Targeting Hodgkin and Reed–Sternberg Cells with an Inhibitor of Heat-Shock Protein 90: Molecular Pathways of Response and Potential Mechanisms of Resistance

 , ,
, ,
Abstract
:1. Introduction
2. Results
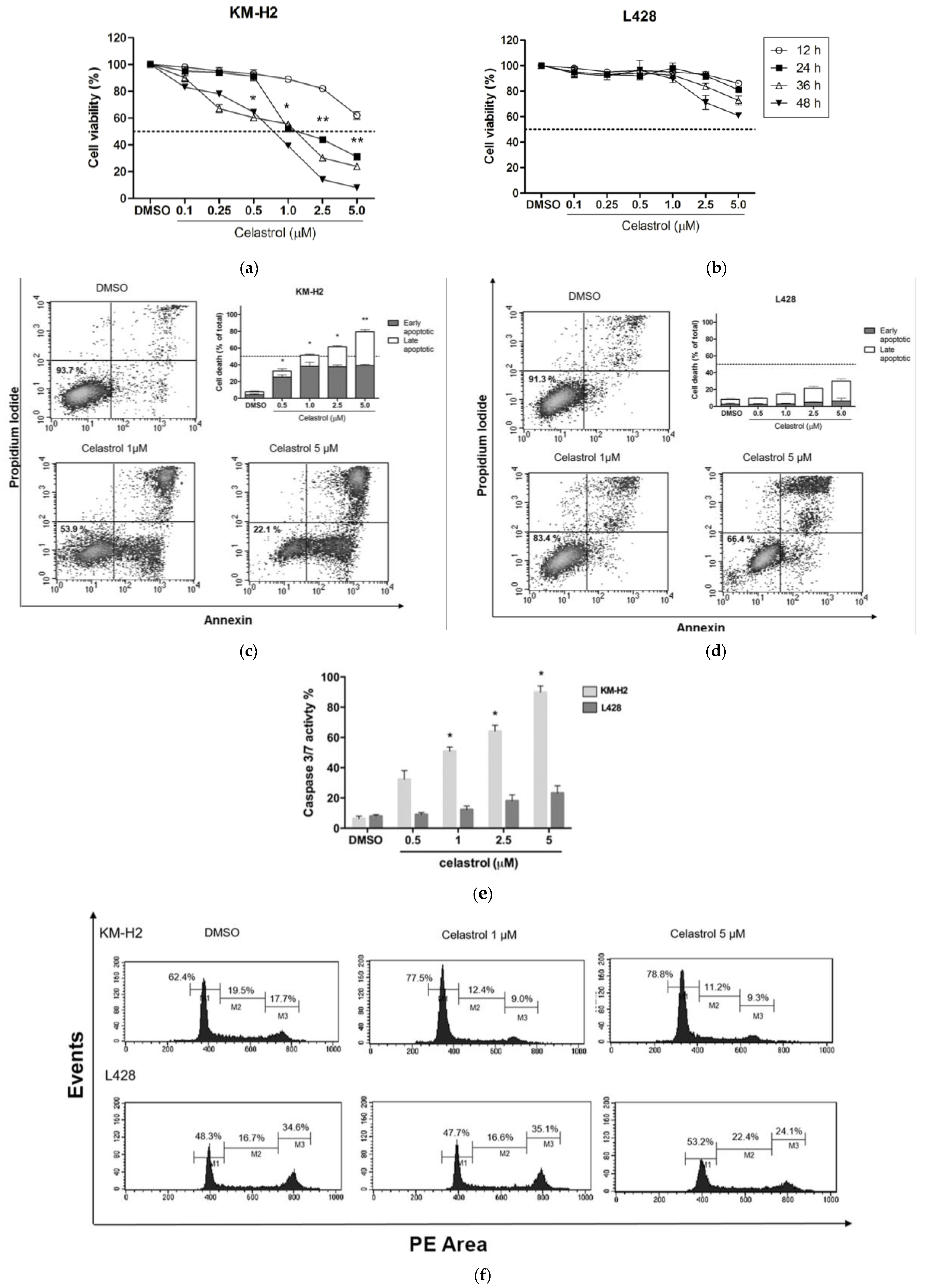
2.1. Effect of Celastrol on the Viability of KM-H2 and L428 Cells
2.2. Celastrol Induces Apoptosis and Changes in Cell Cycle in KM-H2 but Not in L428 Cells
2.3. Celastrol Induces Changes in the Proteome of Hodgkin’s Lymphoma Cell Lines
2.4. Overview of Proteins Modulated by Celastrol in KM-H2 and L428 Cells
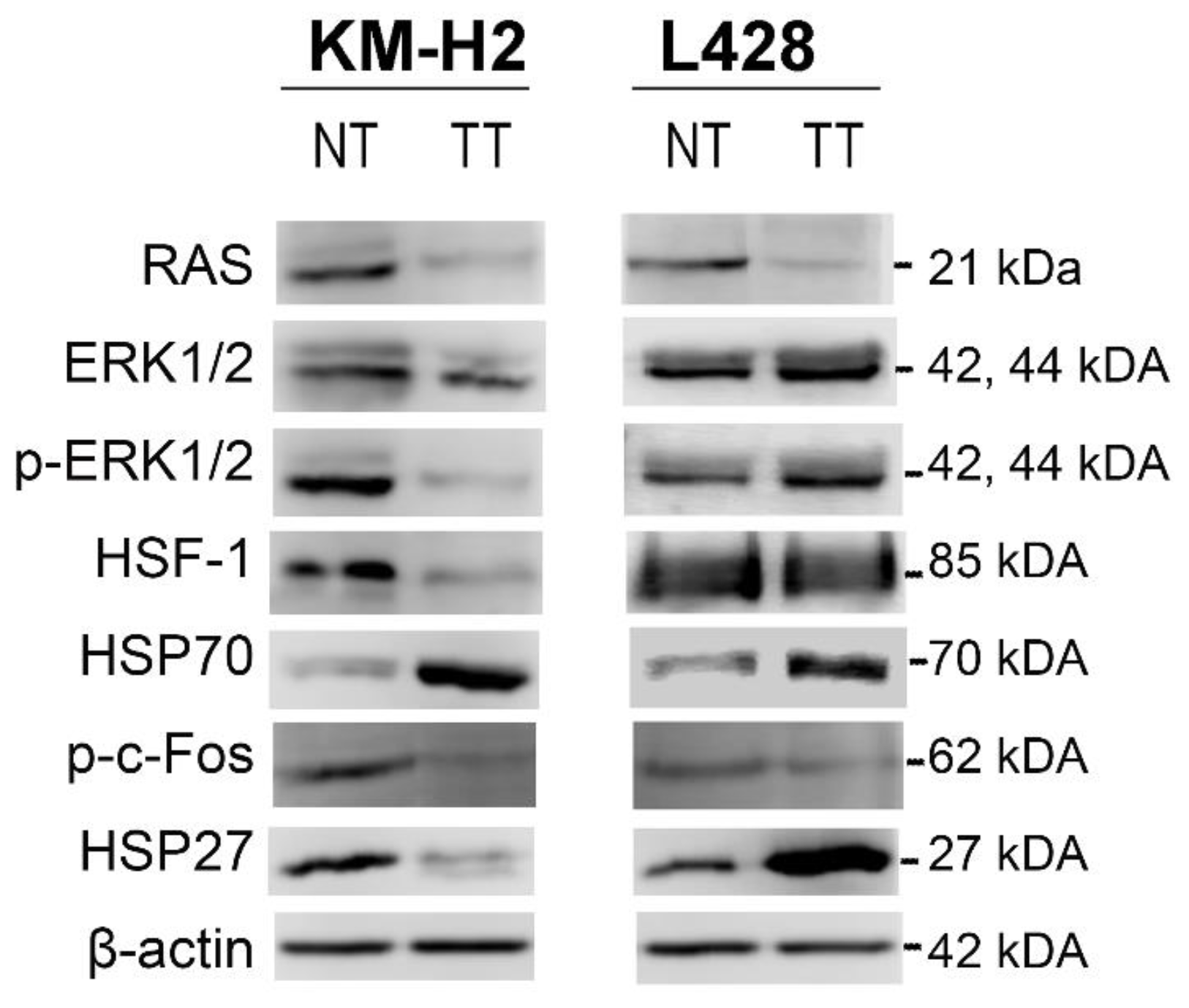
2.5. Validation of the Proteomic Results
3. Discussion
4. Materials and Methods
4.1. Reagents and Drugs
4.2. Cell Culture and Treatments
4.3. Cell Viability Determined by WST-1
4.4. Analysis of Caspase-3 and -7 Activities
4.5. Flow Cytometry Analysis
4.6. Proteomic Studies
4.7. Western Blot Analysis
4.8. qPCR Analysis
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuppers, R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Schwering, I.; Brauninger, A.; Klein, U.; Jungnickel, B.; Tinguely, M.; Diehl, V.; Hansmann, M.L.; Dalla-Favera, R.; Rajewsky, K.; Kuppers, R. Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 101, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Durkop, H.; Latza, U.; Hummel, M.; Eitelbach, F.; Seed, B.; Stein, H. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell 1992, 68, 421–427. [Google Scholar] [CrossRef]
- Smith, C.A.; Gruss, H.J.; Davis, T.; Anderson, D.; Farrah, T.; Baker, E.; Sutherland, G.R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; et al. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell 1993, 73, 1349–1360. [Google Scholar] [CrossRef]
- Hinz, M.; Lemke, P.; Anagnostopoulos, I.; Hacker, C.; Krappmann, D.; Mathas, S.; Dorken, B.; Zenke, M.; Stein, H.; Scheidereit, C. Nuclear factor κB-dependent gene expression profiling of Hodgkin’s disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J. Exp. Med. 2002, 196, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Molin, D.; Fischer, M.; Xiang, Z.; Larsson, U.; Harvima, I.; Venge, P.; Nilsson, K.; Sundstrom, C.; Enblad, G.; Nilsson, G. Mast cells express functional CD30 ligand and are the predominant CD30L-positive cells in Hodgkin’s disease. Br. J. Haematol. 2001, 114, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Diehl, V.; Thomas, R.K.; Re, D. Part II: Hodgkin’s lymphoma—Diagnosis and treatment. Lancet Oncol. 2004, 5, 19–26. [Google Scholar] [CrossRef]
- Harker-Murray, P.D.; Drachtman, R.A.; Hodgson, D.C.; Chauvenet, A.R.; Kelly, K.M.; Cole, P.D. Stratification of treatment intensity in relapsed pediatric Hodgkin lymphoma. Pediatr. Blood Cancer 2014, 61, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Shanmugam, M.K.; Sethi, G. Molecular targets of celastrol derived from Thunder of God Vine: Potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011, 303, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit HSP90 and Cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Ahn, K.S.; Pandey, M.K.; Aggarwal, B.B. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-κB-regulated gene products and TAK1-mediated NF-κB activation. Blood 2007, 109, 2727–2735. [Google Scholar] [PubMed]
- Yang, H.S.; Kim, J.Y.; Lee, J.H.; Lee, B.W.; Park, K.H.; Shim, K.H.; Lee, M.K.; Seo, K.I. Celastrol isolated from Tripterygium regelii induces apoptosis through both caspase-dependent and -independent pathways in human breast cancer cells. Food Chem. Toxicol. 2011, 49, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Xu, L.; Cao, F.; Wei, T.; Yang, C.; Uzan, G.; Zhang, D. HSP90 inhibitor, celastrol, arrests human monocytic leukemia cell U937 at G0/G1 in thiol-containing agents reversible way. Mol. Cancer 2010, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Palmfeldt, J.; Vang, S.; Corydon, T.J.; Gregersen, N.; Bross, P. Quantitative proteomics reveals cellular targets of celastrol. PLoS ONE 2011, 6, e26634. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakano, K.; Togano, T.; Nakashima, M.; Higashihara, M.; Kadin, M.E.; Watanabe, T.; Horie, R. Targeted repression of overexpressed CD30 downregulates NF-κB and ERK1/2 pathway in Hodgkin lymphoma cell lines. Oncol. Res. 2011, 19, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Boll, B.; Eltaib, F.; Reiners, K.S.; von Tresckow, B.; Tawadros, S.; Simhadri, V.R.; Burrows, F.J.; Lundgren, K.; Hansen, H.P.; Engert, A.; et al. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-κB and sensitizes Hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin. Cancer Res. 2009, 15, 5108–5116. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakano, K.; Kadin, M.E.; Higashihara, M.; Watanabe, T.; Horie, R. CD30 Induces Heat Shock Protein 90 and Signal Integration in Classic Hodgkin Lymphoma Cells. Am. J. Pathol. 2017, 187, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Popowska, U.; Sielicka-Dudzin, A.; Kuban-Jankowska, A.; Sawczuk, W.; Knap, N.; Cicero, G.; Wozniak, F. Geldanamycin and its derivatives as Hsp90 inhibitors. Front. Biosci. 2012, 17, 2269–2277. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Parone, P.A.; Martinou, J.C. Mitochondrial fission and apoptosis: An ongoing trial. Biochim. Biophys. Acta 2006, 1763, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Misso, G.; Giuberti, G.; Lombardi, A.; Grimaldi, A.; Ricciardiello, F.; Giordano, A.; Tagliaferri, P.; Abbruzzese, A.; Caraglia, M. Pharmacological inhibition of HSP90 and ras activity as a new strategy in the treatment of HNSCC. J. Cell. Physiol. 2013, 228, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; An, W.G.; Neckers, L.M. Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem. Biophys. Res. Commun. 1997, 239, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; Blagosklonny, M.V.; Ingui, C.; Neckers, L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J. Biol. Chem. 1995, 270, 24585–24588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Fiumara, P.; Li, Y.V.; Georgakis, G.; Snell, V.; Younes, M.; Vauthey, J.N.; Carbone, A.; Younes, A. MEK/ERK pathway is aberrantly active in Hodgkin disease: A signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood 2003, 102, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones 2006, 11, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Lotz, G.P.; Lin, H.; Harst, A.; Obermann, W.M. Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J. Biol. Chem. 2003, 278, 17228–17235. [Google Scholar] [CrossRef] [PubMed]
- Kai, M.; Nakatsura, T.; Egami, H.; Senju, S.; Nishimura, Y.; Ogawa, M. Heat shock protein 105 is overexpressed in a variety of human tumors. Oncol. Rep. 2003, 10, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Zhang, H.Y.; Hao, F.J.; Meng, X.; Guan, Y.; Du, Z.X. Induction of BAG2 protein during proteasome inhibitor-induced apoptosis in thyroid carcinoma cells. Br. J. Pharmacol. 2008, 155, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Slater, A.A.; Alokail, M.; Gentle, D.; Yao, M.; Kovacs, G.; Maher, E.R.; Latif, F. DNA methylation profiling distinguishes histological subtypes of renal cell carcinoma. Epigenetics 2013, 8, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Gusev, N.B.; Bogatcheva, N.V.; Marston, S.B. Structure and properties of small heat shock proteins (sHsp) and their interaction with cytoskeleton proteins. Biochemistry 2002, 67, 511–519. [Google Scholar] [PubMed]
- Santon, A.; Garcia-Cosio, M.; Cristobal, E.; Pascual, A.; Muriel, A.; Garcia-Larana, J. Expression of heat shock proteins in classical Hodgkin lymphoma: Correlation with apoptotic pathways and prognostic significance. Histopathology 2011, 58, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, G.V.; Li, Y.; Rassidakis, G.Z.; Martinez-Valdez, H.; Medeiros, L.J.; Younes, A. Inhibition of heat shock protein 90 function by 17-allylamino-17-demethoxy-geldanamycin in Hodgkin’s lymphoma cells down-regulates Akt kinase, dephosphorylates extracellular signal-regulated kinase, and induces cell cycle arrest and cell death. Clin. Cancer Res. 2006, 12, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Janz, M.; Stuhmer, T.; Vassilev, L.T.; Bargou, R.C. Pharmacologic activation of p53-dependent and p53-independent apoptotic pathways in Hodgkin/Reed-Sternberg cells. Leukemia 2007, 21, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; Wu, Q.; Yang, X.; Zhang, L.S.; Wu, Y.P.; Lu, C. Effects of Celastrol on growth inhibition of U937 leukemia cells through the regulation of the Notch1/NF-κB signaling pathway in vitro. Chin. J. Cancer 2010, 29, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Manu, K.A.; Chen, L.; Li, F.; Rajendran, P.; Subramaniam, A.; Lam, P.; Kumar, A.P.; Sethi, G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 2011, 16, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, X.; Zhao, M.; Wang, Y.; Cheng, X.; Wang, D.; Xu, Y.; Du, Z.; Yu, X. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer 2011, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Cascao, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.N.; Blower, P.E.; Alvarado, O.; Ravula, R.; Gout, P.W.; Huang, Y. Pharmacogenomic approach reveals a role for the xc− cystine/glutamate antiporter in growth and celastrol resistance of glioma cell lines. J. Pharmacol. Exp. Ther. 2010, 332, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.; Wang, X.; Xu, N.; Zhang, H.; Xu, H. Application of proteomics to determine the mechanism of action of traditional Chinese medicine remedies. J. Ethnopharmacol. 2014, 155, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Sampson, S.B. HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 2016, 26, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Schmitt, E.; Cande, C.; Vahsen, N.; Parcellier, A.; Kroemer, G. HSP27 and HSP70: Potentially oncogenic apoptosis inhibitors. Cell Cycle 2003, 2, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.K.; Parra, I.; Lemieux, P.; Oesterreich, S.; Hilsenbeck, S.G.; Fuqua, S.A. Hsp27 overexpression inhibits doxorubicin-induced apoptosis in human breast cancer cells. Breast Cancer Res. Treat. 1999, 56, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Mounier, N.; Arrigo, A.P. Actin cytoskeleton and small heat shock proteins: How do they interact? Cell Stress Chaperones 2002, 7, 167–176. [Google Scholar] [CrossRef]
- Lamoureux, F.; Thomas, C.; Yin, M.J.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Suppression of heat shock protein 27 using OGX-427 induces endoplasmic reticulum stress and potentiates heat shock protein 90 inhibitors to delay castrate-resistant prostate cancer. Eur. Urol. 2014, 66, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Sokol, E.; Jin, D.; Brune, Z.; Thiru, P.; Ghandi, M.; Garraway, L.A.; Gupta, P.B.; Santagata, S.; Whitesell, L.; et al. Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.L.; Hsu, S.M. Abundance of heat shock proteins (HSP89, HSP60, and HSP27) in malignant cells of Hodgkin’s disease. Cancer Res. 1998, 58, 5507–5513. [Google Scholar] [PubMed]
- Drexler, H.G.; Matsuo, Y. Guidelines for the characterization and publication of human malignant hematopoietic cell lines. Leukemia 1999, 13, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Panis, C.; Pizzatti, L.; Herrera, A.C.; Correa, S.; Binato, R.; Abdelhay, E. Label-free proteomic analysis of breast cancer molecular subtypes. J. Proteome Res. 2014, 13, 4752–4772. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Pathway Name * | FDR | N# | Identified Proteins |
|---|---|---|---|
| KM-H2 cell line | |||
| Development_Ligand-independent activation of ESR1 and ESR2 | 2.5 × 10−5 | 8/44 | p300, ERK1/2, ERK1 (MAPK3), ERK2 (MAPK1), PI3KIA, PI3KIA t class IA (p110-alpha), p90RSK1, CBP |
| NETosis in SLE | 3.9 × 10−4 | 6/31 | ERK1/2, Histone H3, Histone H2, Histone H2A, Histone H1.2, Histone H1 |
| Cell cycle_Role of Nek in cell cycle regulation | 3.9 × 10−4 | 6/32 | Histone H3, PI3K cat class IA, Tubulin, Tubulin beta, Histone H1, Tubulin alpha |
| Cytoskeleton remodeling_Neurofilaments | 1.6 × 10−3 | 5/25 | Vimentin, Tubulin (in microtubules), Tubulin beta, Desmuslin, Tubulin alpha |
| Signal transduction_Additional pathways of NF-kB activation | 2.9 × 10−3 | 5/30 | p300, ERK1/2, Histone H3, p90RSK1, CBP |
| Development_IGF-1 signaling | 2.9 × 10−3 | 6/50 | ERK1/2, ERK1 (MAPK3), ERK2 (MAPK1), PI3K cat class IA, NF-kB, CDC42 |
| Sorafenib-induced inhibition of cell proliferation and angiogenesis in HCC | 2.9 × 10−3 | 4/16 | VEGFR-1, ERK1/2, ERK1 (MAPK3), ERK2 (MAPK1) |
| Cell cycle_Start of DNA replication in early S phase | 2.9 × 10−3 | 5/32 | RPA3, MCM3, Histone H1, MCM5, MCM2 |
| Signal transduction_Activin A signaling regulation | 2.9 × 10−3 | 5/33 | p300, Histone H3, Evi-1, Histone H2, CBP |
| Development_S1P1 receptor signaling via beta-arrestin | 2.9 × 10−3 | 5/34 | ERK1/2, ERK1 (MAPK3), ERK2 (MAPK1), PI3K cat class IA (p110-alpha), p90Rsk |
| L428 cell line | |||
| Regulation of degradation of deltaF508-CFTR in CF | 3.5 × 10−5 | 8/39 | HSP70, HSP105, HSP27, SUMO-2, E2I, Aha1, SAE1, BAG-2 |
| NETosis in SLE | 4.3 × 10−4 | 7/31 | ERK1/2, Histone H3, Histone H2A, Histone H2, Histone H1, Histone H1.2, HMGB1 |
| Transcription_Negative regulation of HIF1A function | 4.9 × 10−4 | 8/66 | HSP70, MCM7, PSMA7, PRDX4, RUVBL2, MCM2, MCM5, PRDX2 |
| Cell cycle_Start of DNA replication in early S phase | 1.2 × 10−3 | 6/32 | MCM4/6/7 complex, RPA3, MCM2, MCM4, Histone H1, MCM5 |
| Development_Regulation of cytoskeleton proteins in oligodendrocyte differentiation and myelination | 1.6 × 10−3 | 7/58 | Tubulin alpha, Tubulin, Actin cytoskeletal, Tubulin beta, Dcc, MRLC, Cortactin |
| Cytoskeleton remodeling_Neurofilaments | 2.5 × 10−3 | 5/25 | Tubulin alpha, Tubulin, Actin cytoskeletal, Tubulin beta, Kinesin heavy chain |
| Immune response_Sublytic effects of membrane attack complex | 3.1 × 10−3 | 7/68 | RK1/2, GRP75, HSP27, Actin cytoskeletal, cPLA2, GRP78, eIF2S1 |
| Development_Slit-Robo signaling | 3.1 × 10−3 | 5/30 | Tubulin, Actin cytoskeletal, Actin, ACTB, Cortactin |
| Transport_The role of AVP in regulation of Aquaporin 2 and renal water reabsorption | 3.5 × 10−3 | 6/50 | ERK1/2, Actin cytoskeletal, ACTB, MRLC2, MRLC, Annexin II |
| Cell cycle_Role of Nek in cell cycle regulation | 3.5 × 10−5 | 5/32 | Tubulin alpha, Tubulin, Histone H3, Tubulin beta, Histone H1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segges, P.; Corrêa, S.; Du Rocher, B.; Vera-Lozada, G.; Krsticevic, F.; Arce, D.; Sternberg, C.; Abdelhay, E.; Hassan, R. Targeting Hodgkin and Reed–Sternberg Cells with an Inhibitor of Heat-Shock Protein 90: Molecular Pathways of Response and Potential Mechanisms of Resistance. Int. J. Mol. Sci. 2018, 19, 836. https://doi.org/10.3390/ijms19030836
Segges P, Corrêa S, Du Rocher B, Vera-Lozada G, Krsticevic F, Arce D, Sternberg C, Abdelhay E, Hassan R. Targeting Hodgkin and Reed–Sternberg Cells with an Inhibitor of Heat-Shock Protein 90: Molecular Pathways of Response and Potential Mechanisms of Resistance. International Journal of Molecular Sciences. 2018; 19(3):836. https://doi.org/10.3390/ijms19030836
Chicago/Turabian StyleSegges, Priscilla, Stephany Corrêa, Bárbara Du Rocher, Gabriela Vera-Lozada, Flavia Krsticevic, Debora Arce, Cinthya Sternberg, Eliana Abdelhay, and Rocio Hassan. 2018. "Targeting Hodgkin and Reed–Sternberg Cells with an Inhibitor of Heat-Shock Protein 90: Molecular Pathways of Response and Potential Mechanisms of Resistance" International Journal of Molecular Sciences 19, no. 3: 836. https://doi.org/10.3390/ijms19030836