [Google Scholar] [PubMed]
- McCawley, L.J.; Matrisian, L.M. Matrix metalloproteinases: Multifunctional contributors to tumor progression. Mol. Med. Today 2000, 6, 149–156. [Google Scholar] [CrossRef]
- Park, S.K.; Hwang, Y.S.; Park, K.K.; Park, H.J.; Seo, J.Y.; Chung, W.Y. Kalopanaxsaponin A inhibits PMA-induced invasion by reducing matrix metalloproteinase-9 via PI3K/Akt- and PKCdelta-mediated signaling in MCF-7 human breast cancer cells. Carcinogenesis 2009, 30, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xin, W. Matrine inhibiting pancreatic cells epithelial-mesenchymal transition and invasion through ROS/NF-κB/MMPs pathway. Life Sci. 2018, 192, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Cohen, J.; Arun, P.; Chen, Z.; Van Waes, C. NF-κB in carcinoma therapy and prevention. Expert Opin. Ther. Targets 2008, 12, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Nuclear factor-kappaB: The enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Liu, D.; Zhao, Y.; He, J.; Kang, H.; Dai, Z.; Wang, X.; Zhang, S.; Zan, Y. Sinomenine inhibits breast cancer cell invasion and migration by suppressing NF-kappa B activation mediated by IL-4/miR-324-5p/CUEDC2 axis. Biochem. Biophys. Res. Commun. 2015, 464, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Ren, H.; Lin, H.; Mao, J.; Zhu, T.; Wang, S.; Ye, Z. Sinomenine prevents metastasis of human osteosarcoma cells via S phase arrest and supression of tumor-related neovascularization and osteolysis through the CXCR4-STAT3 pathway. Int. J. Oncol. 2016, 48, 2098–2112. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Agostinis, P.; Vanhoecke, B.; Dewaele, M.; De Meyer, G.R. Autophagy in disease: A double-edged sword with therapeutic potential. Clin. Sci. 2009, 116, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Tuloup-Minguez, V.; Hamaï, A.; Greffard, A.; Nicolas, V.; Codogno, P.; Botti, J. Autophagy modulates cell migration and β1 integrin membrane recycling. Cell Cycle 2013, 12, 3317–3328. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.W. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, Y.; Sheng, G. EMT in developmental morphogenesis. Cancer Lett. 2013, 341, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.W.; Williams, E.D. EMT and MET in carcinoma—Clinical observations, regulatory pathways and new models. Clin. Exp. Metastasis 2008, 25, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Paranandi, K.S.; Sridharan, S.; Basu, A. Autophagy in breast cancer and its implications for therapy. Am. J. Cancer Res. 2013, 3, 251–265. [Google Scholar] [PubMed]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking ER stress to autophagy: Potential implications for cancer therapy. Int. J. Cell Biol. 2010, 2010, 930509. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIFα phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, J.M.; Xiong, X.X.; Qiu, X.Y.; Pan, F.; Liu, D.; Lan, S.J.; Jin, S.; Yu, S.B.; Chen, X.Q. Piperlongumine selectively kills hepatocellular carcinoma cells and preferentially inhibits their invasion via ROS-ER-MAPKs-CHOP. Oncotarget 2015, 6, 6406–6421. [Google Scholar] [CrossRef] [PubMed]
- Nami, B.; Donmez, H.; Kocak, N. Tunicamycin-induced endoplasmic reticulum stress reduces in vitro subpopulation and invasion of CD44+/CD24- phenotype breast cancer stem cells. Exp. Toxicol. Pathol. 2016, 68, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiao, Y.; Wang, Z.; Li, T.; Liu, Y.; Li, Y.; Zhao, X.; Wang, D. Sinomenine Hydrochloride Inhibits Human Glioblastoma Cell Growth through Reactive Oxygen Species Generation and Autophagy-Lysosome Pathway Activation: An In Vitro and In Vivo Study. Int. J. Mol. Sci. 2017, 18, E1945. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.S.; Zhou, B.H.; Li, C.L.; Zhang, F.; Wang, X.F.; Zhang, G.; Bu, X.Z.; Cai, S.H.; Du, J. Epithelial-mesenchymal transition (EMT) induced by TNF-alpha requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. PLoS ONE 2013, 8, e56664. [Google Scholar]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Mokhdomi, T.A.; Bukhari, S.; Wani, S.H.; Wafai, A.H.; Lone, G.N.; Qadri, A.; Qadri, R.A. Tectorigenin ablates the inflammation-induced epithelial-mesenchymal transition in a co-culture model of human lung carcinoma. Pharmacol. Rep. 2015, 67, 382–387. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Andreu, P.; Coussens, L.M. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, K.A.; Muir, B.; Reinhardt, F.; Carpenter, A.E.; Sgroi, D.C.; Weinberg, R.A. The Spemann organizer gene, goosecoid, promotes tumor metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 18969–18974. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.Y.; Hsu, Y.L.; Chen, Y.H.; Chen, T.C.; Wang, J.Y.; Kuo, P.L. Gemifloxacin, a fluoroquinolone antimicrobial drug, inhibits migration and invasion of human colon cancer cells. Biomed. Res. Int. 2013, 2013, 159786. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Liu, L.; Qi, C.; Deng, B.; Cai, X. Sinomenine versus NSAIDs for the treatment of rheumatoid arthritis: A systematic review and meta-analysis. Planta Med. 2008, 74, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, D. Research progress on antitumor mechanisms of sinomenine. Drugs Clin. 2016, 31, 1866–1870. [Google Scholar]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013, 138, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Bendris, N.; Lemmers, B.; Blanchard, J.M. Cell cycle, cytoskeleton dynamics and beyond: The many functions of cyclins and CDK inhibitors. Cell Cycle 2015, 14, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Ma, Y.; Li, W.; Liu, X.; Ying, G.; Fu, L.; Gu, F. Role of aquaporin-4 in the regulation of migration and invasion of human glioma cells. Int. J. Oncol. 2011, 38, 1521–1531. [Google Scholar] [PubMed]
- Polette, M.; Nawrocki-Raby, B.; Gilles, C.; Clavel, C.; Birembaut, P. Tumour invasion and matrix metalloproteinases. Crit. Rev. Oncol. Hematol. 2004, 49, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Cai, J.; Zhang, N.; Wu, J.; Yuan, J.; Li, J.; Li, M. Sp1 is upregulated in human glioma, promotes MMP-2-mediated cell invasion and predicts poor clinical outcome. Int. J. Cancer 2012, 130, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Shiras, A.; Shanmuganandam, K.; Shastry, P. Rictor regulates MMP-9 activity and invasion through Raf-1-MEK-ERK signaling pathway in glioma cells. Mol. Carcinog. 2011, 50, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell. Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Huang, Y.; Guo, B.; Liang, B.; Liu, X.; Ou, H.; Jiang, C.; Li, X.; Yang, D. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 125–139. [Google Scholar] [PubMed]
- Zhang, Z.; Liu, T.; Yu, M.; Li, K.; Li, W. The plant alkaloid tetrandrine inhibits metastasis via autophagy-dependent Wnt/β-catenin and metastatic tumor antigen 1 signaling in human liver cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 7. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/β-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, R.L.; Timpson, P.; Thorburn, J.; Anderson, K.I.; Thorburn, A.; Ryan, K.M. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012, 11, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Zou, J.; Yan, L.; Du, W.; Zhang, Y.; Sun, H.; Lu, P.; Geng, S.; Gu, R.; et al. Tetrahydrocurcumin induces mesenchymal-epithelial transition and suppresses angiogenesis by targeting HIF-1α and autophagy in human osteosarcoma. Oncotarget 2017, 8, 91134–91149. [Google Scholar] [PubMed]
- Hardy, S.D.; Shinde, A.; Wang, W.H.; Wendt, M.K.; Geahlen, R.L. Regulation of epithelial-mesenchymal transition and metastasis by TGF-β, P-bodies, and autophagy. Oncotarget 2017, 8, 103302–103314. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.E.; Tang, B.L. Linking membrane dynamics and trafficking to autophagy and the unfolded protein response. J. Cell. Physiol. 2013, 228, 1638–1640. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, Y.; Zhang, R.; Tu, X.; Gong, X. Ursolic acid induces autophagy in U87MG cells via ROS-dependent endoplasmic reticulum stress. Chem.-Biol. Interact. 2014, 218, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Folch-Puy, E.; Panisello, A.; Oliva, J.; Lopez, A.; Castro Benítez, C.; Adam, R.; Roselló-Catafau, J. Relevance of Endoplasmic Reticulum Stress Cell Signaling in Liver Cold Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2016, 17, 807. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Kim, H.; Ahn, J.; Jung, S.K.; Um, M.Y.; Son, K.H.; Kim, T.W.; Ha, T.Y. Anthricin isolated from Anthriscus sylvestris (L.) Hoffm. inhibits the growth of breast cancer cells by inhibiting Akt/mTOR signaling, and its apoptotic effects are enhanced by autophagy inhibition. Evid. Based Complement. Altern. Med. 2013, 2013, e385219. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lin, Z.; Lamb, R.; Hulit, J.; Howell, A.; Sotgia, F.; Rubin, E.; Lisanti, M.P. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism implications for breast cancer prevention. Cell Cycle 2013, 12, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-kappaB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.Y.; Hu, Y.; Que, Z.Y.; Wang, P.; Liu, Y.H.; Wang, Z.H.; Xue, Y.X. Shikonin inhibits the migration and invasion of human glioblastoma cells by targeting phosphorylated β-catenin and phosphorylated PI3K/Akt: A potential mechanism for the anti-glioma efficacy of a traditional chinese herbal medicine. Int. J. Mol. Sci. 2015, 16, 23823–23848. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, Q.; Zeng, Y.; Li, J.; Wang, L.; Ai, P. Evodiamine inhibits the migration and invasion of nasopharyngeal carcinoma cells in vitro via repressing MMP-2 expression. Cancer Chemother. Pharmacol. 2015, 76, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Z.; Liu, Y.; Liu, X.; Chang, Q.; Liao, Y.; Pan, R. Neuroprotective effect of water extract of Panax ginseng on corticosterone-induced apoptosis in PC12 cells and its underlying molecule mechanisms. J. Ethnopharmacol. 2015, 159, 102–112. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Jiao, Y.; Liu, Y.; Zhang, M.; Wang, Z.; Li, Y.; Li, T.; Zhao, X.; Wang, D. Sinomenine Hydrochloride Inhibits the Metastasis of Human Glioblastoma Cells by Suppressing the Expression of Matrix Metalloproteinase-2/-9 and Reversing the Endogenous and Exogenous Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2018, 19, 844. https://doi.org/10.3390/ijms19030844
Jiang Y, Jiao Y, Liu Y, Zhang M, Wang Z, Li Y, Li T, Zhao X, Wang D. Sinomenine Hydrochloride Inhibits the Metastasis of Human Glioblastoma Cells by Suppressing the Expression of Matrix Metalloproteinase-2/-9 and Reversing the Endogenous and Exogenous Epithelial-Mesenchymal Transition. International Journal of Molecular Sciences. 2018; 19(3):844. https://doi.org/10.3390/ijms19030844
Chicago/Turabian StyleJiang, Yumao, Yue Jiao, Yang Liu, Meiyu Zhang, Zhiguo Wang, Yujuan Li, Tao Li, Xiaoliang Zhao, and Danqiao Wang. 2018. "Sinomenine Hydrochloride Inhibits the Metastasis of Human Glioblastoma Cells by Suppressing the Expression of Matrix Metalloproteinase-2/-9 and Reversing the Endogenous and Exogenous Epithelial-Mesenchymal Transition" International Journal of Molecular Sciences 19, no. 3: 844. https://doi.org/10.3390/ijms19030844