Neutrophils: Beneficial and Harmful Cells in Septic Arthritis
by
 and
and
Daiane Boff
1,2, 
Helena Crijns
1,2,
Mauro M. Teixeira
1,
Flavio A. Amaral
1,† and
Paul Proost
2,*,† 1
Imunofarmacologia, Department of Biochemistry and Immunology, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte 31270-901, Brazil
2
Laboratory of Molecular Immunology, Department of Microbiology and Immunology, Rega Institute for Medical Research, KU Leuven, B-3000 Leuven, Belgium
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2018, 19(2), 468; https://doi.org/10.3390/ijms19020468
Submission received: 31 December 2017
/
Revised: 30 January 2018
/
Accepted: 1 February 2018
/
Published: 5 February 2018
(This article belongs to the Special Issue Molecular Mechanism of Infectious Disease)
Abstract
:Septic arthritis is an inflammatory joint disease that is induced by pathogens such as Staphylococcus aureus. Infection of the joint triggers an acute inflammatory response directed by inflammatory mediators including microbial danger signals and cytokines and is accompanied by an influx of leukocytes. The recruitment of these inflammatory cells depends on gradients of chemoattractants including formylated peptides from the infectious agent or dying cells, host-derived leukotrienes, complement proteins and chemokines. Neutrophils are of major importance and play a dual role in the pathogenesis of septic arthritis. On the one hand, these leukocytes are indispensable in the first-line defense to kill invading pathogens in the early stage of disease. However, on the other hand, neutrophils act as mediators of tissue destruction. Since the elimination of inflammatory neutrophils from the site of inflammation is a prerequisite for resolution of the acute inflammatory response, the prolonged stay of these leukocytes at the inflammatory site can lead to irreversible damage to the infected joint, which is known as an important complication in septic arthritis patients. Thus, timely reduction of the recruitment of inflammatory neutrophils to infected joints may be an efficient therapy to reduce tissue damage in septic arthritis.
1. Introduction
Septic arthritis can be defined as an inflammatory disease of the joints, induced by an infectious agent [1,2]. Bacteria, viruses, fungi and protozoa may invade joints and cause injury. However, Gram-positive bacteria, especially Staphylococcus aureus (S. aureus), are the most prevalent microorganisms causing septic arthritis [3]. In addition, S. aureus is responsible for the most severe cases of septic arthritis. Any synovial joint can be involved; however, most frequently one large joint such as the knee or hip is affected [1,4]. Invasion of bacteria into the synovial space can occur predominantly by two routes: either through hematogenous spread (most common) or by direct invasion [5] as shown in Figure 1A. The synovium is extremely vascularized and contains no limiting basement membrane, facilitating the access to the synovial space. Thus, bacteria may spread directly from adjacent osteomyelitis or from a local soft tissue infection and could reach the joint during diagnostic or therapeutic procedures, penetrating trauma, or prosthetic surgery, or, less commonly, by animal bites [2,6,7].
Septic arthritis patients typically present with a single swollen, warm and painful joint with a decreased range of motion. Fever is present in only 30–40% of cases [8]. Normally a single synovial joint is affected such as the knee, hip, ankle or elbow. The hip is the more frequently affected joint in children. Atypical joint infection, including the sternoclavicular, costochondral and sacroiliac joints, may be common in intravenous drug users [9]. Polyarticular septic arthritis is not common and usually accompanied by a number of risk factors. The articular damage is an important feature and a challenge in this disease, since about 25–50% of patients have irreversible articular damage with total loss of joint function [1,10].
Once microorganisms have gained entry into the joint, the low fluid shear conditions in the joint space allow adherence and infection. The attachment of S. aureus to the joint extracellular matrix or to implanted medical devices, such as prosthetic joints, is mediated by microbial surface component recognizing adhesive matrix molecules (MSCRAMMs). After colonizing the joint, the bacteria can rapidly proliferate and trigger an acute inflammatory response [11]. The synovium responds with a proliferative lining-cell hyperplasia and there is an influx of inflammatory cells [12]. Phagocytes, including neutrophils and macrophages, chemotactically migrate to the infected joint, directed by gradients of bacterial products displaying chemotactic activity and mediators of the immune response [13]. Neutrophils play a major role in the first-line defense against invading pathogens, including bacteria, and these leukocytes are the first to migrate to the site of infection. Activated macrophages are recruited to the joint slightly later and they are followed by T lymphocytes [1,14,15,16]. In this manuscript, we will review current knowledge on septic arthritis with an emphasis on the role of neutrophils. We will discuss neutrophil activation and recruitment, not only resulting in their beneficial role in the elimination of microbial infection, but also regularly causing tissue damage and permanent joint dysfunction.
2. Septic Arthritis
Septic arthritis is an uncommon pathology of which the yearly incidence is estimated to be 3 to 12 cases per 100,000 people in industrialized countries [17,18,19,20]. Septic arthritis can affect people at any age, but elderly people and very young children are more frequently affected [21,22]. Approximately half of the patients are younger than three years and one-third are under the age of two. The incidence is low in children younger than three months. Furthermore, males are slightly more susceptible than females [2,23]. The incidence of septic arthritis appears to decrease in children in the United States [23]. However, several factors including an ageing population, a growing resistance to antibiotics, an increase in infections related to orthopedic procedures and an enhanced use of immune modulating agents, contribute to an increase of septic arthritis in the general population [7,24,25,26]. Furthermore, the presence of previous joint diseases, such as rheumatoid arthritis (RA), osteoarthritis, crystal arthropathies and other forms of inflammatory arthritis is a predisposing factor for the development of infectious arthritis (Figure 1B). In particular, the incidence of septic arthritis is approximately 10-fold higher in patients with RA, in comparison to the general population [27,28]. The incidence of septic arthritis increases not only in previous arthritic patients, but also in people who suffer from other chronic and immunosuppressive diseases, such as diabetes, leukemia, cirrhosis, granulomatous diseases, cancer, hypogammaglobulinemia, human immunodeficiency virus (HIV)-infected patients and intravenous drug users [29,30,31]. Hemodialysis has been reported as an important risk factor for septic arthritis [32]. Also, penetrating trauma, including animal bites and local therapeutic intra-articular corticosteroid injections may cause septic arthritis in atypical joints [11,33,34]. Recent joint surgery is also associated with an increased risk of infection [35,36]. In addition, several cases of joint infections have been reported in patients that received immunosuppressive therapy and/or glucocorticoids [37]. In this context, the use of classic disease modifying anti-rheumatic drugs (DMARDs) in RA patients can be an additional risk factor that facilitates the development of infectious arthritis [38,39]. Although data from observational registers have suggested an increased incidence of joint infections in patients receiving anti-tumor necrosis factor (TNF) therapy, the incidence does not seem to be different from the incidence in patients treated with classical DMARDs [40].
Septic arthritis is associated with significant mortality and morbidity. Moreover, it is a rheumatologic emergency, since irreversible joint destruction and consequently loss of function of the joint can occur rapidly [24,25,41]. Septic arthritis has a mortality ranging from approximately 10% to, depending on the report, more than 50% in case of polyarticular disease [3,7,41]. Persisting joint damage occurs in more than 30% of the patients [41]. Early diagnosis and immediate and effective treatment are essential to prevent severe outcomes such as irreversible joint destruction or death [2,23]. Furthermore, the general state of the patient and the number, type and resistance pattern of the causing agent are also of significance to the outcome [42].
The most common causative agent associated with septic arthritis is S. aureus, which accounts for about 50% of cases [43]. Recently, an increase in methicillin-resistant S. aureus (MRSA) infections has been reported in several health-care systems, particularly in the elderly and intravenous drug abuser populations as well as in patients who underwent orthopedic procedures [44]. MRSA has been associated with 18% and 41% of septic arthritis cases in studies in São Paulo, Brazil and Tainan, Taiwan, respectively [44,45]. Other bacteria such as group B streptococci, Streptococcus pneumoniae, Neisseria gonorhoeae, Pseudomonas aeruginosa, Escherichia coli, Proteus genus and Klebsiella species can be associated with septic arthritis, but are less frequent [46]. Common causative agents in children include S. aureus, Streptococcus pneumonia and Kingella kingae [47]. The infectious capacity of S. aureus in different tissues is provided by the presence of several virulence factors [48].
S. aureus has a capsule composed of polysaccharides, which acts as a physical barrier that protects the bacteria from phagocytosis by immune cells [49]. Peptidoglycan (PGN) is the major component of the cell wall of Gram-positive bacteria. Bacterial PGN was detected in synovial tissue of patients with septic arthritis [50] and studies demonstrated that intra-articular injection of PGN in mice can cause arthritis [51]. S. aureus is a bone pathogen because it possesses several cell-surface adhesion molecules that facilitate its binding to the bone matrix [52]. Binding involves a family of adhesins that interact with extracellular matrix components and these adhesins have been termed MSCRAMMs [53]. Specific MSCRAMMs are needed for the colonization of specific tissues. Particular MSCRAMMs include fibronectin-binding proteins, fibrinogen-binding proteins, elastin-binding and collagen-binding adhesion molecules. Once the bacteria adhere to and colonize bone matrix, they elaborate several virulence factors such as proteases, which can break down matrix components [54]. Further experimental studies demonstrated that collagen adhesin is an important virulence determinant in S. aureus-induced arthritis [55].
S. aureus secretes a large number of enzymes and toxins, many of which have been implicated as potential virulence factors. Alpha and gamma toxins are lytic to red blood cells and various leukocytes, but not to neutrophils [56]. The combination of these two toxins has been experimentally demonstrated to be important for the development of septic arthritis [57]. Another toxin is Panton–Valentine leukocidin (PVL, consisting of the LukS and LukF proteins) that can lyse leukocytes, especially human neutrophils, and is related to fulminant cases of septic arthritis [58]. Enterotoxins, such as the superantigen toxic shock syndrome toxin-1 (TSST-1) can cause shock by stimulating the release of interleukin (IL)-1, IL-2, TNF and other cytokines [59]. Experimentally, the presence of TSST-1 favors the development of septic arthritis [60]. Another important virulence factor is bacterial deoxyribonucleic acid (DNA) with non-methylated CpG motifs, which is considerably less frequent in vertebrate DNA [61]. The CpG DNA can bind to Toll-like receptor 9 (TLR9) in immune cells, leading to the production of cytokines such as IL-1β, TNF, IL-6 and IL-12 [62,63]. Some studies showed that intra-articular injection of S. aureus CpG DNA can induce arthritis in mice [64,65].
3. Diagnosis and Treatment of Septic Arthritis
Gram staining and cultures of synovial fluid should be investigated in any case of suspected septic arthritis. Antibiotic therapy is started ideally after synovial fluid samples have been obtained [64]. Gram stains of synovial fluid are helpful when positive, but they are not always sensitive enough for the diagnosis of septic arthritis [65]. Patients should be treated empirically for septic arthritis when synovial fluid leukocyte counts exceed 50,000 cells/mm3, although gout and pseudogout also commonly present with leukocyte counts of this magnitude [66]. Thus, the analysis of the presence of urate crystals in synovial fluid by polarized light microscopy is very important for the exclusion of a gouty attack [67,68,69]. Furthermore, the analysis of the delta neutrophil index (DNI) could be a valuable tool to distinguish septic arthritis and gout. DNI is a value that corresponds to the fraction of circulating immature granulocytes, reflecting a burden of infection. In this context, a study demonstrated that septic arthritic patients presented with a significantly higher DNI as compared to acute gouty attack patients, suggesting DNI as complementary predicting tool for septic arthritis diagnosis [70]. However, the serum procalcitonin level also appears to be a promising marker for septic arthritis [71]. On the other hand, mono-arthritis can also be misdiagnosed with cases of SAPHO (Synovitis-acne-pustulosis-hyperostosis-osteitis) syndrome, characterized by a combination of skin and osteoarticular manifestations [72]. Although S. aureus and other pathogens have been isolated from affected tissues [73], radiology features, such as radiography and MRI mainly in sternoclavicular joints are necessary for SAPHO syndrome diagnosis, especially in the absence of dermatological clinical manifestations [72]. Blood cultures should be obtained in all patients with suspected septic arthritis. However, the cultures must be obtained before starting antibiotic treatment to optimize the possibility of isolating the causative bacteria [74]. DNA-based techniques, hybridization probes, polymerase chain reaction (PCR)-based techniques and detection of typical bacterial compounds by mass spectrometry provide quick results [75]. The detection of microorganisms by PCR has shown more accurate results [76]. However, the risk of contamination, the presence of background DNA, the lack of a gold standard and the fact that PCR techniques detect DNA instead of living pathogens make the interpretation of these tests difficult [77].
Imaging can be used as complementary diagnosis since a computed tomography (CT) scan may not depict abnormalities during the early stages of infection. However, CT is a better imaging technique for visualization of local edema, bone erosions, osteitic foci and sclerosis [77]. Magnetic resonance imaging (MRI) provides better resolution for the detection of joint effusion and for differentiation between bone and soft-tissue infections. MRI findings in patients with septic arthritis include joint effusion, cartilage and bone destruction, soft-tissue abscesses, bone edema and cortical interruption [78].
Septic arthritis is so rapidly destructive that broad-spectrum antibiotics are usually warranted until culture data are available or bacteria have been identified by mass spectrometry. Given the increasing importance of MRSA as a cause of septic arthritis, initial antibiotic regimens should generally include an antibiotic active against MRSA, such as vancomycin [79]. Cefazolin is a reasonable alternative in areas with a low prevalence of MRSA. If serious vancomycin allergy is present, empiric therapy utilizing linezolid or daptomycin must be considered [80]. Septic arthritis associated with animal bites should be treated with agents such as ampicillin-sulbactam, which are active against oral microbiota [81].
In general, septic arthritis in adults should be treated for at least 3 weeks, which may include a period of step-down oral therapy [25]. In children with uncomplicated septic arthritis, as few as 10 days of antibiotic therapy may be sufficient [82]. Septic arthritis can be managed with antibiotics combined with joint drainage by arthroscopy, arthrocentesis, or arthrotomy [83,84,85]. Joint drainage decompresses the joint, improves blood flow, and removes bacteria, toxins, and proteases [84]. Arthrocentesis should be repeated daily until effusions resolve and cultures are negative. Aggressive rehabilitation is essential to prevent joint contractures and muscle atrophy [2].
4. Immune Response against S. aureus
4.1. Introduction
Pathogens are controlled by innate and adaptive immune responses and the recognition of microorganisms is the first step in host defense [86]. In the joint, resident cells, such as synoviocytes, can recognize S. aureus through pattern recognition receptors (PRRs). In that way, those cells produce inflammatory mediators such as cytokines, chemokines, complement proteins and lipids that will attract neutrophils and macrophages [87]. The complement system plays an important role in host defense against infection. Products of complement activation affect many functions of neutrophils in host defense. The complement system can opsonize microorganisms, thereby stimulating phagocytosis. Phagocytosis of S. aureus by neutrophils is of major importance for the outcome in the early stage of septic arthritis. Moreover, chemotaxis of neutrophils to the site of inflammation is facilitated by complement factors such as C5a. Complement depletion, by using cobra venom factor, in a murine model of hematogenously induced S. aureus septic arthritis caused an aggravation of septicemia and arthritis [88]. The prevalence and severity of septic arthritis and septicemia-induced mortality were augmented upon complement depletion. Manifestations of the disease, such as synovitis and destruction of cartilage and/or bone, occurred earlier and were more common and severe in the decomplemented mice compared to the control group. Altogether, complement depletion disturbed phagocytosis by impairing opsonization of bacteria, and interfered with the extravasation and migration of neutrophils, leading to a deterioration of the disease [88]. During the onset of the inflammatory process, neutrophils are the main cells recruited to the site of infection and they play a fundamental role in both the phagocytosis and killing of the microorganism [85]. The importance of neutrophils in controlling S. aureus in the joint was demonstrated in a study in which neutrophils were depleted. This caused the impairment of bacterial control [15]. Other immune cells such as macrophages [6], natural killer (NK) cells [6] and B lymphocytes [89] are described to have a role in experimental models of septic arthritis. Dendritic cells in S. aureus-induced arthritis are fundamental for the activation of the adaptive immune response. The depletion of dendritic cells during S. aureus infection in the lungs showed an increase in bacterial load and mortality [90]. During S. aureus infection, dendritic cells can induce a Th1 response probably through IL-12 production. Experimentally, the lack of systemic IL-12 increased the bacterial load in the joint during S. aureus-induced septic arthritis [91]. Dendritic cells can also stimulate Th17 activation, an important source of IL-17. The cytokine IL-17 has been shown to be important for bacterial clearance and to prevent tissue damage in experimental S. aureus-induced arthritis [92].
4.2. Neutrophils
Neutrophils are continuously generated in the bone marrow from myeloid precursors. Humans and mice differ in their numbers of circulating neutrophils. In humans, 50–70% of circulating leukocytes are neutrophils, whereas this number drops to only 10–25% in mice [93]. In the circulation, mature neutrophils have a segmented nucleus and their cytoplasm is enriched with granules and secretory vesicles. After the first moments following infection, neutrophils can be recruited from blood vessels to the site of infection, a process that involves a close interaction between neutrophils and endothelial cells and is mediated by different chemotactic agents that activate the cells and guide their migration [94]. Chemotactic factors for neutrophils include bacterial peptides [95], products of complement activation (such as C5a) [96], extracellular matrix degradation products (laminin digests) [97], arachidonic acid metabolites (leukotriene B4/LTB4) [98], other lipid mediators such as platelet activating factors (PAF) [99] and chemokines [100].
Neutrophils are recruited in a cascade of events that involves the following commonly recognized steps that precede the transmigration: tethering, rolling, adhesion, and crawling on the endothelial cell surface [101,102]. Neutrophil recruitment is initiated by changes on endothelial cells during the early steps of inflammation. Endothelial cells can be activated directly by pathogens through PRR activation, causing an increase of the expression and exposure of adhesion molecules on their surface. Once on the endothelial surface, P selectin and E selectin bind to their glycosylated ligands on leukocytes, leading to the tethering (capturing) of free-flowing neutrophils to the surface of the endothelium and subsequent rolling of neutrophils along the vessel in the direction of the blood flow [103]. Rolling requires rapid formation and breakage of adhesive bonds. The rolling of neutrophils facilitates their contact with chemokine-decorated endothelium to induce activation. Full activation may be a two-step process initiated by specific priming by pro-inflammatory cytokines, such as TNF and IL-1β, or by contact with activated endothelial cells followed by an exposure to pathogen-associated molecular patterns (PAMPs), chemoattractants or growth factors [104,105]. The adhesion step of the recruitment cascade prepares neutrophils for transmigration, but migration does not necessarily occur at the initial site of their arrest on the endothelium. Some of the adherent neutrophils reveal so called crawling behavior as they elongate and continue to send out pseudopods, apparently actively scanning and probing the surroundings while remaining firmly attached to a single location within the microvasculature [106]. During the transmigration process, neutrophils cross the endothelium in a process dependent on integrins. The migration across the endothelial cell layer occurs either paracellularly (between endothelial cells) or transcellularly (through an endothelial cell without mixing the cytoplasmic content of both cells). Next, neutrophils migrate towards the infectious/inflammatory focus in the tissue [107].
4.3. Neutrophil Functions during Infections
In order to kill microorganisms, neutrophils can phagocyte, secrete the content of their granules, produce reactive oxygen species (ROS) and antimicrobial peptides, and release neutrophil extracellular traps (NETs) as demonstrated in Figure 2A [108]. S. aureus may produce several virulence factors that neutralize neutrophil-dependent killing. These include the pore-forming toxin Panton-Valentine leukocidin, antioxidants staphyloxanthin, catalase and superoxide dismutase and the surface factor promoting resistance to oxidative killing (SOK) to neutralize the action of ROS [58,109,110,111] (Figure 2B). Neutrophil defensin-dependent killing of bacteria is inhibited by the binding of neutrophil defensins to staphylokinase [112]. In addition, the neutrophil-derived antibacterial peptide and neutrophil attractant LL37 may be degraded by the S. aureus metalloproteinase aureolysin [113,114]. Finally, NETs may be degraded by a S. aureus nuclease, resulting in diminished antibacterial efficiency of NETs [115].
Once at the site of infection, the neutrophils bind and ingest invading microorganisms by phagocytosis, a critical first step in the removal of bacteria during infection. Neutrophils recognize numerous surface-bound and freely secreted bacterial products such as PGN, lipoproteins, lipopolysaccharide, CpG-containing DNA, and flagellin [116]. Such conserved bacterial PAMPs are recognized directly by PRRs expressed on the extracellular membrane or on organelles in the cytosol of the neutrophil [117]. The process of neutrophil phagocytosis triggers synthesis of a number of immunomodulatory factors that will recruit additional neutrophils, modulates subsequent neutrophil responses, and coordinates early responses of other cell types such as monocytes, macrophages, dendritic cells and lymphocytes, thereby providing an important link between innate and acquired immune responses [118].
Phagocytosis is accompanied by the generation of microbicidal ROS (oxygen-dependent) and fusion of cytoplasmic granules with microbe-containing phagosomes (degranulation). Degranulation enriches the phagosome lumen with antimicrobial peptides and proteases (oxygen-independent process), which in combination with ROS create an environment non-conducive to survival of the ingested microbe [119]. In the most classical sense, neutrophil activation is intimately linked with the production of superoxide and other secondarily derived ROS, an oxygen-dependent process known as the oxidative or respiratory burst. High levels of superoxide are generated upon full assembly of the multi-subunit nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidase in both the plasma- and phagosomal membranes [120,121].
Neutrophils present three fundamental types of granules: primary or azurophilic, secondary or specific and tertiary or gelatinase-containing granules [122,123]. Primary granules are the largest and are formed first during neutrophil maturation. They are named after their ability to take up the basic dye azure A and contain myeloperoxidase (MPO), defensins, lysozyme, bactericidal/permeability-increasing protein (BPI), and a number of serine proteases such as neutrophil elastase, proteinase 3 and cathepsin G [124]. Granules of the second class are smaller, do not contain MPO and are characterized by the presence of the glycoprotein lactoferrin and antimicrobial compounds including neutrophil gelatinase-associated lipocalin, human cationic antimicrobial protein-18 and lysozyme [125]. The gelatinase granules are also MPO-negative, are smaller than specific granules and contain few antimicrobials, but they serve as a storage location for a number of metalloproteases, such as gelatinase and leukolysin [123]. Neutrophils also present secretory vesicles that serve as a reservoir for a number of important membrane-bound molecules active during neutrophil migration. As a neutrophil proceeds through the activation process, granules are mobilized and fuse with either the plasma membrane or the phagosome, releasing their content into the respective environments [126].
Neutrophils produce peptides and proteins that directly or indirectly kill microbes. There are three main types of antimicrobials: cationic peptides and proteins that bind to microbial membranes, enzymes, and proteins that deprive microorganisms of essential nutrients [127]. Many of these peptides disrupt the membrane integrity, whereas some antimicrobials are thought to disrupt essential microbial functions, such as DNA replication, transcription or production of energy [128]. In addition, some of these neutrophil-derived antimicrobial peptides also attract additional leukocytes to the inflammatory site [113,129]. Recently, it was demonstrated that neutrophils can produce neutrophil extracellular traps (NETs) that contain decondensed chromatin, bound histones, azurophilic granule proteins and cytosolic proteins. They have a demonstrated capacity to bind to and kill a variety of pathogens including S. aureus [130]. Extrusion of such structures by neutrophils is predicted to limit microbial spread and dissemination, while enhancing effective local concentrations of extruded microbicidal agents, thereby promoting synergistic killing of attached microorganisms [131].
Several mechanisms used by neutrophils to eliminate pathogens can also cause host tissue damage [132]. In that way, recruitment of inflammatory neutrophils needs to be tightly controlled and such neutrophils must be removed before they have serious, detrimental effects on inflamed tissues. Once neutrophils have executed their antimicrobial function, they die via a built-in cell-death program. However, not only does apoptosis reduce the number of neutrophils present, it also produces signals that abrogate further neutrophil recruitment [133]. In addition, evidence is accumulating for the existence of anti-inflammatory neutrophils that produce IL-10 [103]. Indeed, different neutrophil populations were collected from MRSA-resistant versus MRSA-sensitive mice [134]. It is not clear whether these are generated as different populations or evolve separately as a consequence of stimulation with microorganisms, different growth factors or cytokines.
4.4. The Chemokine System in Neutrophil Recruitment
Chemokines are small proteins with molecular masses of ~7–12 kDa that belong to the family of chemotactic cytokines. Chemokines are the only group of cytokines that bind to G protein-coupled receptors (GPCRs) [135]. Chemokines were named based on their chemoattractant property, described first in 1987 when CXCL8 was shown to be involved in chemotaxis of neutrophils in vitro [136,137]. Additionally, chemokines were described to be involved in other processes such as embryogenesis, homeostasis, angiogenesis and inflammation [138,139]. Chemokines can be divided into 4 subfamilies based on the position of the two cysteine residues in their N-terminal amino acid sequence: (1) CC chemokines have two adjacent cysteines; (2) CXC chemokines present with one amino acid between the two cysteines; (3) the CX3C chemokine has 3 amino acids between the cysteines, and; (4) C chemokines lack one of the two N-terminal cysteines [140]. The ELR+ CXC chemokines that have a specific amino acid sequence of glutamic acid-leucine-arginine (ELR) immediately before the first cysteine of the CXC motif, are associated with neutrophil recruitment and include CXCL1, 2, 3, 5, 6, 7 and 8. Those without an ELR motif rather recruit T and B lymphocytes, monocytes or hematopoietic precursor cells [141,142,143,144,145,146,147].
Chemokines can bind to two types of receptors: GPCRs and atypical chemokine receptors (ACKRs) that do not signal through G proteins and lack chemotactic activity. GPCRs are classified as CCR, CXCR, CX3CR and XCR according to the cysteine motif in their ligands [148,149]. The interactions of human and murine chemokines with GPCRs reported to be expressed on neutrophils are shown in Table 1. CXCR1 and CXCR2 are the abundantly expressed receptors on circulating neutrophils. However, under inflammatory condition, neutrophils in tissues have been reported to express multiple other CXC and CC chemokine receptors including CXCR3, CCR1, CCR2 and CCR3 [150,151]. CXCR4 expression on neutrophils enhances upon aging of neutrophils and has been suggested to be linked to resolution of inflammation [152,153]. As can be seen, one chemokine (e.g., CXCL8) can bind to several receptors and one receptor (e.g., CXCR2) may transduce signals for different ligands. The chemokine interactions that at the first moment were considered as “redundant” gave rise to the term “promiscuity” of the chemokine system. However, much attention is given now to the “bias of the chemokine system”, including ligand bias, receptor bias and tissue bias, which tend to explain and allow us to understand how those chemokines bind to their receptors and promote different responses in different situations [154]. For instance the chemokines CXCL4 and CXCL7 are typical platelet products [155]. In contrast, CCL3, CCL3L1 and CCL4 are primarily produced in leukocytes [156]. Other chemokines such as the major human neutrophil attractant CXCL8 or IL-8 may be induced in almost any cell type [157].
GPCRs have seven transmembrane helices with three extra and three intracellular loops, an extracellular N-terminus and intracellular C-terminus. Chemokines bind to the extracellular domain and to a pocket in the transmembrane area and the signal is transmitted to the intracellular compartment. Cells are activated by the direct coupling to G proteins or β arrestins [154,158,159]. The intracellular signaling in the chemokine receptors is related to second messengers such as calcium, cyclic adenosine monophosphate (cAMP) and GTPases (Ras and Rac). The GPCRs can also signal through β arrestins, a pathway that can regulate the receptor signal through the desensitization process [159,160]. β arrestins can block the binding to the phosphorylated G proteins and they are responsible for internalization of receptors to endosomes and degradation. Desensitization may be critical for maintaining the capacity of the cell to sense a chemoattractant gradient [161]. Multiple ACKRs, which fail to signal through the G proteins, have been reported to signal through β arrestins [154,162,163].
In total, 20 chemokine receptors are described and they are all expressed on leukocytes. Based on their functions, they can be divided into constitutive and inducible or homeostatic and inflammatory receptors. Initially, inflammatory chemokines and their receptors were only studied in the context of inflammation, but some receptors were identified as co-receptors for HIV entrance into the cell and others are associated with tumor metastasis [164,165,166,167,168]. Regarding homeostasis, the chemokine system is involved in embryogenesis, leukocyte trafficking to lymphoid organs, tissue/organ development and angiogenesis. For instance, much attention has been given to the contribution of the CXCR4 receptor to embryogenesis, hematopoiesis, and leukocyte trafficking from bone marrow. The importance of CXCR4 in this condition is critical for survival, since the deletion of CXCR4 or its ligand CXCL12 in mice is embryonically lethal [169,170]. Already at its discovery it was recognized that CXCR4 is expressed on neutrophils [171]. CXCR4 upregulated on “aging” neutrophils was shown to induce reverse migration of senescent neutrophils from the circulation to bone marrow [172]. CXCR1 and CXCR2 were the first members of the chemokine receptor family to be cloned, sharing a high degree of homology with formyl peptide receptors (FPRs) [173,174]. Inflammatory neutrophils express high levels of CXCR1 and CXCR2 on their surface once activated and the receptors and their ligands have an important role in neutrophil recruitment [175].
The atypical chemokine receptors (ACKRs) are related to classical chemokine receptors and also have seven transmembrane domains but they are not able to activate G proteins [176,177,178,179]. These receptors are expressed on leukocytes and non-hematopoietic cells. ACKRs signal through the β arrestin pathway, but they also work as scavenger receptors, since they can internalize the bound chemokines without chemotactic actions. Neutrophils express ACKR2 (or D6), a receptor for most inflammatory CC chemokines [180,181]. ACKR2 is supposed to restrict migration of CCR1 expressing neutrophils to its ligands including the highly potent CC chemokine CCL3 [177]. CCRL2, a receptor with high homology with chemokine receptors and with chemerin as identified ligand, on the other hand was shown to heterodimerize with CXCR2 and to promote neutrophil migration in mice [178,182].
4.5. Regulation of Chemokine-Dependent Neutrophil Recruitment
Chemokine activity can be regulated at multiple levels, including gene duplication, gene transcription and translation. Upon stimulation with PAMPs several connective tissue cells will produce the inflammatory cytokine IL-1β. IL-1β induces the production of chemokines, CXCL8 being the most potent neutrophil attracting chemokine in human. In addition, neutrophils that are recruited to the inflammatory joint may enhance the response through the secretion of additional active IL-1β further enhancing the production of CXCR1/CXCR2 ligands and neutrophil accumulation [183]. Some pre-formed chemokines are stored in endothelial cells, inside secretory granules including Weibel-Palade bodies, and are quickly released upon cell insult [184]. Once produced, chemokine activity can be regulated by binding to glycosaminoglycans on endothelial cell layers of lymph and blood vessels, by binding to and expression of GPCRs and ACKRs on cells, or by receptor-mediated synergy and antagonism among chemokines [184,185,186]. Recently, microRNAs, regulating the chemokine and chemokine receptor mRNA levels, were discovered as a novel mechanism for fine-tuning chemokine and chemokine receptor expression [187,188]. Finally, chemokines and their receptors become post-translationally modified. Chemokines can be modified post-translationally through: (1) proteolytic cleavage by enzymes such as metalloproteinases, CD26 and enzymes from pathogens [189,190,191,192]; (2) citrullination, that is the formation of citrulline by the deimination of arginine by peptidylarginine deiminases (PAD) [193,194,195]; (3) N-glycosylation on asparagine within an Asn-Xaa-Ser/Thr motif, or O-glycosylation on serine (Ser) or threonine (Thr) residues [196]; and (4) nitration, where peroxynitrite, produced during oxidative stress, can selectively oxidize and nitrate several residues, including the oxidation of histidine and the nitration of tyrosine and tryptophan [197,198,199]. Reduced or enhanced receptor affinity and chemokine activity have been reported, depending on the chemokine and on the type of posttranslational modification [189]. Most posttranslational modifications of inflammatory chemokines are dependent on proteolytic cleavage, mainly affecting the N-terminal region of the protein with highly specific proteases.
In addition to specific GPCRs, glycosaminoglycans (GAGs) play an important role in the regulation of neutrophil migration. GAGs are linear carbohydrate structures, consisting of a repeating disaccharide unit, that comprises a hexuronic acid linked to an N-acetyl-hexosamine that can be sulfated at different positions [200,201,202]. GAGs are negatively charged and can be divided into six groups: heparan sulfate, heparin, chondroitin sulfate, dermatan sulfate, keratan sulfate and hyaluronic acid. These sugar units can bind or attach to protein cores of proteoglycans or can be found associated with the extracellular matrix. GAGs are heterogeneous in length and composition and they can bind to a huge number of proteins. GAGs have fundamental roles in cell signaling and development, angiogenesis, tumor progression, embryogenesis, wound healing, and have anti-coagulant properties [203,204]. Interestingly, GAGs can interact directly with pathogens. Particularly related to this study, hyaluronic acid favors to increase lubrication in synovial joints. The loss of hyaluronic acid in osteoarthritic patients is associated with an increase of pain and stiffness [205].
Each tissue produces specific GAG repertoires and cells can alter their GAG expression in response to specific stimuli or in pathologic states. GAGs are important in cell recruitment during homeostatic and inflammatory processes by their direct interaction with chemokines [202,206]. The binding of chemokines to GAGs can generate an immobilized chemokine gradient that directs cell migration, as shown in Figure 3. Cell surface immobilization of chemokines enables them to act locally rather than as paracrine molecules, and likely prevents inappropriate activation and desensitization of receptors on cells outside the region of interest for a given physiological situation [207]. Moreover, GAG expression on the leukocyte also influences the chemokine interaction with GPCRs on the same cell [208,209].
Almost all chemokines are basic proteins, often with a pI of 10 or higher, with many Arg, Lys and His residues and GAGs bind to proteins with positive charges. The epitopes for GAG binding on chemokines are described to be BBXB and BBBXXB motifs, where B represents a basic amino acid [210]. It has been shown that some chemokines act as monomers, whereas many chemokines can oligomerize and form diverse quaternary structures including dimers, tetramers and polymers, increasing the number of epitopes that bind to GAGs [211,212]. Oligomerization increases the affinity of chemokines for GAGs through an avidity effect and this interaction also stabilizes the chemokine oligomers. Moreover, oligomerization may have a dramatic effect on GAG affinity and specificity [213,214].
4.6. The 5-Lipoxygenase Pathway: Mechanisms of Neutrophil Recruitment and Inflammation
At the onset of inflammation, classic lipid mediators are produced, including LTB4, which activate and amplify the cardinal signs of inflammation [215]. 5-lipoxygenase (5-LO) is a main enzyme involved in the production of these lipid mediators. This enzyme is expressed in leukocytes such as neutrophils, macrophages, dendritic cells, B cells and T cells [216]. During the inflammatory process, another class of arachidonic acid (AA)-derived lipids, prostaglandins E2 and D2, induce the switch of leukotriene synthesis to pro-resolving lipid production, including lipoxin A4 (LXA4) [189,217,218]. The generation of anti-inflammatory resolvins assists in the control of the inflammatory process [219]. The synthesis of LXA4 is also dependent on 5-LO. LXA4 has an important role in the resolution of inflammation by decreasing neutrophil migration. On the other hand, LXA4 increases the recruitment of macrophages. Additionally, LXA4 increases the phagocytosis of apoptotic neutrophils by macrophages, a process named efferocytosis, to avoid tissue damage [220].
4.6.1. Leukotriene B4
LTB4 is a very potent chemoattractant for neutrophils. LTB4 is produced from AA in a pathway dependent on lipoxygenases (LO) [98]. AA is a 20-carbon fatty acid that is present in all cells and it is the main eicosanoid precursor. Some stimuli such as N-formyl-methionyl-leucyl-phenylalanine (fMLF), CXCL8, microorganisms, phagocytic particles and damage or injury can activate phospholipases and release AA from the cell membrane [221]. In the cytosol AA can be metabolized into leukotrienes and lipoxins via a pathway dependent on LO. The main LO enzymes are 5-LO, that is expressed in leukocytes, and 12/15-LO, expressed in reticulocytes, eosinophils, immature dendritic cells (DCs), epithelial and airway cells, pancreatic islets and resident peritoneal macrophages [222]. The first step in leukotriene biosynthesis is the conversion of AA into a hydroperoxide, named 5-hydroperoxyeicosatetraenoic acid (5-HPETE), by the insertion of an oxygen at position 5. In this step the activation of 5-LO is dependent on the 5-LO activating protein (FLAP). 5-HPETE can be reduced to 5-hydroxyeicosatetraenoic acid (5-HETE) or can be converted in a 5,6-epoxide containing a conjugated triene structure, named leukotriene A4 (LTA4) by removal of a water molecule [223]. LTA4 is instable and can be converted into LTB4 by insertion of a hydroxyl group at carbon 12 by the enzyme LTA4 hydrolase. Another possibility is the conversion in leukotriene C4 (LTC4) by addition of a glutathionyl group at carbon 6 by γ-glutamyl-S-transferase [224]. LTB4 is produced and released within minutes by neutrophils, macrophages, and mast cells and is an important element of the immediate inflammatory response [225].
Leukotrienes bind to extracellular GPCRs, which are members of the rhodopsin-like receptors family and related to chemokine receptors. LTB4 is known to bind to two LTB4 receptors, BLT1 and BLT2 [226]. BLT1 is a 43 kDa GPCR expressed in inflammatory cells, such as neutrophils, alveolar macrophages, eosinophils, differentiated T cells, dendritic cells and osteoclasts, and has a high affinity for LTB4. The BLT2 receptor has low affinity for LTB4 and is expressed more ubiquitously [227]. BLT1 is widely related to chemotaxis. The axis LTB4/BLT1 is needed for neutrophil recruitment in arthritis and for the recruitment of neutrophils to lymph nodes during bacterial infection. On the other hand, the axis LTB4/BLT2 is involved in the generation of reactive oxygen species and can enhance wound healing [225,228,229].
4.6.2. Lipoxin A4
Lipoxins can be generated by three main pathways. In the first one, AA is released from the cell membrane and one oxygen is inserted at carbon 15 by 15-LO in eosinophils, monocytes or epithelial cells, resulting in the intermediate 15S-HPETE. 15S-HPETE can be taken up by neutrophils or monocytes and converted in the 5,6-epoxytetraene by 5-LO and then is hydrolyzed by LXA4 or lipoxin B4 (LXB4) hydrolases in LXA4 and LXB4 [230,231]. The second route involves the interaction between leukocytes and platelets. The 5-LO present in leukocytes, such as neutrophils, converts AA into LTA4 as described before. The LTA4 is released and taken up by adherent platelets. These express 12-LO that converts LTA4 in LXA4 and LXB4 [232]. The third route occurs after the exogenous administration of aspirin. In this case, aspirin triggers the formation of the 15R-epimer of lipoxins, 15-epi-LXA4 and 15-epi-LXB4. These epimers carry a carbon 15 alcohol group in the R configuration. They arise from aspirin-acetylated by cyclooxygenase-2 (COX-2) and share the actions of LXA4 [233].
LXA4 binds to the GPCR receptor ALX/FPR2. ALX is expressed on leukocytes, astrocytoma cells, epithelial cells, hepatocytes, microvascular endothelial cells and neuroblastoma cells. Unlike classic GPCRs for chemoattractants that mobilize intracellular Ca2+ to evoke chemotaxis, lipoxins instead induce changes in the phosphorylation of proteins of the cytoskeleton, resulting in β arrestin activation [234,235]. LXA4 presents pro-resolving actions such as decreased neutrophil infiltration, increased recruitment of mononuclear cells and an increase in the uptake of apoptotic neutrophils by macrophages. LXA4 has also effects on the return of vascular permeability to normal levels [236].
Both lipids LTB4 and LXA4 have been described to be involved in articular diseases. LTB4 and 5-LO mRNA was found in synovial tissue of patients with rheumatoid arthritis [237,238]. LTB4 is also associated with pathogenesis in the collagen-induced arthritis model, the K/BxN serum transfer arthritis model [239,240,241] and the experimental model of gout [242]. LXA4 was also detected in synovial tissue of patients with rheumatoid arthritis [243]. Nonetheless, in zymosan-induced arthritis, LXA4 was related to attenuation of the disease [244]. During infection, LTB4 and LXA4 are related to clearance of pathogens and improvement of the disease. Some studies show that LTB4 has a role in the control of lung Paracoccidioidomycosis and is important for phagocytosis and killing of Borrelia burgdorferi [245,246]. In lung infection by Cryptococcus neoformans and sepsis, LXA4 is associated with the control of infection and an increase in survival [247,248]. However, in pneumosepsis induced by Klebsiella pneumoniae, the LXA4 in the early stage of the disease is associated with systemic infection-induced mortality and can improve survival at a late stage of the disease [249].
5. Dual Functions of Neutrophils during Septic Arthritis
Based on the aforementioned mechanisms of the pathology of septic arthritis, this disease is associated with severe articular damage and pain. During the immune response against S. aureus infection, neutrophils are the main cells recruited to the joint. As previously mentioned, neutrophils contain potent antimicrobial molecules that are important in the control of infection, including joint infections. The depletion of neutrophils prior to the systemic injection of S. aureus in mice impaired the bacterial control and increased mortality. Furthermore, the absence of neutrophils increased the circulating levels of pro-inflammatory cytokines and the frequency of arthritis after three days of infection, suggesting that a systemic inflammatory response against a high titer of S. aureus could also cause arthritis independent of the toxic effects of neutrophils [15]. Similarly, the increased concentration of neutrophils in the joint due to a photodynamic therapy applied locally improves the clearance of MRSA and decreases tissue damage [250]. These examples highlight the importance of neutrophils to control S. aureus-induced arthritis. However, neutrophils are associated with articular damage and pain development [251,252]. Lögters et al. identified increased NETosis in synovial fluid of septic arthritis patients, mainly patients infected with S. aureus, compared to noninfectious or osteoarthritic joints. Importantly, there was a positive correlation with levels of IL-6 and IL-1 in the joints [253].
The blockade of neutrophil migration and activity could be an interesting strategy to avoid excessive articular damage and pain during S. aureus-induced septic arthritis in mice [254,255]. Formylated peptides are potent neutrophil chemoattractants. In mice, the intravenous injection of S. aureus carrying a mutation that prevents the synthesis of formylated peptides decreases the accumulation of neutrophils in the joint when compared to wild type S. aureus injection. In mice injected with mutated bacteria, the incidence of arthritis was lower, associated with decreased synovitis and cartilage damage as compared to injection with wild type S. aureus. However, the bacterial load analyzed in the joints is comparable between the two bacteria at seven days after injection, suggesting that the neutrophils guided by formylated peptides are important for joint inflammation and damage [13].
The specific blockage of neutrophil recruitment in order to avoid or reduce tissue damage was also successful using CXCR1/2 inhibitors in some animal models of inflammatory articular diseases such as antigen-induced arthritis, collagen-induced arthritis, autoantibody-mediated arthritis and gout [254,255,256,257,258]. Since neutrophils have a crucial role during the infection, the blockade of these cells in septic arthritis can be protective or detrimental. CXCR2-binding chemokines are present in the joint of Brucella mellitensis-infected mice, guiding the recruitment of neutrophils. Neutrophil counts correlated with joint inflammation and damage. Indeed, CXCR2-deficient mice displayed delayed incidence, reduced clinical scores, and decreased swelling as compared to WT mice, although there is no difference on bacterial load between both groups [259]. We previously demonstrated in a septic arthritis model induced by the intra-articular injection of S. aureus that the treatment with an antagonist of CXCR1/2 starting from the beginning of the infection was able to decrease neutrophil recruitment, articular damage and hypernociception. Nonetheless, the bacterial load increased, showing that neutrophils are important for bacterial control [24]. In addition, we also evaluated if the blockage of neutrophils at a later time point of the infection could be effective. The treatment starting 3 days after the infection, partially reduced hypernociception, prevented the increase in bacterial load, but failed in inhibiting articular damage. Besides the blockage of neutrophil migration, the decrease of neutrophil activation could also be useful to control joint inflammation and damage in septic arthritis. Activated neutrophils produce hypochlorous acid (HOCl) by a reaction of myeloperoxidase with hydrogen peroxide. The accumulation of HOCl is toxic and its blockage could be beneficial to avoid excessive tissue damage. Taurine is an amino acid found abundantly in the cytosol of neutrophils and acts as scavenger of HOCl. Interestingly, the injection of taurine chloramine (a product of taurine and HOCl) in the joint at the same time of S. aureus significantly reduced the histopathological score, especially cartilage and bone destruction [260]. Thus, the benefits of targeting neutrophil activation or migration during septic arthritis still need to be better clarified, considering effects of different bacterial strains and disease progression. Therefore, it is clear that the neutrophils recruited to the joint in the initial phase of the infection play a role in the control of infection, but can cause articular damage. Thus the blockage of neutrophils as a potential therapy for septic arthritis needs to be carefully evaluated in order to create a balance between control of infection (with or without co-treatment with antibiotics) and induction of articular damage.
6. Conclusions
In most cases, the host has the ability to induce a protective inflammatory response resulting in elimination of the invading pathogen and subsequent resolution of infection. However, if the infection cannot be controlled and persists, strong activation of the immune response can cause destruction of the joint [11]. Accordingly, most of the detrimental effects of infection result from the exaggerated immune response of the host rather than from direct cytotoxicity of bacteria [6].
The role of neutrophils in the pathogenesis of septic arthritis is dual. On the one hand, they are indispensable in the early phase of disease for effective defense against bacteria and consequently for host survival. On the other hand, these leukocytes act as mediators of tissue-destructive events. The massive infiltration of neutrophils into the infected joint can contribute to cartilage and bone destruction by the release of free radicals and bacteria- and tissue-degrading enzymes, including products of the NADPH oxidase complex and proteolytic enzymes targeting collagen and/or proteoglycans. Permanent destruction of cartilage and subchondral bone loss can occur within three days after infection [11,13,15]. Elimination of neutrophils from the site of inflammation is a prerequisite for resolution of the acute inflammatory response. This implicates that prolonged presence of neutrophils at the site of inflammation can lead to persistence of the inflammatory response, associated with complications such as tissue damage [261,262].
Other factors playing a role in joint damage include: high levels of cytokines, which enhance the release of MMPs (such as MMP-2, MMP-3 and MMP-9) and other enzymes degrading collagen, and bacterial toxins and enzymes [11]. Furthermore, the inflammatory process following infection alters the synovium as well as the composition and cellular content of the synovial fluid. These changes in the synovial fluid might affect the articular cartilage, since synovial fluid is indispensable for lubrication and nutrition of the articular cartilage. Moreover, synovial fluid dynamics can be disrupted by the infectious process, leading to a fluid effusion in the joint. Subsequently, intra-articular pressure increases, preventing the supply of blood and nutrients to the joint, thereby resulting in destruction of the synovium and cartilage [2,11].
Early and effective treatment may enable resolution of the inflammatory process. In case of unsuccessful or no treatment, articular cartilage may be lost entirely, resulting in fibrous or bony joint ankylosis. When inhibition of neutrophil infiltration is considered as a treatment option, co-treatment with antibiotics will probably be essential to combine diminished tissue destruction with efficient clearance of the microorganisms.
Acknowledgments
This work was supported by the Brazilian National Council for Scientific and Technological Development (CNPq), the “Fundação de Amparo a Pesquisa de Minas Gerais” (FAPEMIG—APQ 03072-15), INCT in dengue and host pathogen interactions, the Research Foundation—Flanders (FWO-Vlaanderen, grants G.0D66.13, G.0764.14 and G.0808.18), the Interuniversity Attraction Poles Programme initiated by the Belgian Science Policy Office (I.A.P. Project 7/40), and C1 funding (C16/17/010) of the KU Leuven. Helena Crijns holds an FWO-SB PhD scholarship from FWO-Vlaanderen.
Author Contributions
Daiane Boff and Helena Crijns wrote the initial text of the manuscript and Mauro M. Teixeira, Flavio A. Amaral and Paul Proost further modified the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| AA | Arachidonic acid |
| ACKR | Atypical chemokine receptor |
| DC | Dendritic cell |
| FPR | Formyl peptide receptor |
| GAG | glycosaminoglycan |
| GPCR | G protein-coupled receptor |
| HETE | hydroxyeicosatetraenoic acid |
| HPETE | hydroperoxyeicosatetraenoic acid |
| IL- | Interleukin- |
| LTA4 | Leukotriene A4 |
| LTB4 | Leukotriene B4 |
| LTC4 | Leukotriene C4 |
| LO | Lipoxygenase |
| LXA4 | Lipoxin A4 |
| MPO | myeloperoxidase |
| MRSA | methicillin-resistant Staphylococcus aureus |
| MSCRAMM | microbial surface component recognizing adhesive matrix molecules |
| NETs | neutrophil extracellular traps |
| PAMP | pathogen-associated molecular pattern |
| PGN | peptidoglycan |
| PRR | pattern recognition receptor |
| RA | rheumatoid arthritis |
| ROS | reactive oxygen species |
| S. aureus | Staphylococcus aureus |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
References
- Colavite, P.M.; Sartori, A. Septic arthritis: Immunopathogenesis, experimental models and therapy. J. Venom. Anim. Toxins Incl. Trop. Dis. 2014, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nade, S. Septic arthritis. Best Pract. Res. Clin. Rheumatol. 2003, 17, 183–200. [Google Scholar] [CrossRef]
- Lieber, S.B.; Fowler, M.L.; Zhu, C.; Moore, A.; Shmerling, R.H.; Paz, Z. Clinical characteristics and outcomes in polyarticular septic arthritis. Jt. Bone Spine 2017. [Google Scholar] [CrossRef] [PubMed]
- Garcia-De La Torre, I.; Nava-Zavala, A. Gonococcal and nongonocollal arthritis. Rheum. Dis. Clin. N. Am. 2009, 35, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kherani, R.B.; Shojania, K. Septic arthritis in patients with pre-existing inflammatory arthritis. CMAJ 2007, 176, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Role of macrophages in Staphylococcus aureus—Induced arthritis and sepsis. Arthritis Rheum. 2000, 43, 2276–2282. [Google Scholar] [CrossRef]
- Mathews, C.J.; Weston, V.C.; Jones, A.; Field, M.; Coakley, G. Bacterial septic arthritis in adults. Lancet 2010, 375, 846–855. [Google Scholar] [CrossRef]
- Ross, J.J. Septic arthritis of native joints. Infect. Dis. Clin. N. Am. 2017, 31, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.J.; Lowy, F.D. Bacterial infections in drug users. N. Engl. J. Med. 2005, 353, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.L. Acute bacterial arthritis. Ann. Clin. Lab. Sci. 1975, 5, 452–455. [Google Scholar] [PubMed]
- Shirtliff, M.E.; Mader, J.T. Acute septic arthritis. Clin. Microbiol. Rev. 2002, 15, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.L. Septic arthritis. Lancet 1998, 351, 197–202. [Google Scholar] [CrossRef]
- Gjertsson, I.; Jonsson, I.M.; Peschel, A.; Tarkowski, A.; Lindholm, C. Formylated peptides are important virulence factors in Staphylococcus aureus arthritis in mice. J. Infect. Dis. 2012, 205, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, A.; Bokarewa, M.; Collins, L.V.; Gjertsson, I.; Hultgren, O.H.; Jin, T.; Jonsson, I.M.; Jesefsson, E.; Sakiniene, E.; Verdrengh, M. Current status of pathogenetic mechanisms in staphylococcal arthritis. FEMS Microbiol. Lett. 2002, 217, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect. Immun. 1997, 65, 2517–2521. [Google Scholar] [PubMed]
- Boff, D.; Oliveira, V.L.S.; Queiroz, C.M., Jr.; Silva, T.A.; Allegretti, M.; Verri, W.A., Jr.; Proost, P.; Teixeira, M.M.; Amaral, F.A. CXCR2 is critical for bacterial control and development of joint damage and pain in Staphylococcus aureus-induced septic arthritis in mouse. Eur. J. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Weston, V.C.; Jones, A.C.; Bradbury, N.; Fawthrop, F.; Doherty, M. Clinical features and outcome of septic arthritis in a single UK Health District 1982–1991. Ann. Rheum. Dis. 1999, 58, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Geirsson, A.J.; Statkevicius, S.; Vikingsson, A. Septic arthritis in Iceland 1990–2002: Increasing incidence due to iatrogenic infections. Ann. Rheum. Dis. 2007, 67, 638–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, N.; Chambers, S.T.; Nolan, I.; Gallagher, K.; Werno, A.; Browne, M.; Stamp, L.K. Native joint septic arthritis: Epidemiology, clinical features, and microbiological causes in a New Zealand population. J. Rheumatol. 2015, 42, 2392–2397. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.Y.; Abu-Khattab, M.; Baagar, K.; Mohamed, S.F.; Elgendy, I.; Anand, D.; Malallah, H.; Sanjay, D. Characteristics of patients with definite septic arthritis at Hamad General Hospital, Qatar: A hospital-based study from 2006 to 2011. Clin. Rheumatol. 2013, 32, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; Wang, S.M.; Yang, Y.J.; Tsai, C.H.; Liu, C.C. Septic arthritis in children: Relationship of causative pathogens, complications, and outcome. J. Microbiol. Immunol. Infect. 2003, 36, 41–46. [Google Scholar] [PubMed]
- Gavet, F.; Tournadre, A.; Soubrier, M.; Ristori, J.M.; Dubost, J.J. Septic Arthritis in Patients Aged 80 and Older : A Comparison with Younger Adults. J. Am. Geriatr. Soc. 2005, 53, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Nochioka, K.; Marcia, T. Nationwide survey of pediatric septic arthritis in the United States. J. Orthop. 2017, 14, 342–346. [Google Scholar] [CrossRef] [PubMed]
- García-Arias, M.; Balsa, A.; Mola, E.M. Septic arthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Sharff, K.A.; Richards, E.P.; Townes, J.M. Clinical management of septic arthritis. Curr. Rheumatol. Rep. 2013, 15, 332. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Yu, S. The burden of septic arthritis on the U.S. inpatient care: A national study. PLoS ONE 2017, 12, e0182577. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.; Schiavon, F.; Riato, L.; Carraro, V.; Punzi, L. Rheumatoid arthritis is the major risk factor for septic arthritis in rheumatological settings. Autoimmun. Rev. 2008, 8, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Spear, J.; Nathanson, L.A.; McCauley, J.; Edlow, J.A. Does the presence of crystal arthritis rule out septic arthritis? J. Emerg. Med. 2007, 32, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Laupland, K.B.; Church, D.L.; Mucenski, M.; Sutherland, L.R.; Davies, H.D. Population-based study of the epidemiology of and the risk factors for invasive Staphylococcus aureus infections. J. Infect. Dis. 2003, 187, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Kak, V.; Chandrasekar, P.H. Bone and joint infections in injection drug users. Infect. Dis. Clin. N. Am. 2002, 16, 681–695. [Google Scholar] [CrossRef]
- Saraux, A.; Taelman, H.; Blanche, P.; Batungwanayo, J.; Clerinx, J.; Kagame, A.; Kabagabo, L.; Ladner, J.; van de Perre, P.; le Goff, P.; et al. HIV infection as a risk factor for septic arthritis. Br. J. Rheumatol. 1997, 36, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Al-Nammari, S.S.; Gulati, V.; Patel, R.; Bejjanki, N.; Wright, M. Septic arthritis in haemodialysis patients: A seven-year multi-centre review. J. Orthop. Surg. 2008, 16, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.W.; Piercy, E.A. Infectious Arthritis. Clin. Infect. Dis. 1995, 20, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Pioro, M.H.; Mandell, B.F. Septic arthritis. Rheum. Dis. Clin. N. Am. 1997, 23, 239–258. [Google Scholar] [CrossRef]
- Berbari, E.F.; Hanssen, A.D.; Duffy, M.C.; Steckelberg, J.M.; Ilstrup, D.M.; Harmsen, W.S.; Osmon, D.R. Risk factors for prosthetic joint infection: Case-control study. Clin. Infect. Dis. 1998, 27, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, C.P.; Tryfonidis, M.; Sadiq, S.; Hirst, P.; Paul, A. Septic arthritis following intra-articular steroid injection of the knee—A survey of current practice regarding antiseptic technique used during intra-articular steroid injection of the knee. Clin. Rheumatol. 2003, 22, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Farrow, L. A systematic review and meta-analysis regarding the use of corticosteroids in septic arthritis. BMC Musculoskelet. Disord. 2015, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Hudson, M.; Suissa, S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology 2007, 46, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Salar, O.; Baker, B.; Kurien, T.; Taylor, A.; Moran, C. Septic arthritis in the era of immunosuppressive treatments. Ann. R. Coll. Surg. Engl. 2014, 96, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.B.; Hyrich, K.L.; Mercer, L.K.; Dixon, W.G.; Ustianowski, A.P.; Helbert, M.; Watson, K.D.; Lunt, M.; Symmons, D.P.; BSR Biologics Register. Risk of septic arthritis in patients with rheumatoid arthritis and the effect of anti-TNF therapy: Results from the British Society for Rheumatology Biologics Register. Ann. Rheum. Dis. 2011, 70, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- García-De La Torre, I. Advances in the management of septic arthritis. Rheum. Dis. Clin. N. Am. 2003, 2, 61–75. [Google Scholar] [CrossRef]
- Ateschrang, A.; Albrecht, D.; Schroeter, S.; Weise, K.; Dolderer, J. Current concepts review: Septic arthritis of the knee pathophysiology, diagnostics, and therapy. Wien. Klin. Wochenschr. 2011, 123, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.J.; Davidson, L. Methicillin-resistant Staphylococcus aureus septic arthritis: An emerging clinical syndrome. Rheumatology 2005, 44, 1197–1198. [Google Scholar] [CrossRef] [PubMed]
- Helito, C.P.; Zanon, B.B.; Miyahara, H.S.; Pecora, J.R.; Munhoz Lima, A.L.; Oliveira, P.R.; Vicente, J.R.; Demange, M.K.; Camanho, G.L. Clinical and epidemiological differences between septic arthritis of the knee and hip caused by oxacillin-sensitive and -resistant S. aureus. Clinics 2015, 70, 30–33. [Google Scholar] [CrossRef]
- Lin, W.T.; Wu, C.D.; Cheng, S.C.; Chiu, C.C.; Tseng, C.C.; Chan, H.T.; Chen, P.Y.; Chao, C.M. High Prevalence of Methicillin-Resistant Staphylococcus aureus among Patients with Septic Arthritis Caused by Staphylococcus aureus. PLoS ONE 2015, 10, e0127150. [Google Scholar] [CrossRef] [PubMed]
- Bouza, E.; Muñoz, P. Micro-organisms responsible for osteo-articular infections. Baillieres Best Pract. Res. Clin. Rheumatol. 1999, 13, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, N.I.; Epps, H.R. Pediatric Septic Arthritis. Orthop. Clin. N. Am. 2017, 48, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Oogai, Y.; Matsuo, M.; Hashimoto, M.; Kato, F.; Sugai, M.; Komatsuzawa, H. Expression of virulence factors by Staphylococcus aureus grown in Serum. Appl. Environ. Microbiol. 2011, 77, 8097–8105. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, K.; Lee, J.C. Staphylococcus aureus Capsular Polysaccharides. Clin. Microbiol. Rev. 2004, 17, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, I.M.; Wilbrink, B.; Tchetverikov, I.; Schrijver, I.A.; Schouls, L.M.; Hazenberg, M.P.; Breedveld, F.C.; Tak, P.P. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000, 43, 593–598. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Deng, G.M.; Foster, S.; Tarkowski, A. Staphylococcal peptidoglycans induce arthritis. Arthritis Res. 2001, 3, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.C.; Ramp, W.K.; Frankenburg, K.P. Staphylococcus aureus adhesion to bone matrix and bone-associated biomaterials. FEMS Microbiol. Lett. 1999, 173, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.A.; Nair, S.P. Interaction of staphylococci with bone. Int. J. Med. Microbiol. 2010, 300, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Höök, M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998, 6, 484–488. [Google Scholar] [CrossRef]
- Patti, J.M.; Bremell, T.; Krajewska-Pietrasik, D.; Tarkowski, A.; Ryden, C.; Höök, M. The Staphylococcus aureus collagen adhesin is a virulence determinant in experimental septic arthritis. Infect. Immun. 1994, 62, 152–161. [Google Scholar] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.; Hartford, O.; Foster, T. Alpha-toxin and gamma-toxin jointly promote Staphylococcus aureus virulence in murine septic arthritis. Infect. Immun. 1999, 67, 1045–1049. [Google Scholar] [PubMed]
- Loffler, B.; Hussain, M.; Grundmeier, M.; Bruck, M.; Holzinger, D.; Varga, G.; Roth, J.; Kahl, B.C.; Proctor, R.A.; Peters, G. Staphylococcus aureus panton-valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog. 2010, 6, e1000715. [Google Scholar] [CrossRef] [PubMed]
- Bratton, D.L.; May, K.R.; Kailey, J.M.; Doherty, D.E.; Leung, D.Y. Staphylococcal toxic shock syndrome toxin-1 inhibits monocyte apoptosis. J. Allergy Clin. Immunol. 1999, 103, 895–900. [Google Scholar] [CrossRef]
- Abdelnour, A.; Bremell, T.; Tarkowski, A. Toxic shock syndrome toxin 1 contributes to the arthritogenicity of Staphylococcus aureus. J. Infect. Dis. 1994, 170, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; de Waaij, D.J.; Morré, S.A.; Ouburg, S. CpG DNA analysis of bacterial STDs. BMC Infect. Dis. 2015, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002, 20, 709–760. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Kirschning, C.J.; Häcker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.B.; Hsu, J.L. Septic arthritis in the native joint. Curr. Infect. Dis. Rep. 2012, 14, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.; Tahir, M.; Atkinson, H.D. The limitations of gram-stain microscopy of synovial fluid in concomitant septic and crystal arthritis. Curr. Rheumatol. Rev. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Margaretten, M.E.; Kohlwes, J.; Moore, D. Does this adult patient have septic arthritis ? JAMA 2010, 297, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.; Kabrhel, C. Sensitivity of erythrocyte sedimentation rate and C-reactive protein for the exclusion of septic arthritis in emergency department patients. J. Emerg. Med. 2011, 40, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Talebi-Taher, M.; Shirani, F.; Nikanjam, N.; Shekarabi, M. Septic versus inflammatory arthritis: Discriminating the ability of serum inflammatory markers. Rheumatol. Int. 2013, 33, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Cassidy, C.; Chang, C.; Gharib, S.; Torres, J. Diagnostic utility of laboratory tests in septic arthritis. Emerg. Med. J. 2007, 24, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.Y.; Kim, D.S.; Jung, S.M.; Song, J.J.; Park, Y.B.; Lee, S.W. Clinical significance of delta neutrophil index in the differential diagnosis between septic arthritis and acute gout attack within 24 hours after hospitalization. Medicine 2017, 96, e7431. [Google Scholar] [CrossRef] [PubMed]
- Maharajan, K.; Patro, D.K.; Menon, J.; Hariharan, A.P.; Parija, S.C.; Poduval, M.; Thimmaiah, S. Serum procalcitonin is a sensitive and specific marker in the diagnosis of septic arthritis and acute osteomyelitis. J. Orthop. Surg. Res. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Rukavina, I. SAPHO syndrome: A review. J. Child. Orthop. 2015, 9, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rozin, A.P.; Nahir, A.M. Is SAPHO syndrome a target for antibiotic therapy? Clin. Rheumatol. 2007, 26, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Henderson, J.; Dickman, E.; Darzynkiewicz, R. Laboratory tests in adults with monoarticular arthritis: Can they rule out a septic joint? Acad. Emerg. Med. 2004, 11, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, H.; Kepley, R.; Pawlak, J.; Belian, B.; Raynor, A.; Saravolatz, L.D. Rapid diagnosis of septic arthritis using 16S rDNA PCR: A comparison of 3 methods. Diagn. Microbiol. Infect. Dis. 2011, 69, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Canvin, J.M.; Goutcher, S.C.; Hagig, M.; Gemmell, C.G.; Sturrock, R.D. Persistence of Staphylococcus aureus as detected by polymerase chain reaction in the synovial fluid of a patient with septic arthritis. Br. J. Rheumatol. 1997, 36, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Fenollar, F.; Roux, V.; Stein, A.; Drancourt, M.; Raoult, D. Analysis of 525 samples to determine the usefulness of PCR amplification and sequencing of the 16S rRNA gene for diagnosis of bone and joint infections analysis of 525 samples. J. Clin. Microbiol. 2006, 44, 1018. [Google Scholar] [CrossRef] [PubMed]
- Karchevsky, M.; Schweitzer, M.E.; Morrison, W.B.; Parellada, J.A. MRI findings of septic arthritis and associated osteomyelitis in adults. Am. J. Roentgenol. 2004, 182, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.J.; Kingsley, G.; Field, M.; Jones, A.; Weston, V.C.; Phillips, M.; Walker, D.; Coakley, G. Management of septic arthritis: A systematic review. Ann. Rheum. Dis. 2007, 66, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Coakley, G. What’s new in the management of bacterial septic arthritis? Curr. Infect. Dis. Rep. 2011, 13, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Dendle, C.; Woolley, I.J.; Korman, T.M. Rat-bite fever septic arthritis: Illustrative case and literature review. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.C. Orthopedic emergencies. Preface. Emerg. Med. Clin. N. Am. 2010, 28. [Google Scholar] [CrossRef]
- Flores-Robles, B.J.; Jiménez Palop, M.; Sanabria Sanchinel, A.A.; Andrus, R.F.; Royuela Vicente, A.; Sanz Pérez, M.I.; Espinosa Malpartida, M.; Ramos Giráldez, C.; Merino Argumanez, C.; Villa Alcázar, L.F.; et al. Initial Treatment in Septic Arthritis: Medical Versus Surgical Approach: An 8-Year, Single Center in Spain Experience. J. Clin. Rheumatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Stutz, G.; Kuster, M.S.; Kleinstück, F.; Gächter, A. Arthroscopic management of septic arthritis: Stages of infection and results. Knee Surg. Sport Traumatol. Arthrosc. 2000, 8, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sharma, A. Neutrophils: Cinderella of innate immune system. Int. Immunopharmacol. 2010, 10, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian-Ding, I.; Stein, C.; Uebele, J. The innate immune response against Staphylococcus aureus. Curr. Top. Microbiol. Immunol. 2015, 6, 23–27. [Google Scholar] [CrossRef]
- Sakiniene, E.; Bremell, T.; Tarkowski, A. Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin. Exp. Immunol. 1999, 115, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gjertsson, I.; Hultgren, O.H.; Stenson, M.; Holmdahl, R.; Tarkowski, A. Are B lymphocytes of importance in severe Staphylococcus aureus infections ? Infect. Immun. 2000, 68, 2431–2434. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.J.; Parker, D.; Harfenist, B.S.; Soong, G.; Prince, A. Participation of CD11c+ leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect. Immun. 2011, 79, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Hultgren, O.H.; Stenson, M.; Tarkowski, A. Role of IL-12 in Staphylococcus aureus-triggered arthritis and sepsis. Arthritis Res. 2001, 3, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Henningsson, L.; Jirholt, P.; Lindholm, C.; Eneljung, T.; Silverpil, E.; Iwakura, Y.; Linden, A.; Gjertsson, I. Interleukin-17A during local and systemic Staphylococcus aureus-induced arthritis in mice. Infect. Immun. 2010, 78, 3783–3790. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Azcutia, V.; Newton, G.; Alcaide, P.; Luscinskas, F.W. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 2011, 32, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Dorward, D.A.; Lucas, C.D.; Chapman, G.B.; Haslett, C.; Dhaliwal, K.; Rossi, A.G. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am. J. Pathol. 2015, 185, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Manthey, H.D.; Woodruff, T.M.; Taylor, S.M.; Monk, P.N. Complement component 5a (C5a). Int. J. Biochem. Cell Biol. 2009, 41, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Mydel, P.; Shipley, J.M.; Adair-Kirk, T.L.; Kelley, D.G.; Broekelmann, T.J.; Mecham, R.P.; Senior, R.M. Neutrophil elastase cleaves laminin-332 (laminin-5) generating peptides that are chemotactic for neutrophils. J. Biol. Chem. 2008, 283, 9513–9522. [Google Scholar] [CrossRef] [PubMed]
- Afonso, P.V.; Janka-Junttila, M.; Lee, Y.J.; McCann, C.P.; Oliver, C.M.; Aamer, K.A.; Losert, W.; Cicerone, M.T.; Parent, C.A. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev. Cell 2012, 22, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Montrucchio, G.; Alloatti, G.; Mariano, F.; Comino, A.; Cacace, G.; Polloni, R.; de Filippi, P.G.; Emanuelli, G.; Camussi, G. Role of platelet-activating factor in polymorphonuclear neutrophil recruitment in reperfused ischemic rabbit heart. Am. J. Pathol. 1993, 142, 471–480. [Google Scholar] [PubMed]
- Sanz, M.J.; Kubes, P. Neutrophil-active chemokines in in vivo imaging of neutrophil trafficking. Eur. J. Immunol. 2012, 42, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Voyich, J.M.; Burlak, C.; DeLeo, F.R. Neutrophils in the innate immune response. Arch. Immunol. Ther. Exp. 2005, 53, 505–517. [Google Scholar]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Santoso, S.; Chavakis, T. Mechanisms of neutrophil transendothelial migration. Front. Biosci. 2009, 14, 1596–1605. [Google Scholar] [CrossRef]
- Muller, W.A. Getting leucocytes to the sites of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. How human neutrophils kill and degrade microbes: An integrated view. Immunol. Rev. 2007, 219, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Clauditz, A.; Resch, A.; Wieland, K.P.; Peschel, A.; Götz, F. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect. Immun. 2006, 74, 4950–4953. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Saha, S.S.; Bishayi, B. Intracellular survival of Staphylococcus aureus: Correlating production of catalase and superoxide dismutase with levels of inflammatory cytokines. Inflamm. Res. 2008, 57, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Kohler, P.L.; Schlievert, P.M.; Chuang, O.N.; Dunny, G.M.; Kobayashi, S.D.; Miedzobrodzki, J.; Bohach, G.A.; Seo, K.S. Characterization of a Staphylococcus aureus surface virulence factor that promotes resistance to oxidative killing and infectious endocarditis. Infect. Immun. 2011, 79, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Bokarewa, M.; Foster, T.; Mitchell, J.; Higgins, J.; Tarkowski, A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J. Immunol. 2004, 172, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wójcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J.; et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 2004, 48, 4673–4679. [Google Scholar] [CrossRef] [PubMed]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Köckritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microbes Infect. 2003, 5, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.M. Reactive oxygen species in phagocytic leukocytes. Histochem. Cell Biol. 2008, 130, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Van Kessel, K.P.; Bestebroer, J.; van Strijp, J.A. Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front. Immunol. 2014, 5, 467. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Sørensen, O.E.; Theilgaard-Mönch, K. Neutrophil granules: A library of innate immunity proteins. Trends Immunol. 2007, 28, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Van den Steen, P.E.; Dubois, B.; Nelissen, I.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Biol. 2002, 37, 375–536. [Google Scholar] [CrossRef] [PubMed]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A.; Sendo, F. Neutrophil secretory vesicles are the intracellular reservoir for GPI-80, a protein with adhesion-regulating potential. J. Leukoc. Biol. 2001, 69, 57–62. [Google Scholar] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Territo, M.C.; Ganz, T.; Selsted, M.E.; Lehrer, R. Monocyte-chemotactic activity of defensins from human neutrophils. J. Clin. Investig. 1989, 84, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Missiakas, D.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The balancing act of neutrophils. Cell Host Microbe 2014, 15, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Takahashi, H.; Kobayashi, M.; Hanafusa, T.; Herndon, D.N.; Suzuki, F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 2004, 21, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Yoshimura, T.; Matsushima, K.; Tanaka, S.; Robinson, E.A.; Appella, E.; Oppenheim, J.J.; Leonard, E.J. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc. Natl. Acad. Sci. USA 1987, 84, 9233–9237. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, J.; Van Beeumen, J.; Opdenakker, G.; Billiau, A. A novel NH2-terminal sequence-characterized human monokine possessing neutrophil chemotactic, skin-reactive, and granulocytosis-promoting activity. J. Exp. Med. 1988, 167, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, B.; Keane, M.P.; Strieter, R.M. Chemokines as mediators of angiogenesis. Thromb. Haemost. 2007, 97, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Struyf, S.; Van Damme, J.; Fiten, P.; Ugarte-Berzal, E.; Opdenakker, G. Chemokine isoforms and processing in inflammation and immunity. J. Autoimmun. 2017, 85, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Borish, L.C.; Steinke, J.W. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 111, S460–S475. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Beccari, A.R.; Bertini, R.; Cavicchia, M.R.; Giorgini, S.; Allegretti, M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Ther. 2006, 112, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Moser, B. CXCR5, the defining marker for follicular B helper T (TFH) cells. Front. Immunol. 2015, 6, 296. [Google Scholar] [CrossRef] [PubMed]
- Maravillas-Montero, J.L.; Burkhardt, A.M.; Hevezi, P.A.; Carnevale, C.D.; Smit, M.J.; Zlotnik, A. Cutting edge: GPR35/CXCR8 is the receptor of the mucosal chemokine CXCL17. J. Immunol. 2015, 194, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Struyf, S.; Proost, P. The unique structural and functional features of CXCL12. Cell. Mol. Immunol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Vanheule, V.; Janssens, R.; Struyf, S.; Proost, P. Overview of the mechanisms that may contribute to the non-redundant activities of interferon-inducible CXC chemokine receptor 3 ligands. Front. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Kunkel, E.J.; Boisvert, J.; Johnston, B.; Campbell, J.J.; Genovese, M.C.; Greenberg, H.B.; Butcher, E.C. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J. Clin. Investig. 2001, 107, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2013, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Krauss-Etschmann, S.; Koller, B.; Hordijk, P.L.; Kuijpers, T.W.; Hoffmann, F.; Hector, A.; Eber, E.; Marcos, V.; Bittmann, I.; et al. Infiltrated neutrophils acquire novel chemokine receptor expression and chemokine responsiveness in chronic inflammatory lung diseases. J. Immunol. 2008, 181, 8053–8067. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, A.; Kuba, K.; Morita, M.; Chida, S.; Tezuka, H.; Hara, H.; Sasaki, T.; Ohteki, T.; Ranieri, V.M.; dos Santos, C.C.; et al. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 2013, 187, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Wolach, B.; van der Laan, L.J.; Maianski, N.A.; Tool, A.T.; van Bruggen, R.; Roos, D.; Kuijpers, T.W. Growth factors G-CSF and GM-CSF differentially preserve chemotaxis of neutrophils aging in vitro. Exp. Hematol. 2007, 35, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K.C.; Bautz, F.; Seitz, G.; Yildirim, S.; Kanz, L.; Möhle, R. Modulation of CXC chemokine receptor expression and function in human neutrophils during aging in vitro suggests a role in their clearance from circulation. Mediat. Inflamm. 2009, 2009, 790174. [Google Scholar] [CrossRef] [PubMed]
- Steen, A.; Larsen, O.; Thiele, S.; Rosenkilde, M.M. Biased and G protein-independent signaling of chemokine receptors. Front. Immunol. 2014, 5, 277. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.; Petersen, F.; Ludwig, A.; Ehlert, J.E.; Bock, L.; Flad, H.D. The β-thromboglobulins and platelet factor 4: Blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J. Leukoc. Biol. 2000, 67, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Menten, P.; Wuyts, A.; Van Damme, J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002, 13, 455–481. [Google Scholar] [CrossRef]
- Proost, P.; Wuyts, A.; Van Damme, J. The role of chemokines in inflammation. Int. J. Clin. Lab. Res. 1996, 26, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, E.; Kumari, P.; Jaiman, D.; Shukla, A.K. Methodological advances: The unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. Cell Biol. 2015, 16, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Lefkowitz, R.J. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [PubMed]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Mortier, A.; Boff, D.; Ruytinx, P.; Gouwy, M.; Vantilt, B.; Larsen, O.; Daugvilaite, V.; Rosenkilde, M.M.; Parmentier, M.; et al. Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4- and atypical chemokine receptor 3-dependent activity on endothelial cells and lymphocytes. Biochem. Pharmacol. 2017, 132, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Vacchini, A.; Locati, M.; Borroni, E.M. Overview and potential unifying themes of the atypical chemokine receptor family. J. Leukoc. Biol. 2016, 99, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. Chemokine control of HIV-1 infection: Beyond a binding competition. Retrovirology 2010, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M. Chemokines and the molecular basis of cancer metastasis. N. Engl. J. Med. 2001, 345, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Locati, M.; Mantovani, A. Chemokines and cancer: A fatal attraction. Cancer Cell 2011, 19, 434–435. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, K.A.; Zinszner, H.; Kunwar, P.S.; Schaible, K.; Stebler, J.; Sunshine, M.J.; O’Brien, W.; Raz, E.; Littman, D.; Wylie, C.; et al. The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development 2003, 130, 4279–4286. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, M.; Geiser, T.; O'Reilly, T.; Zwahlen, R.; Baggiolini, M.; Moser, B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J. Biol. Chem. 1994, 269, 232–237. [Google Scholar] [PubMed]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef]
- Murphy, P.M.; Tiffany, H.L. Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 1991, 253, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.E.; Lee, J.; Kuang, W.J.; Rice, G.C.; Wood, W.I. Structure and functional expression of a human interleukin-8 receptor. Science 1991, 253, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y. Neutrophil infiltration and chemokines. Crit. Rev. Immunol. 2006, 26, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Graham, G.J.; Locati, M.; Mantovani, A.; Murphy, P.M.; Nibbs, R.; Rot, A.; Sozzani, S.; Thelen, M. An atypical addition to the chemokine receptor nomenclature: IUPHAR Review 15. Br. J. Pharmacol. 2015, 172, 3945–3949. [Google Scholar] [CrossRef] [PubMed]
- Rot, A.; McKimmie, C.; Burt, C.L.; Pallas, K.J.; Jamieson, T.; Pruenster, M.; Horuk, R.; Nibbs, R.J.B.; Graham, G.J. Cell-autonomous regulation of neutrophil migration by the D6 chemokine decoy receptor. J. Immunol. 2013, 190, 6450–6456. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Martínez-Muñoz, L.; Mazzon, C.; Toffali, L.; Sozio, F.; Za, L.; Bosisio, D.; Gazzurelli, L.; Salvi, V.; Tiberio, L.; et al. The atypical receptor CCRL2 is required for CXCR2-dependent neutrophil recruitment and tissue damage. Blood 2017, 130, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, R.J.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Savino, B.; Borroni, E.M.; Torres, N.M.; Proost, P.; Struyf, S.; Mortier, A.; Mantovani, A.; Locati, M.; Bonecchi, R. Recognition versus adaptive up-regulation and degradation of CC chemokines by the chemokine decoy receptor D6 are determined by their N-terminal sequence. J. Biol. Chem. 2009, 284, 26207–26215. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.J.; Locati, M.; Mantovani, A.; Rot, A.; Thelen, M. The biochemistry and biology of the atypical chemokine receptors. Immunol. Lett. 2012, 145, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Salvi, V.; Sozio, F.; Sozzani, S.; del Prete, A. Role of atypical chemokine receptors in microglial activation and polarization. Front. Aging Neurosci. 2017, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Sadik, C.D.; Kim, N.D.; Luster, A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011, 32, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Øynebråten, I.; Barois, N.; Hagelsteen, K.; Johansen, F.E.; Bakke, O.; Haraldsen, G. Characterization of a novel chemokine-containing storage granule in endothelial cells: Evidence for preferential exocytosis mediated by protein kinase A and diacylglycerol. J. Immunol. 2005, 175, 5358–5369. [Google Scholar] [CrossRef] [PubMed]
- Gouwy, M.; Struyf, S.; Proost, P.; Van Damme, J. Synergy in cytokine and chemokine networks amplifies the inflammatory response. Cytokine Growth Factor Rev. 2005, 16, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Uguccioni, M. Modulation of chemokine responses: Synergy and cooperativity. Front. Immunol. 2016, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Mirolo, M.; Renzi, T.A.; Rossato, M.; Bazzoni, F.; Locati, M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc. Natl. Acad. Sci. USA 2013, 110, 11499–11504. [Google Scholar] [CrossRef] [PubMed]
- Elmesmari, A.; Fraser, A.R.; Wood, C.; Gilchrist, D.; Vaughan, D.; Stewart, L.; McSharry, C.; McInnes, I.B.; Kurowska-Stolarska, M. MicroRNA-155 regulates monocyte chemokine and chemokine receptor expression in rheumatoid arthritis. Rheumatology 2016, 55, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Van Damme, J.; Proost, P. Regulation of chemokine activity by posttranslational modification. Pharmacol. Ther. 2008, 120, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Van Damme, J.; Mortier, A.; Proost, P. Regulation of chemokine activity - A focus on the role of dipeptidyl peptidase IV/CD26. Front. Immunol. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Moelants, E.A.; Loozen, G.; Mortier, A.; Martens, E.; Opdenakker, G.; Mizgalska, D.; Szmigielski, B.; Potempa, J.; Van Damme, J.; Teughels, W.; et al. Citrullination and proteolytic processing of chemokines by Porphyromonas gingivalis. Infect. Immun. 2014, 82, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczyk-Pawlinska, J.; Travis, J.; Potempa, J. Modulation of interleukin-8 activity by gingipains from Porphyromonas gingivalis: Implications for pathogenicity of periodontal disease. FEBS Lett. 1998, 440, 282–286. [Google Scholar] [CrossRef]
- Proost, P.; Loos, T.; Mortier, A.; Schutyser, E.; Gouwy, M.; Noppen, S.; Dillen, C.; Ronsse, I.; Conings, R.; Struyf, S.; et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J. Exp. Med. 2008, 205, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Loos, T.; Mortier, A.; Gouwy, M.; Ronsse, I.; Put, W.; Lenaerts, J.P.; Van Damme, J.; Proost, P. Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: A naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood 2008, 112, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Loos, T.; Gouwy, M.; Ronsse, I.; Van Damme, J.; Proost, P. Posttranslational modification of the NH2-terminal region of CXCL5 by proteases or peptidylarginine Deiminases (PAD) differently affects its biological activity. J. Biol. Chem. 2010, 285, 29750–29759. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, P.; Flati, S.; di Cioccio, V.; Maurizi, G.; Macchia, G.; Facchin, A.; Anacardio, R.; Maras, A.; Lucarelli, M.; Boraschi, D. Glycosylation enhances functional stability of the chemotactic cytokine CCL2. Eur. Cytokine Netw. 2003, 14, 91–96. [Google Scholar] [PubMed]
- Barker, C.E.; Ali, S.; O’Boyle, G.; Kirby, J.A. Transplantation and inflammation: Implications for the modification of chemokine function. Immunology 2014, 143, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Mortier, A.; Boff, D.; Vanheule, V.; Gouwy, M.; Franck, C.; Larsen, O.; Rosenkilde, M.M.; Van Damme, J.; Amaral, F.A.; et al. Natural nitration of CXCL12 reduces its signaling capacity and chemotactic activity in vitro and abrogates intra-articular lymphocyte recruitment in vivo. Oncotarget 2016, 7, 62439–62459. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.E.; Thompson, S.; O’Boyle, G.; Lortat-Jacob, H.; Sheerin, N.S.; Ali, S.; Kirby, J.A. CCL2 nitration is a negative regulator of chemokine-mediated inflammation. Sci. Rep. 2017, 7, 44384. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Sugahara, K.; Özbek, S. Evolution of glycosaminoglycans: Comparative biochemical study. Commun. Integr. Biol. 2011, 4, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I. Chemokines and glycosaminoglycans. Front. Immunol. 2015, 6, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, J.; Cao, R.; Morita, H.; Soininen, R.; Chan, K.M.; Liu, B.; Cao, Y.; Tryggvason, K. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004, 64, 4699–4702. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, F.M.; Vitale, D.L.; Demarchi, G.; Cristina, C.; Alaniz, L. The immunological effect of hyaluronan in tumor angiogenesis. Clin. Transl. Immunol. 2015, 4, e52. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.C. Role of intra-articular hyaluronic acid preparations in medical management of osteoarthritis of the knee. Semin. Arthritis Rheum. 2000, 30, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Monneau, Y.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. The sweet spot: How GAGs help chemokines guide migrating cells. J. Leukoc. Biol. 2015, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hamel, D.J.; Sielaff, I.; Proudfoot, A.E.I.; Handel, T.M. Interactions of chemokines with glycosaminoglycans. Methods Enzymol. 2009, 461, 71–102. [Google Scholar] [CrossRef] [PubMed]
- Connell, B.J.; Sadir, R.; Baleux, F.; Laguri, C.; Kleman, J.P.; Luo, L.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. Heparan sulfate differentially controls CXCL12α- and CXCL12γ-mediated cell migration through differential presentation to their receptor CXCR4. Sci. Signal. 2016, 9, ra107. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Mortier, A.; Janssens, R.; Boff, D.; Vanbrabant, L.; Lamoen, N.; Van Damme, J.; Teixeira, M.M.; De Meester, I.; Amaral, F.A.; et al. Glycosaminoglycans regulate CXCR3 ligands at distinct levels: Protection against processing by dipeptidyl peptidase IV/CD26 and interference with receptor signaling. Int. J. Mol. Sci. 2017, 18, 1513. [Google Scholar] [CrossRef] [PubMed]
- Kuschert, G.S.; Coulin, F.; Power, C.A.; Proudfoot, A.E.I.; Hubbard, R.E.; Hoogewerf, A.J.; Wells, T.N. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 1999, 38, 12959–12968. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Watson, C.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Triandafillou, C.G.; Huang, T.Y.; Zulueta, M.M.; Banerjee, S.; Dinner, A.R.; Hung, S.C.; Tang, W.J. Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc. Natl. Acad. Sci. USA 2016, 113, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 2015, 26, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Migliorini, E.; Salanga, C.L.; Thakar, D.; Handel, T.M.; Richter, R.P. Differential structural remodelling of heparan sulfate by chemokines: The role of chemokine oligomerization. Open Biol. 2017, 7, pii: 160286. [Google Scholar] [CrossRef]
- Le Bel, M.; Brunet, A.; Gosselin, J. Leukotriene B4, an endogenous stimulator of the innate immune response against pathogens. J. Innate Immun. 2014, 6, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase: Regulation of expression and enzyme activity. Trends Biochem. Sci. 2007, 32, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Pro-resolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Gilroy, D.W. Lipid Mediators in Inflammation. Microbiol. Spectr. 2016, 4, 1–21. [Google Scholar] [CrossRef]
- Peters-Golden, M.; Henderson, W.R., Jr. Leukotrienes. N. Engl. J. Med. 2007, 357, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Canetti, C.; Mancuso, P.; Coffey, M.J. Leukotrienes: Underappreciated mediators of innate immune responses. J. Immunol. 2005, 174, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Crooks, S.; Stockley, R. Leukotriene B4. Int. J. Biochem. Cell Biol. 1998, 30, 173–178. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr. The role of leukotrienes in inflammation. Ann. Intern. Med. 1994, 121, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, T. Leukotriene B4 receptors: Novel roles in immunological regulations. Adv. Enzyme Regul. 2011, 51, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J. Biochem. 2015, 157, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sadik, C.D.; Luster, A.D. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J. Leukoc. Biol. 2012, 91, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Claesson, H.E.; Odlander, B.; Jakobsson, P.J. Leukotriene B4 in the immune system. Int. J. Immunopharmacol. 1992, 14, 441–449. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Resolution phase of inflammation: Novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, J.A.; Hofheinz, K.; Zaiss, M.M.; Krönke, G. The double-edged role of 12/15-lipoxygenase during inflammation and immunity. Biochim. Biophys. Acta 2017, 1862, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N.; Dahlen, S.E.; Drazen, J.M.; Hay, D.W.; Rovati, G.E.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor ALX: Potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Maderna, P.; Cottell, D.C.; Toivonen, T.; Dufton, N.; Dalli, J.; Perretti, M.; Godson, C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010, 24, 4240–4249. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Serhan, C.N. Lipoxins and new lipid mediators in the resolution of inflammation. Curr. Opin. Pharmacol. 2006, 6, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Korotkova, M.; Jakobsson, P.J. Persisting eicosanoid pathways in rheumatic diseases. Nat. Rev. Rheumatol. 2014, 10, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Endo, H.; Hayashi, I.; Murakami, Y.; Kitasato, H.; Kono, S.; Matsui, T.; Tanaka, S.; Nishimura, A.; Urabe, K.; et al. Differential expression of leukotriene B4 receptor subtypes (BLT1 and BLT2) in human synovial tissues and synovial fluid leukocytes of patients with rheumatoid arthritis. J. Rheumatol. 2003, 30, 1712–1718. [Google Scholar] [PubMed]
- Yousefi, B.; Jadidi-Niaragh, F.; Azizi, G.; Hajighasemi, F.; Mirshafiey, A. The role of leukotrienes in immunopathogenesis of rheumatoid arthritis. Mod. Rheumatol. 2013, 7595, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Jala, V.R.; Haribabu, B. Role of leukotriene B4 receptors in rheumatoid arthritis. Autoimmun. Rev. 2007, 7, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lam, B.K.; Kanaoka, Y.; Nigrovic, P.A.; Audoly, L.P.; Austen, K.F.; Lee, D.M. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 2006, 203, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.A.; Costa, V.V.; Tavares, L.D.; Sachs, D.; Coelho, F.M.; Fagundes, C.T.; Soriani, F.M.; Silveira, T.N.; Cunha, L.D.; Zamboni, D.S.; et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B4 in a murine model of gout. Arthritis Rheum. 2012, 64, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Hayashi, I.; Murakami, Y.; Sato, Y.; Kitasato, H.; Matsushita, R.; Iizuka, N.; Urabe, K.; Itoman, M.; Hirohata, S.; et al. Antiinflammatory mediator lipoxin A4 and its receptor in synovitis of patients with rheumatoid arthritis. J. Rheumatol. 2007, 34, 2144–2153. [Google Scholar] [PubMed]
- Conte, F.P.; Menezes-De-Lima, O., Jr.; Verri, W.A., Jr.; Cunha, F.Q.; Penido, C.; Henriques, M.G. Lipoxin A 4 attenuates zymosan-induced arthritis by modulating endothelin-1 and its effects. Br. J. Pharmacol. 2010, 161, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.C.; Santos, D.A.; Ribeiro, L.S.; Fagundes, C.T.; de Paula, T.P.; Avila, T.V.; Baltazar Lde, M.; Madeira, M.M.; Cruz Rde, C.; Dias, A.C.; et al. The pivotal role of 5-lipoxygenase-derived LTB4 in controlling pulmonary Paracoccidioidomycosis. PLoS Negl. Trop. Dis. 2013, 7, e2390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Olson, R.M.; Brown, C.R. Macrophage LTB4 drives efficient phagocytosis of Borrelia burgdorferi via BLT1 or BLT2. J. Lipid Res. 2017, 58, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Colby, J.K.; Gott, K.M.; Wilder, J.A.; Levy, B.D. Lipoxin signaling in murine lung host responses to Cryptococcus neoformans infection. Am. J. Respir. Cell Mol. Biol. 2016, 54, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Capilato, J.; Pham, M.P.; Walker, J.; Spur, B.; Rodriguez, A.; Perez, L.J.; Yin, K. Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. FASEB J. 2016, 30, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Sordi, R.; Menezes-De-Lima, O., Jr.; Horewicz, V.; Scheschowitsch, K.; Santos, L.F.; Assreuy, J. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int. Immunopharmacol. 2013, 17, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Mroz, P.; Dai, T.; Huang, L.; Morimoto, Y.; Kinoshita, M.; Yoshihara, Y.; Nemoto, K.; Shinomiya, N.; Seki, S.; et al. Photodynamic therapy can induce a protective innate immune response against murine bacterial arthritis via neutrophil accumulation. PLoS ONE 2012, 7, e39823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, F.M.; Pinho, V.; Amaral, F.A.; Sachs, D.; Costa, V.V.; Rodrigues, D.H.; Vieira, A.T.; Silva, T.A.; Souza, D.G.; Bertini, R.; et al. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheum. 2008, 58, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Sachs, D.; Coelho, F.M.; Costa, V.V.; Lopes, F.; Pinho, V.; Amaral, F.A.; Silva, T.A.; Teixeira, A.L.; Souza, D.G.; Teixeira, M.M. Cooperative role of tumour necrosis factor-α, interleukin-1β and neutrophils in a novel behavioural model that concomitantly demonstrates articular inflammation and hypernociception in mice. Br. J. Pharmacol. 2011, 162, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Lögters, T.; Paunel-Görgülü, A.; Zilkens, C.; Altrichter, J.; Scholz, M.; Thelen, S.; Krauspe, R.; Margraf, S.; Jeri, T.; Windolf, J.; et al. Diagnostic accuracy of neutrophil-derived circulating free DNA (cf-DNA/NETs) for septic arthritis. J. Orthop. Res. 2009, 27, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Barsante, M.M.; Cunha, T.M.; Allegretti, M.; Cattani, F.; Policani, F.; Bizzarri, C.; Tafuri, W.L.; Poole, S.; Cunha, F.Q.; Bertini, R.; et al. Blockade of the chemokine receptor CXCR2 ameliorates adjuvant-induced arthritis in rats. Br. J. Pharmacol. 2008, 153, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Haringman, J.J.; Tak, P.P. Chemokine blockade: A new era in the treatment of rheumatoid arthritis? Arthritis Res. Ther. 2004, 6, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podolin, P.L.; Bolognese, B.J.; Foley, J.J.; Schmidt, D.B.; Buckley, P.T.; Widdowson, K.L.; Jin, Q.; White, J.R.; Lee, J.M.; Goodman, R.B.; et al. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. J. Immunol. 2002, 169, 6435–6444. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Wang, Y.; Gonsiorek, W.; Anilkumar, G.; Kozlowski, J.; Lundell, D.; Fine, J.S.; Grant, E.P. Pharmacological targeting reveals distinct roles for CXCR2/CXCR1 and CCR2 in a mouse model of arthritis. Biochem. Biophys. Res. Commun. 2010, 391, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; Ortiz-Lopez, A.; Campbell, J.J.; Gerard, C.J.; Mathis, D.; Benoist, C. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 2010, 62, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.A.; Keleher, L.L.; Mitchell, W.J.; Brown, C.R.; Skyberg, J.A. CXCR2 mediates Brucella-induced arthritis in interferon γ-deficient mice. J. Infect. Dis. 2016, 214, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Inhibition of septic arthritis by local administration of taurine chloramine, a product of activated neutrophils. J. Rheumatol. 2005, 32, 1513–1517. [Google Scholar] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Routes of bacterial infection and risk factors for septic arthritis development. (A) Bacteria can access the joint through 5 routes: (1) by hematogenous spread; (2) from an adjacent infected tissue; (3) through infected bones; (4) as a consequence of trauma or (5) during diagnostic procedures. (B) Additionally, some risk factors are related to septic arthritis such as presence of other rheumatic or immunosuppressive diseases, prosthetic surgery and higher age.
Figure 1.
Routes of bacterial infection and risk factors for septic arthritis development. (A) Bacteria can access the joint through 5 routes: (1) by hematogenous spread; (2) from an adjacent infected tissue; (3) through infected bones; (4) as a consequence of trauma or (5) during diagnostic procedures. (B) Additionally, some risk factors are related to septic arthritis such as presence of other rheumatic or immunosuppressive diseases, prosthetic surgery and higher age.

Figure 2.
Killing of S. aureus by neutrophils and immune evasion. (A) Neutrophils phagocytose S. aureus and kill the bacteria by the production of ROS, liberation of lytic enzymes from granules and production of NETs. (B) S. aureus may possess virulence factors including enzymes that kill neutrophils or allow the bacteria to evade killing by neutrophils. ROS: reactive oxygen species; NET: neutrophil extracellular traps; SOK: surface factor promoting resistance to oxidative killing; SOD: superoxide dismutase.
Figure 2.
Killing of S. aureus by neutrophils and immune evasion. (A) Neutrophils phagocytose S. aureus and kill the bacteria by the production of ROS, liberation of lytic enzymes from granules and production of NETs. (B) S. aureus may possess virulence factors including enzymes that kill neutrophils or allow the bacteria to evade killing by neutrophils. ROS: reactive oxygen species; NET: neutrophil extracellular traps; SOK: surface factor promoting resistance to oxidative killing; SOD: superoxide dismutase.

Figure 3.
Neutrophil recruitment by chemokines and leukotriene B4. Neutrophils are recruited to the tissue through chemoattractants such as chemokines and LTB4. Chemokines bind to GAGs, which are expressed on endothelial cells and tissue. The retention of chemokines on GAGs generates a chemokine gradient that favors the binding of chemokines to their GPCR. LTB4 binds to specific GPCRs on the neutrophil to induce firm adhesion of the cell to the endothelium. GAGs: glycosaminoglycans, LTB4: leukotriene B4, GPCR: G protein-coupled receptor.
Figure 3.
Neutrophil recruitment by chemokines and leukotriene B4. Neutrophils are recruited to the tissue through chemoattractants such as chemokines and LTB4. Chemokines bind to GAGs, which are expressed on endothelial cells and tissue. The retention of chemokines on GAGs generates a chemokine gradient that favors the binding of chemokines to their GPCR. LTB4 binds to specific GPCRs on the neutrophil to induce firm adhesion of the cell to the endothelium. GAGs: glycosaminoglycans, LTB4: leukotriene B4, GPCR: G protein-coupled receptor.


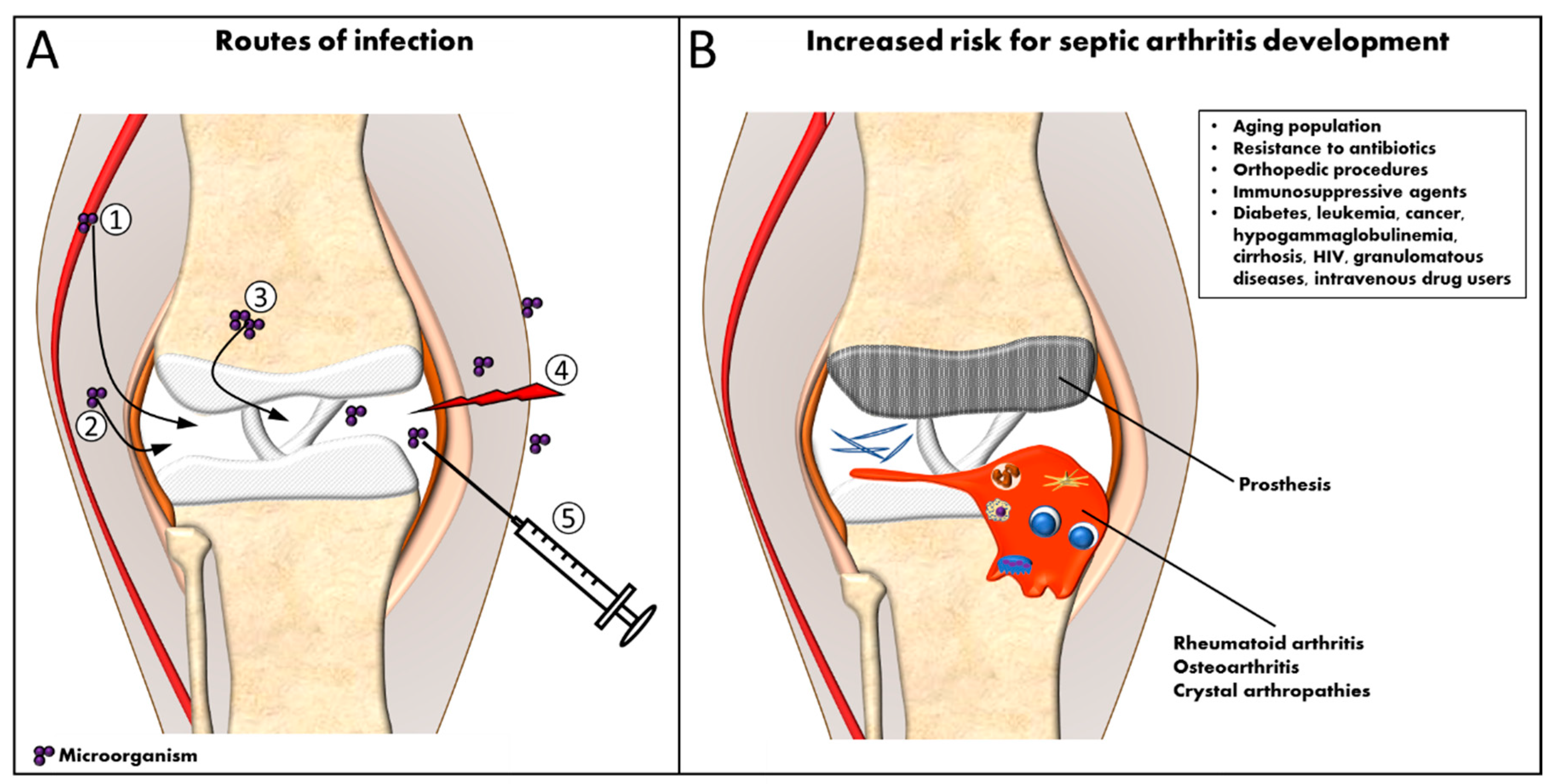{kind=link}
{kind=link}
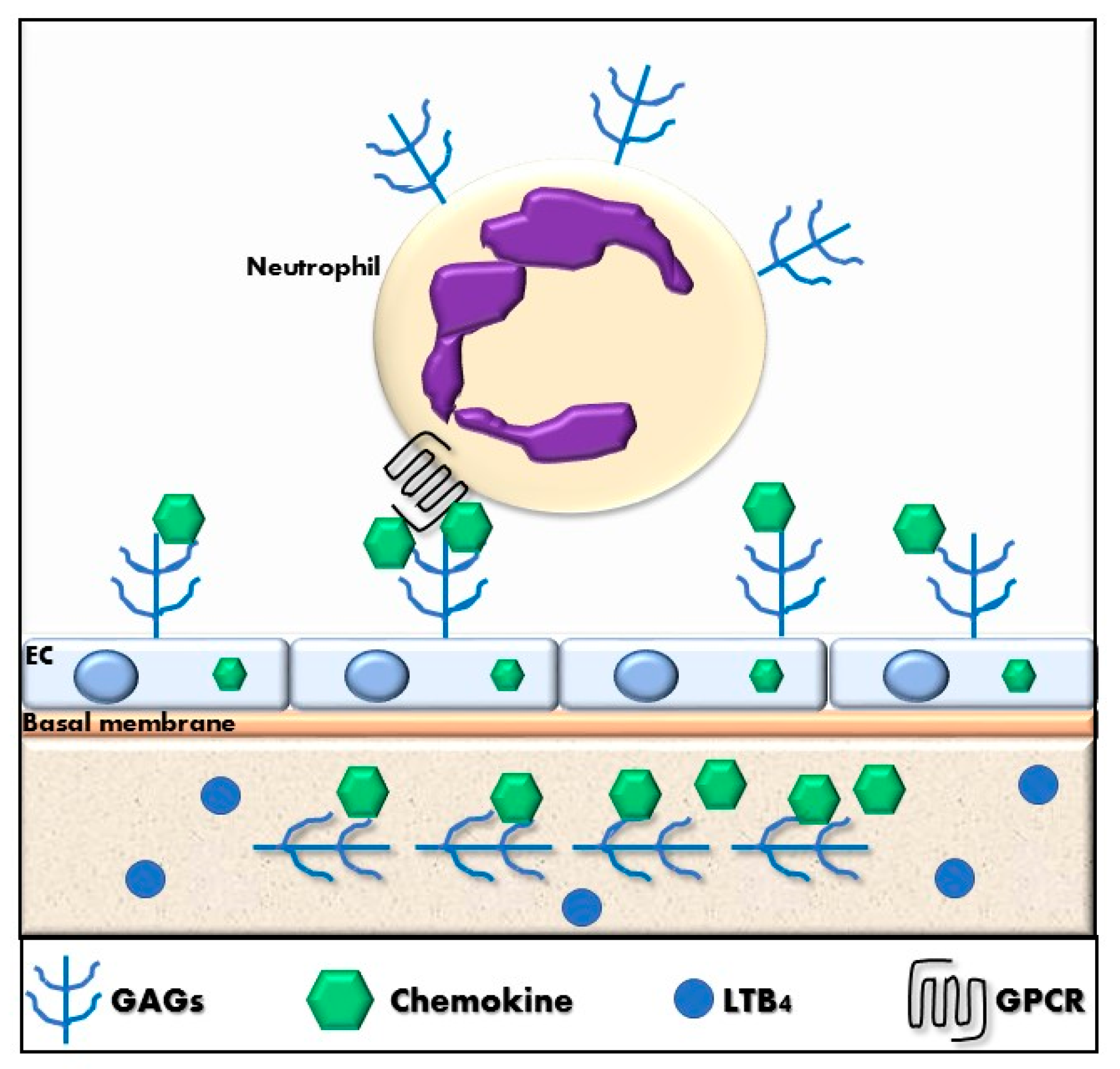{kind=link}
Table 1.
Chemokine receptors expressed on neutrophils and their human and murine ligands 1.
| Receptor | Human Ligand(s) | Murine Ligand(s) |
|---|---|---|
| CXCR1 | CXCL6, CXCL8 | CXCL6 |
| CXCR2 | CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, CXCL8 | CXCL1, CXCL2, CXCL3, CXCL6, CXCL7 |
| CXCR3 | CXCL4, CXCL4L1, CXCL9, CXCL10, CXCL11, | CXCL4, CXCL9, CXCL10, CXCL11 |
| CXCR4 | CXCL12 | CXCL12 |
| CCR1 | CCL3, CCL3L1, CCL4L1, CCL5, CCL7, CCL8, CCL14, CCL15, CCL16, CCL23 | CCL3, CCL5, CCL6, CCL7, CCL9 |
| CCR2 | CCL2, CCL7, CCL8, CCL13, CCL16 | CCL2, CCL7, CCL12 |
| CCR3 | CCL3L1, CCL5, CCL7, CCL8, CCL11, CCL13, CCL15, CCL24, CCL26, CCL28 | CCL5, CCL7, CCL9, CCL11, CCL14, CCL24, CCL26, CCL28 |
| CCR5 | CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL8, CCL11, CCL14, CCL16 | CCL3, CCL4, CCL5 |
| ACKR2 | Inflammatory CC chemokines | Inflammatory CC chemokines |
1 CXCR1 and CXCR2 are highly expressed on circulating neutrophils, CXCR4 is enhanced on “aging” neutrophils and other chemokine receptors may be upregulated on neutrophils in inflamed tissues.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Boff, D.; Crijns, H.; Teixeira, M.M.; Amaral, F.A.; Proost, P. Neutrophils: Beneficial and Harmful Cells in Septic Arthritis. Int. J. Mol. Sci. 2018, 19, 468. https://doi.org/10.3390/ijms19020468
AMA Style
Boff D, Crijns H, Teixeira MM, Amaral FA, Proost P. Neutrophils: Beneficial and Harmful Cells in Septic Arthritis. International Journal of Molecular Sciences. 2018; 19(2):468. https://doi.org/10.3390/ijms19020468
Chicago/Turabian StyleBoff, Daiane, Helena Crijns, Mauro M. Teixeira, Flavio A. Amaral, and Paul Proost. 2018. "Neutrophils: Beneficial and Harmful Cells in Septic Arthritis" International Journal of Molecular Sciences 19, no. 2: 468. https://doi.org/10.3390/ijms19020468
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.