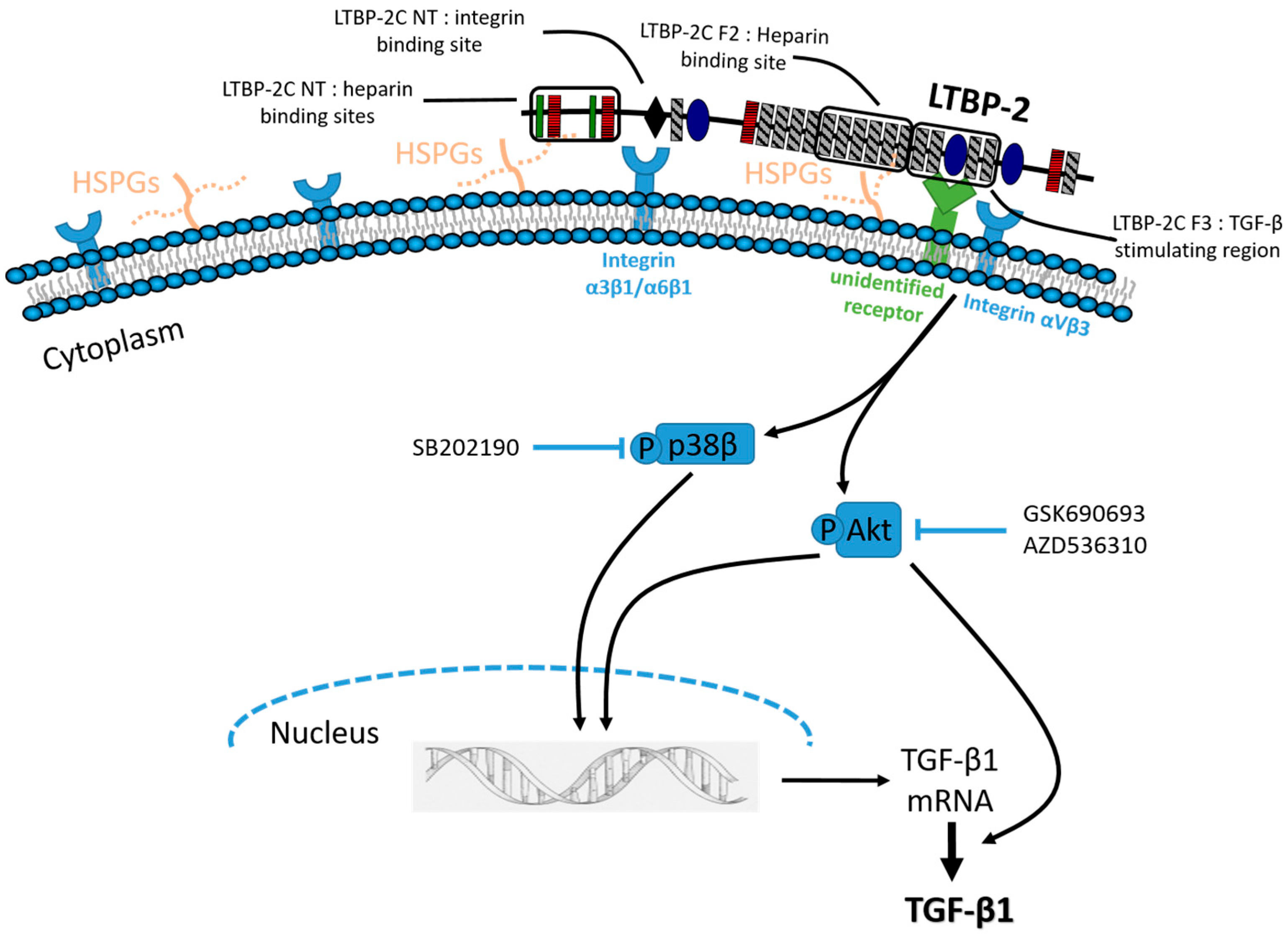
A Central Bioactive Region of LTBP-2 Stimulates the Expression of TGF-β1 in Fibroblasts via Akt and p38 Signalling Pathways

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
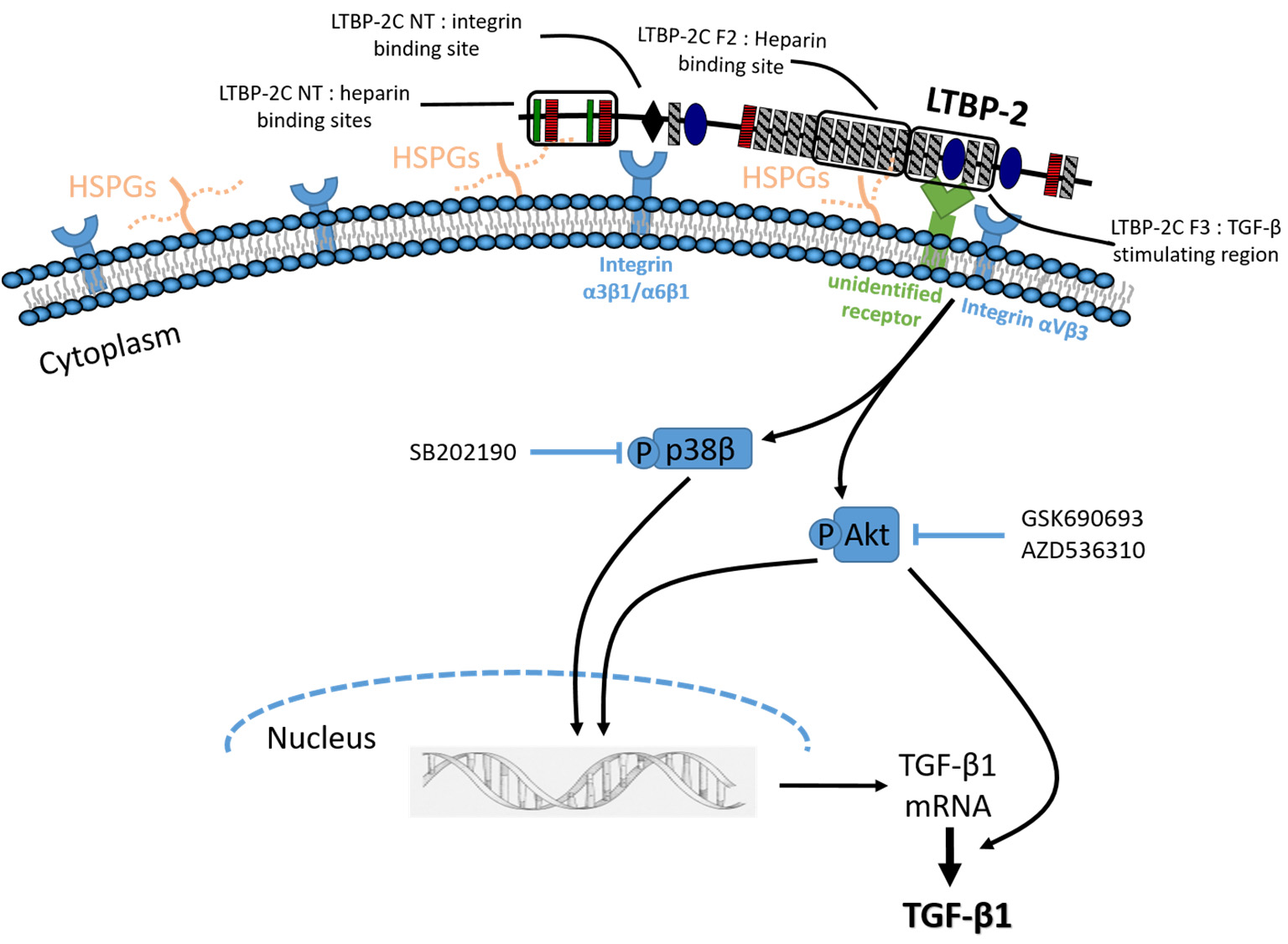
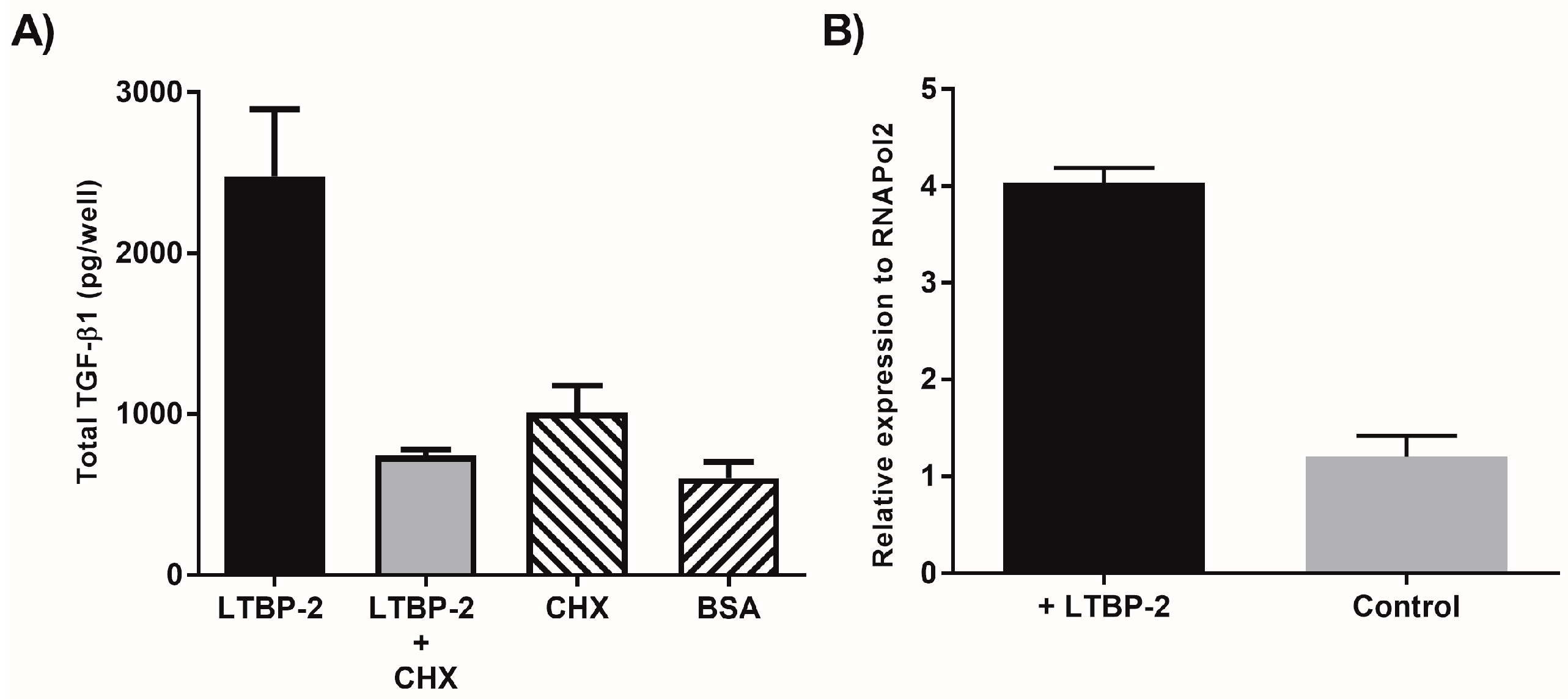
2.1. Exogenous LTBP-2 Stimulates Expression and Secretion of TGF-β1 in MSU-1.1 Fibroblasts
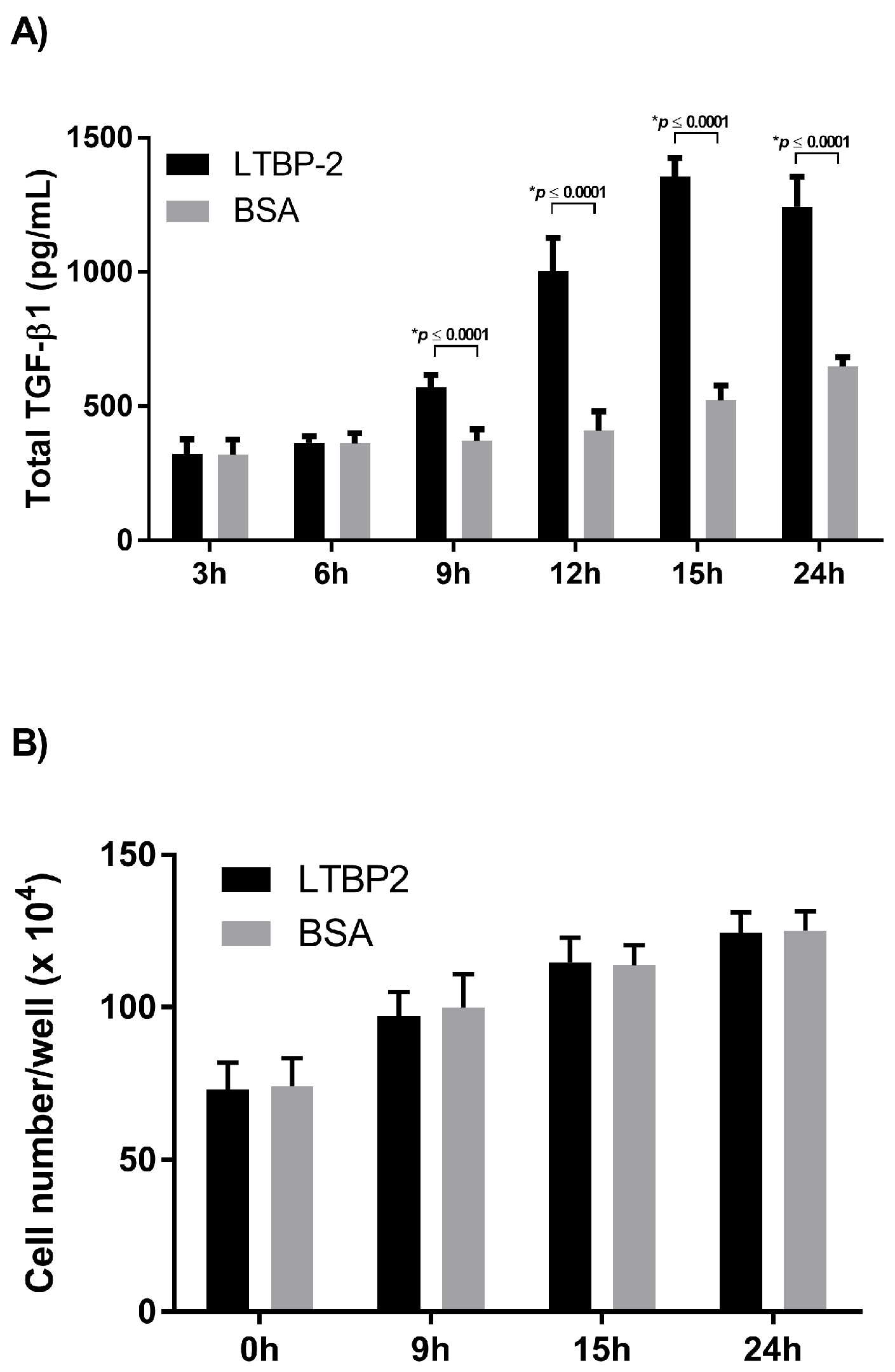
2.2. Time Course for LTBP-2 Stimulation of TGF-β1 Upregulation
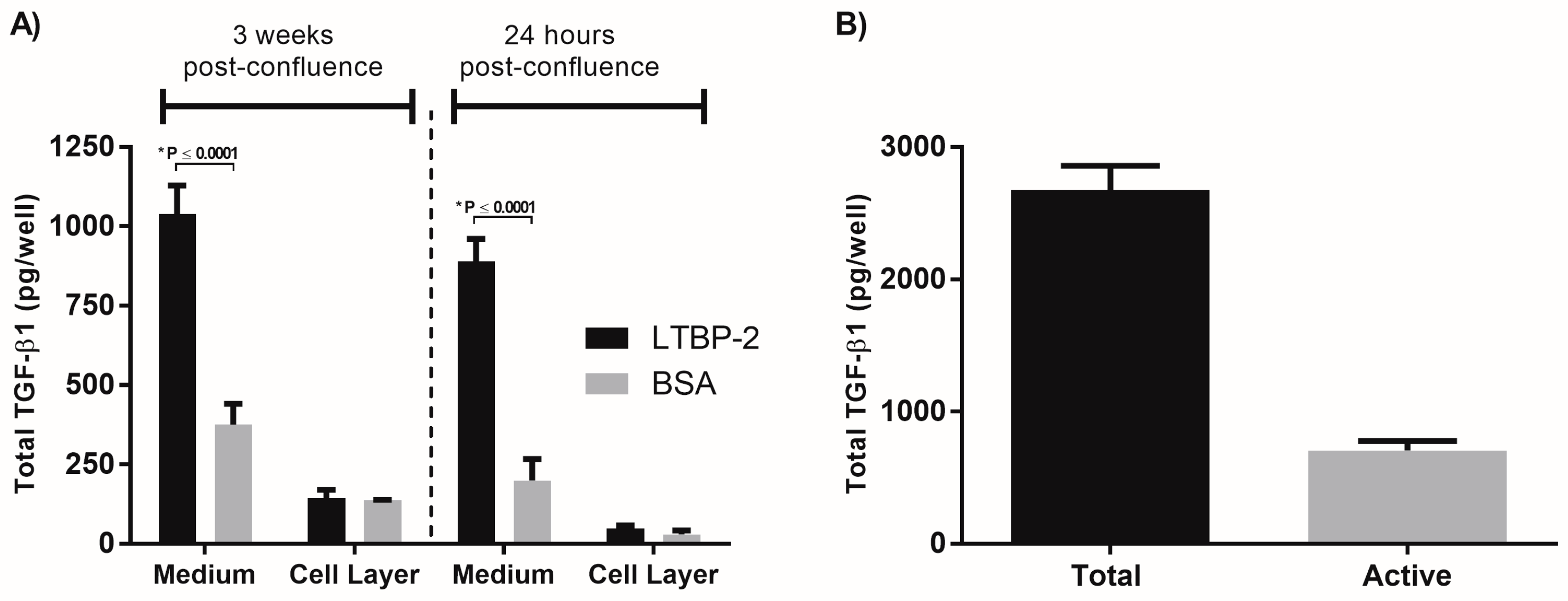
2.3. A Short Exposure of Cells to Exogenous LTBP-2 Is Sufficient to Stimulate TGF-β Upregulation
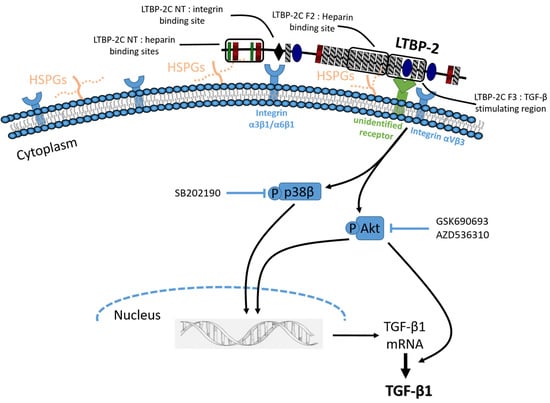
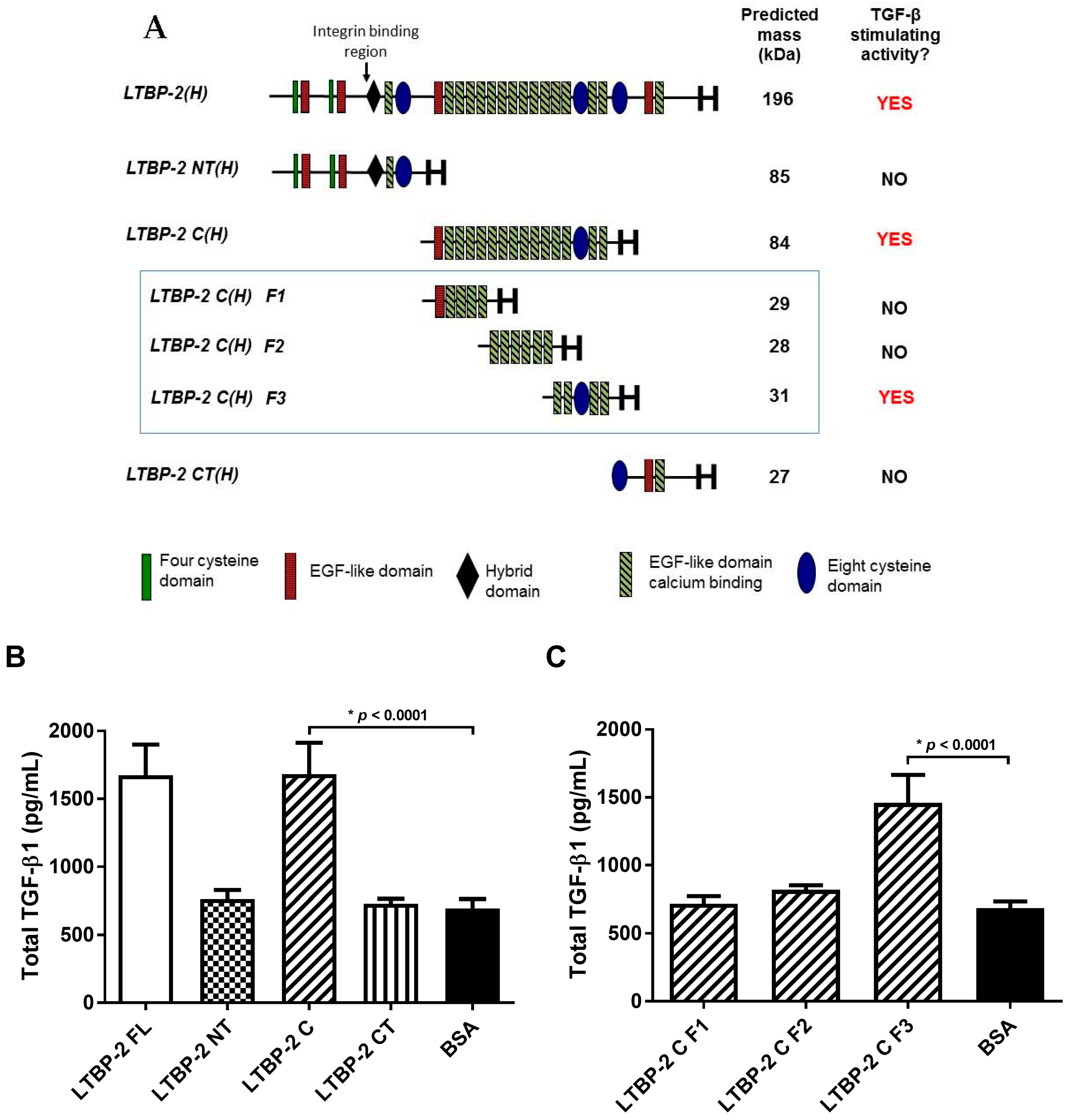
2.4. The TGF-β1 Stimulating Activity Maps to a Central Region of LTBP-2 Consisting of an Eight-Cys Motif Flanked by Pairs of EGF-Like Repeats
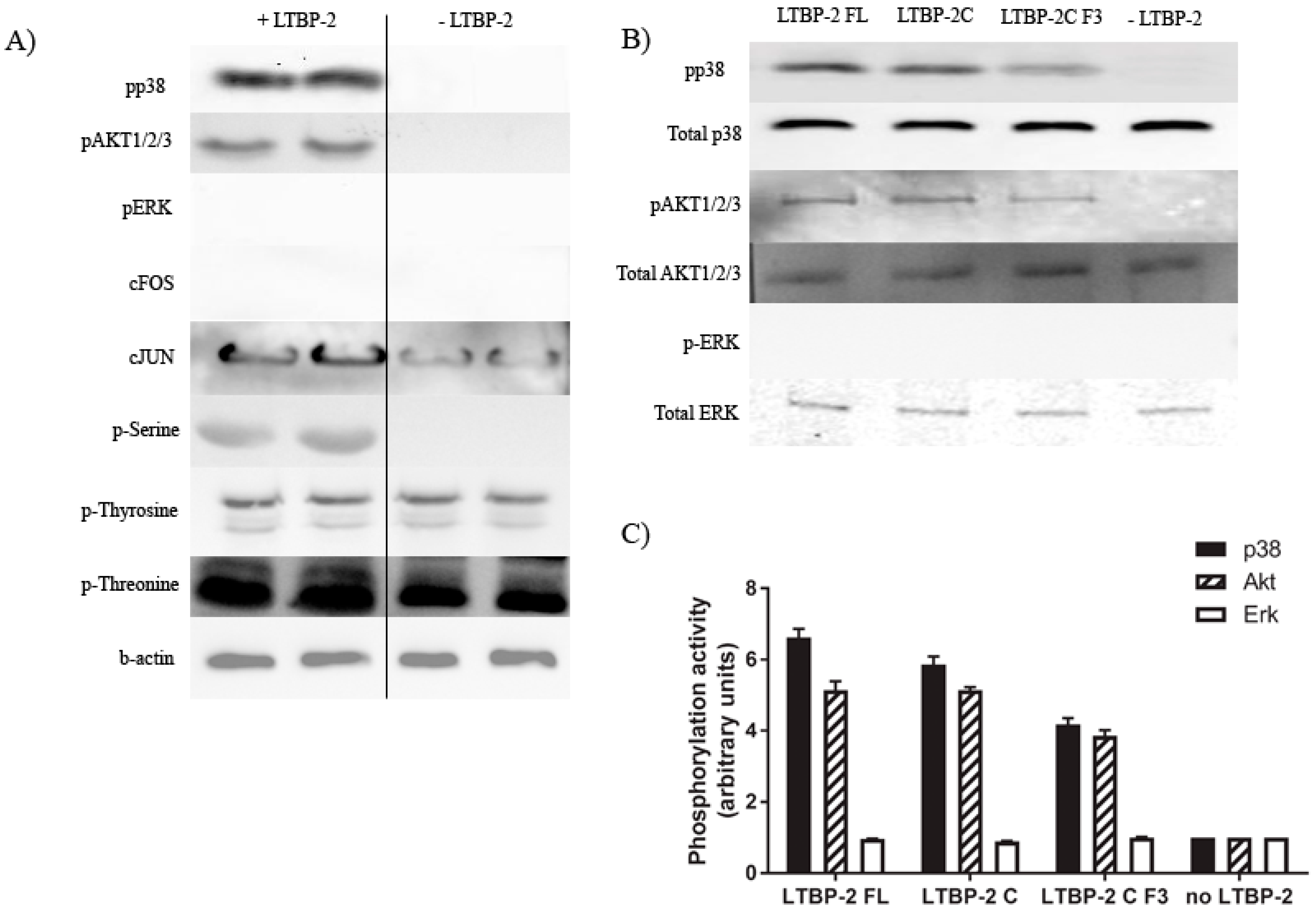
2.5. Induction of Akt and p38 MAPK Phosphorylation by LTBP-2
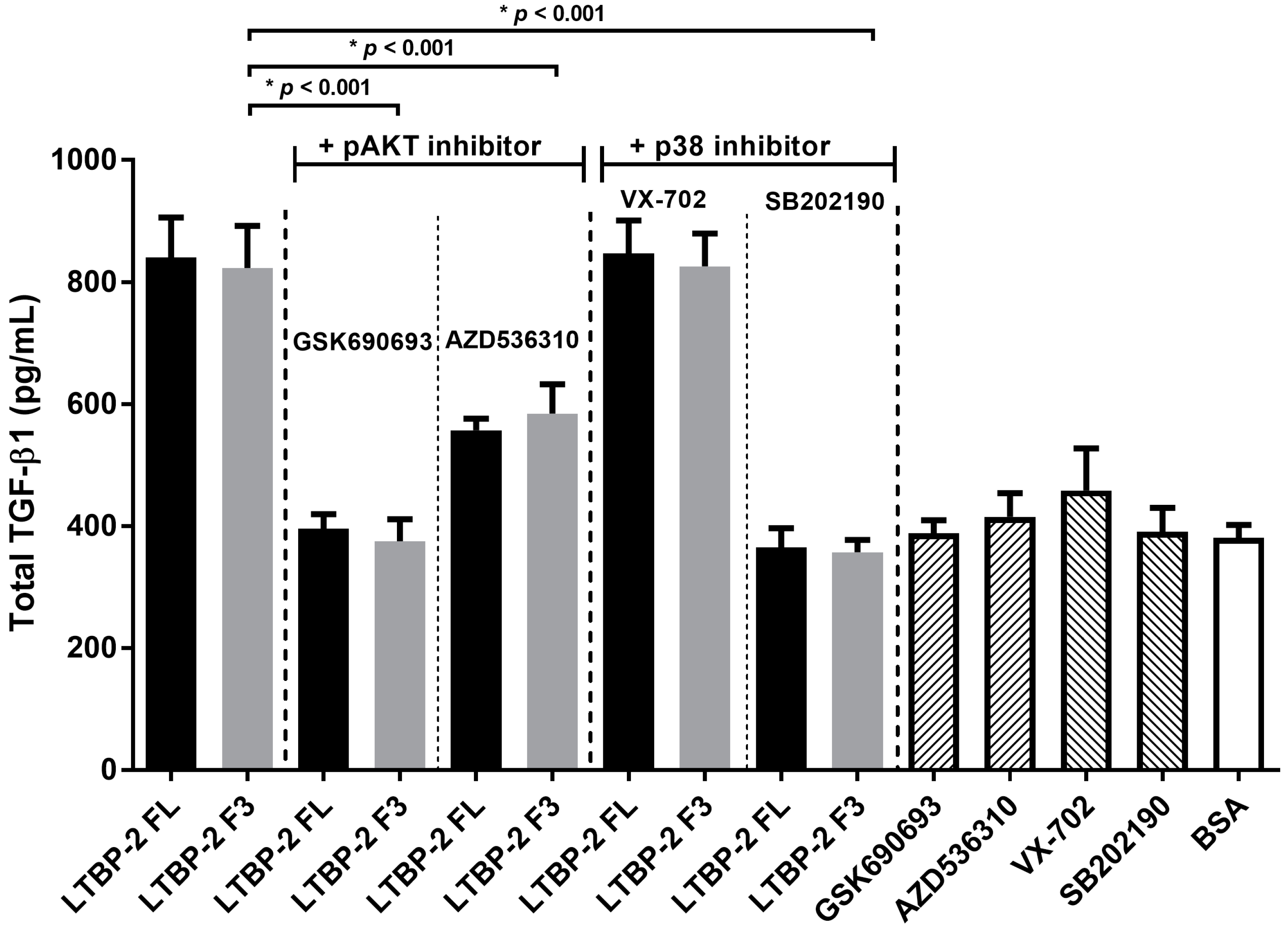
2.6. LTBP-2 Stimulates the Expression of TGF-β1 via Akt and p38 MAPK Signalling Pathways
2.7. Blocking of Integrin αVβ3 Receptors Partially Attenuates TGF-β1 Production by LTBP-2
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Detection and Quantitation of TGF-β
4.4. Quantitative PCR
4.5. Measurement of Signal Phosphorylation
4.6. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LTBP-2 | Latent Transforming Growth Factor β-1 Binding Protein 2 |
| TGF-β | Transforming Growth Factor β |
| EGF | Epidermal Growth Factor |
| FGF | Fibroblast Growth Factor |
| MAPK | Mitogen-Activated Protein Kinase |
| TBS | tris-Buffered Saline |
| PBS | Phosphate-buffered Saline |
| BSA | Bovine Serum Albumin |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| HSPG | Heparan Sulphate Proteoglycan |
| WMS | Weill–Marchesani Syndrome |
References
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGF-β activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.A.; Coker, R. Transforming growth factor-β (TGF-β). Int. J. Biochem. Cell Biol. 1998, 30, 293–298. [Google Scholar] [CrossRef]
- Tatler, A.L.; Jenkins, G. TGF-β activation and lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, S.Y. Transforming growth factor-β and smooth muscle differentiation. World J. Biol. Chem. 2012, 3, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Pohlers, D.; Brenmoehl, J.; Loffler, I.; Muller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-β and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.; Jenkins, G. Role of integrin-mediated TGF-β activation in the pathogenesis of pulmonary fibrosis. Biochem. Soc. Trans. 2009, 37, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Bottinger, E.P.; Letterio, J.J.; Roberts, A.B. Biology of TGF-β in knockout and transgenic mouse models. Kidney Int. 1997, 51, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Heldin, C.H. Latent forms of TGF-β: Molecular structure and mechanisms of activation. Ciba Found. Symp. 1991, 157, 81–89. [Google Scholar] [PubMed]
- Rifkin, D.B. Latent transforming growth factor-β (TGF-β) binding proteins: Orchestrators of TGF-β availability. J. Biol. Chem. 2005, 280, 7409–7412. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Heagerty, A.M.; Shuttleworth, C.A.; Kielty, C.M. Expression of latent TGF-β binding proteins and association with TGF-β 1 and fibrillin-1 following arterial injury. Cardiovasc. Res. 2002, 53, 971–983. [Google Scholar] [CrossRef]
- Saharinen, J.; Keski-Oja, J. Specific sequence motif of 8-Cys repeats of TGF-β binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-β. Mol. Biol. Cell 2000, 11, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.A.; Hatzinikolas, G.; Davis, E.C.; Baker, E.; Sutherland, G.R.; Mecham, R.P. Bovine latent transforming growth factor β 1-binding protein 2: Molecular cloning, identification of tissue isoforms, and immunolocalization to elastin-associated microfibrils. Mol. Cell. Biol. 1995, 15, 6932–6942. [Google Scholar] [CrossRef] [PubMed]
- Hirani, R.; Hanssen, E.; Gibson, M.A. LTBP-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with LTBP-1 for binding to this microfibrillar protein. Matrix Biol. 2007, 26, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Moren, A.; Olofsson, A.; Stenman, G.; Sahlin, P.; Kanzaki, T.; Claesson-Welsh, L.; ten Dijke, P.; Miyazono, K.; Heldin, C.H. Identification and characterization of LTBP-2, a novel latent transforming growth factor-β-binding protein. J. Biol. Chem. 1994, 269, 32469–32478. [Google Scholar] [PubMed]
- Shipley, J.M.; Mecham, R.P.; Maus, E.; Bonadio, J.; Rosenbloom, J.; McCarthy, R.T.; Baumann, M.L.; Frankfater, C.; Segade, F.; Shapiro, S.D. Developmental expression of latent transforming growth factor β binding protein 2 and its requirement early in mouse development. Mol. Cell. Biol. 2000, 20, 4879–4887. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Ohbayashi, T.; Fujikawa, Y.; Yoshida, H.; Akama, T.O.; Noda, K.; Horiguchi, M.; Kameyama, K.; Hata, Y.; Takahashi, K.; et al. Latent TGF-β binding protein-2 is essential for the development of ciliary zonule microfibrils. Hum. Mol. Genet. 2014, 23, 5672–5682. [Google Scholar] [CrossRef] [PubMed]
- Desir, J.; Sznajer, Y.; Depasse, F.; Roulez, F.; Schrooyen, M.; Meire, F.; Abramowicz, M. LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur. J. Hum. Genet. 2010, 18, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; McKibbin, M.; Booth, A.; Parry, D.A.; Jain, P.; Riazuddin, S.A.; Hejtmancik, J.F.; Khan, S.N.; Firasat, S.; Shires, M.; et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet. 2009, 84, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Haji-Seyed-Javadi, R.; Jelodari-Mamaghani, S.; Paylakhi, S.H.; Yazdani, S.; Nilforushan, N.; Fan, J.B.; Klotzle, B.; Mahmoudi, M.J.; Ebrahimian, M.J.; Chelich, N.; et al. LTBP2 mutations cause weill-marchesani and weill-marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum. Mutat. 2012, 33, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.A.; McGovern, A.; Baldwin, A.K.; Baldock, C.; Kielty, C.M. Fibrillin-1 mutations causing weill-marchesani syndrome and acromicric and geleophysic dysplasias disrupt heparan sulfate interactions. PLoS ONE 2012, 7, e48634. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Keene, D.R.; Bruder, S.P.; Saharinen, J.; Sakai, L.Y.; Mundy, G.R.; Bonewald, L.F. Role of the latent transforming growth factor β binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J. Bone Miner. Res. 2000, 15, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Massam-Wu, T.; Chiu, M.; Choudhury, R.; Chaudhry, S.S.; Baldwin, A.K.; McGovern, A.; Baldock, C.; Shuttleworth, C.A.; Kielty, C.M. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF-β. J. Cell Sci. 2010, 123, 3006–3018. [Google Scholar] [CrossRef] [PubMed]
- Krohn, K. TGF-β1-dependent differential expression of a rat homolog for latent TGF-β Binding protein in astrocytes and C6 glioma cells. Glia 1999, 25, 332–342. [Google Scholar] [CrossRef]
- Kettle, S.; Card, C.M.; Hutchinson, S.; Sykes, B.; Handford, P.A. Characterisation of fibrillin-1 cDNA clones in a human fibroblast cell line that assembles microfibrils. Int. J. Biochem. Cell Biol. 2000, 32, 201–214. [Google Scholar] [CrossRef]
- Kielty, C.M.; Shuttleworth, C.A. Synthesis and assembly of fibrillin by fibroblasts and smooth muscle cells. J. Cell Sci. 1993, 106, 167–173. [Google Scholar] [PubMed]
- McKeehan, W.; Hardesty, B. The mechanism of cycloheximide inhibition of protein synthesis in rabbit reticulocytes. Biochem. Biophys. Res. Commun. 1969, 36, 625–630. [Google Scholar] [CrossRef]
- Vehvilainen, P.; Hyytiainen, M.; Keski-Oja, J. Latent transforming growth factor-β-binding protein 2 is an adhesion protein for melanoma cells. J. Biol. Chem. 2003, 278, 24705–24713. [Google Scholar] [CrossRef] [PubMed]
- Menz, C.; Parsi, M.K.; Adams, J.R.; Sideek, M.A.; Kopecki, Z.; Cowin, A.J.; Gibson, M.A. LTBP-2 has a single high-affinity binding site for FGF-2 and blocks FGF-2-induced cell proliferation. PLoS ONE 2015, 10, e0135577. [Google Scholar] [CrossRef] [PubMed]
- Parsi, M.K.; Adams, J.R.; Whitelock, J.; Gibson, M.A. LTBP-2 has multiple heparin/heparan sulfate binding sites. Matrix Biol. 2010, 29, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Damjanov, N.; Kauffman, R.S.; Spencer-Green, G.T. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: Results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheumatol. 2009, 60, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.S.; Cain, S.A.; Morgan, A.; Dallas, S.L.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 regulates the bioavailability of TGF-β1. J. Cell Biol. 2007, 176, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Goel, H.L.; Zarif, M.J.; Butterfield, J.E.; Perkins, H.M.; Sansoucy, B.G.; Sawyer, T.K.; Languino, L.R. The integrin-growth factor receptor duet. J. Cell. Physiol. 2007, 213, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Assoian, R.K.; Schwartz, M.A. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr. Opin. Genet. Dev. 2001, 11, 48–53. [Google Scholar] [CrossRef]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.J.; Grimme, J.M.; Sun, J.; Lu, S.; Gai, L.; Cropek, D.M.; Wang, Y. The antagonistic roles of PDGF and integrin αvβ3 in regulating ROS production at focal adhesions. Biomaterials 2013, 34, 3807–3815. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Couchman, J.R. Syndecan-4 and focal adhesion function. Curr. Opin. Cell Biol. 2001, 13, 578–583. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Xiang, J.; Huang, S.; Lin, A. Induction of apoptosis by SB202190 through inhibition of p38β mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 16415–16420. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Zwerina, J.; Firestein, G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Du, J. Potential role of Akt signaling in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Zhang, Q.G.; Chen, J.W.; Chen, S.Q.; Chen, S.Y. The fibrotic role of phosphatidylinositol-3-kinase/Akt pathway in injured skeletal muscle after acute contusion. Int. J. Sports Med. 2013, 34, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Murthy, S.; Ryan, A.J.; Carter, A.B. Modulation of the mevalonate pathway by Akt regulates macrophage survival and development of pulmonary fibrosis. J. Biol. Chem. 2014, 289, 36204–36219. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the Akt signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, Z.; Xu, H.; Yang, Q. miR-409-3p suppresses breast cancer cell growth and invasion by targeting Akt1. Biochem. Biophys. Res. Commun. 2016, 469, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.J.; Yang, M.; Zheng, J.; Wang, L.Y.; Du, J. Role of Akt1 in cardiac fibrosis induced by angiotensin II. Am. J. Hypertens. 2013, 26, 1635. [Google Scholar] [CrossRef]
- Tsubaki, M.; Yamazoe, Y.; Yanae, M.; Satou, T.; Itoh, T.; Kaneko, J.; Kidera, Y.; Moriyama, K.; Nishida, S. Blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathways by statins reduces the expression of BFGF, HGF, and TGF-β as angiogenic factors in mouse osteosarcoma. Cytokine 2011, 54, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Negishi, Y.; Aramaki, Y. Involvement of phosphatidylinositol-3-kinase and ERK pathways in the production of TGF-β1 by macrophages treated with liposomes composed of phosphatidylserine. FEBS Lett. 2007, 581, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.Q.; Freire-de-Lima, C.G.; Schiemann, W.P.; Bratton, D.L.; Vandivier, R.W.; Henson, P.M. Transcriptional and translational regulation of transforming growth factor-β production in response to apoptotic cells. J. Immunol. 2008, 181, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Peng, L.; Zhu, C.; Zhang, X.; Chen, F.; Liu, T. Knockdown of latent transforming growth factor-β (TGF-β)-binding protein 2 (LTBP2) inhibits invasion and tumorigenesis in thyroid carcinoma cells. Oncol. Res. 2017, 25, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.A.; Teixeira, F.C.; Fontes, M.; Areas, A.L.; Leal, M.G.; Pavao, M.S.; Stelling, M.P. Heparan sulfate proteoglycans may promote or inhibit cancer progression by interacting with integrins and affecting cell migration. Biomed. Res. Int. 2015, 2015, 453801. [Google Scholar] [CrossRef] [PubMed]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Millette, E.; Rauch, B.H.; Kenagy, R.D.; Daum, G.; Clowes, A.W. Platelet-derived growth factor-BB transactivates the fibroblast growth factor receptor to induce proliferation in human smooth muscle cells. Trends Cardiovasc. Med. 2006, 16, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Midgley, A.C.; Rogers, M.; Hallett, M.B.; Clayton, A.; Bowen, T.; Phillips, A.O.; Steadman, R. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, E.G.; Haupt, J.; Dietz, H.C.; Shore, E.M. TGF-β family signaling in connective tissue and skeletal diseases. Cold Spring Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Pakyari, M.; Farrokhi, A.; Maharlooei, M.K.; Ghahary, A. Critical role of transforming growth factor β in different phases of wound healing. Adv. Wound Care 2013, 2, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Sideek, M.A.; Teia, A.; Kopecki, Z.; Cowin, A.J.; Gibson, M.A. Co-localization of LTBP-2 with FGF-2 in fibrotic human keloid and hypertrophic scar. J. Mol. Histol. 2016, 47, 35–45. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sideek, M.A.; Smith, J.; Menz, C.; Adams, J.R.J.; Cowin, A.J.; Gibson, M.A. A Central Bioactive Region of LTBP-2 Stimulates the Expression of TGF-β1 in Fibroblasts via Akt and p38 Signalling Pathways. Int. J. Mol. Sci. 2017, 18, 2114. https://doi.org/10.3390/ijms18102114
Sideek MA, Smith J, Menz C, Adams JRJ, Cowin AJ, Gibson MA. A Central Bioactive Region of LTBP-2 Stimulates the Expression of TGF-β1 in Fibroblasts via Akt and p38 Signalling Pathways. International Journal of Molecular Sciences. 2017; 18(10):2114. https://doi.org/10.3390/ijms18102114
Chicago/Turabian StyleSideek, Mohamed A., Joshua Smith, Clementine Menz, Julian R. J. Adams, Allison J. Cowin, and Mark A. Gibson. 2017. "A Central Bioactive Region of LTBP-2 Stimulates the Expression of TGF-β1 in Fibroblasts via Akt and p38 Signalling Pathways" International Journal of Molecular Sciences 18, no. 10: 2114. https://doi.org/10.3390/ijms18102114