
PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences

Abstract
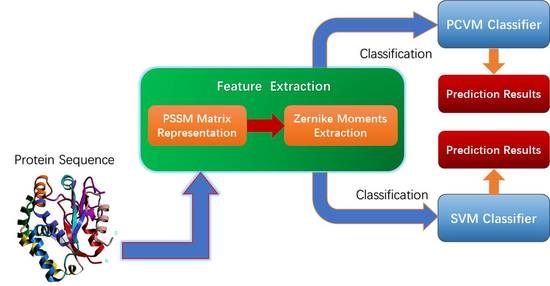
:
1. Introduction
2. Results and Discussion
2.1. Evaluation Measure
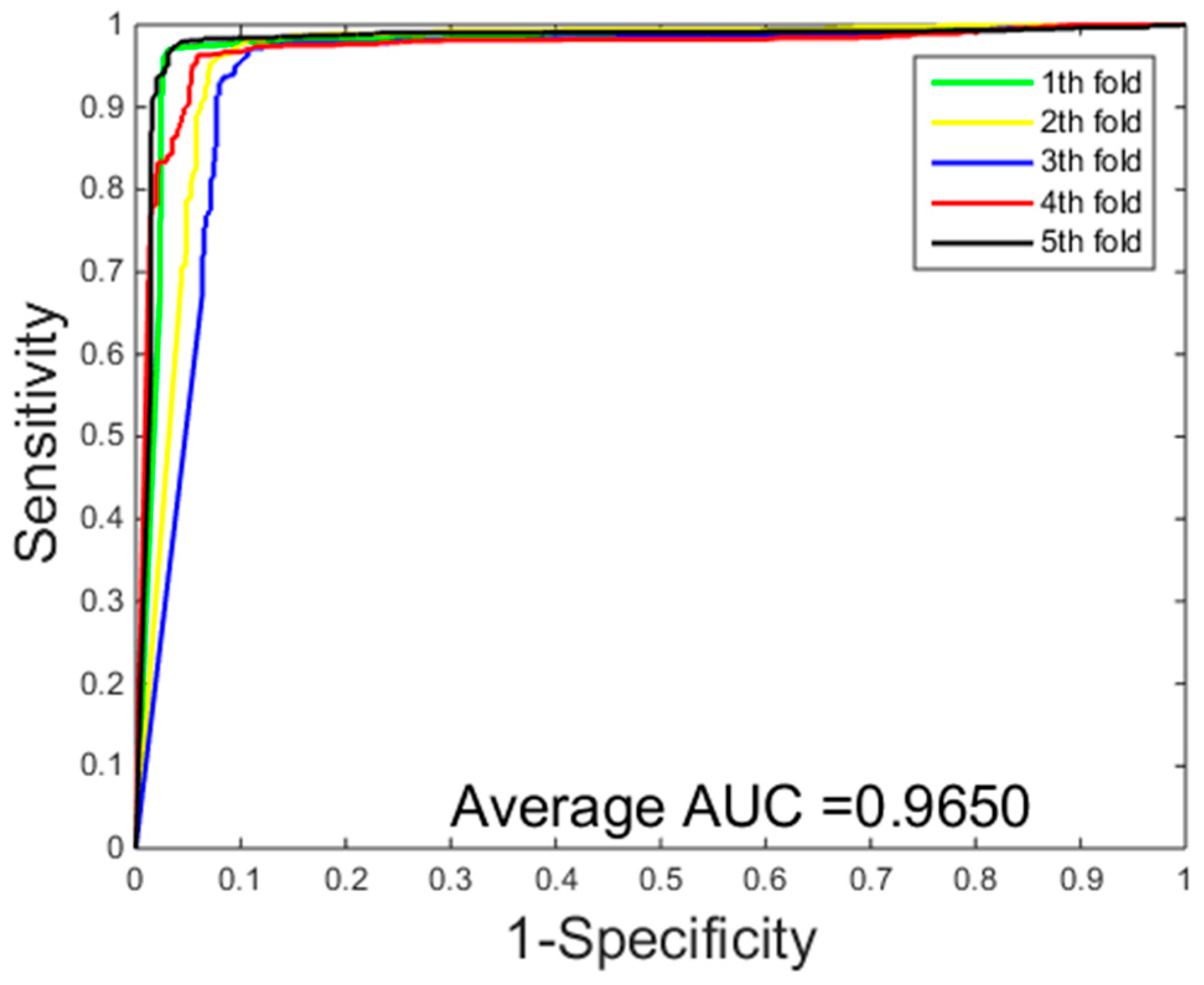
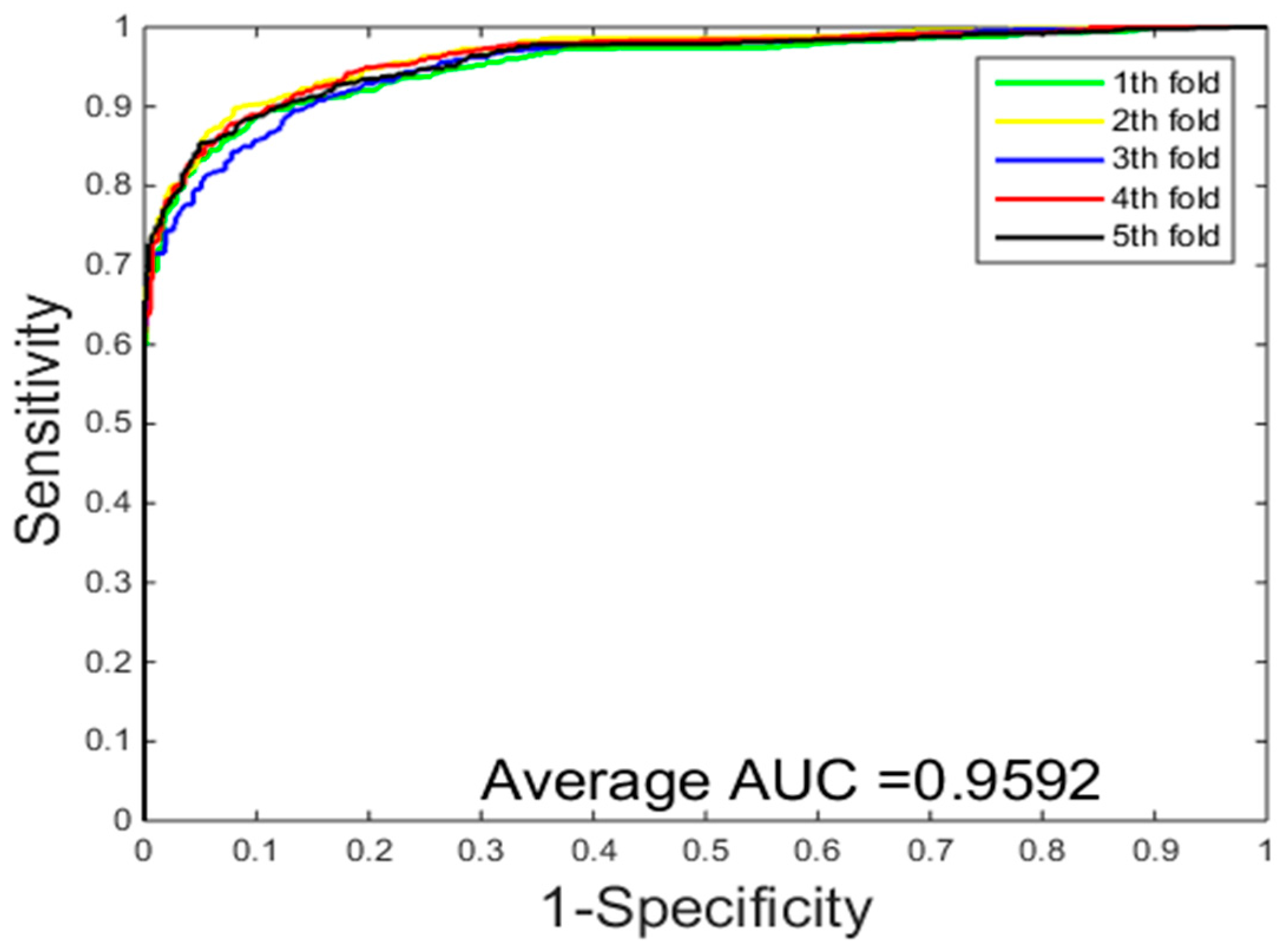
2.2. Assessment of Prediction
2.3. Comparison with the Support Vector Machine (SVM)-Based Method
2.4. Comparison with Other Methods
3. Materials and Methodology
3.1. Dataset
3.2. Position-Specific Scoring Matrix
3.3. Zernike Moments
3.3.1. Invariance of Normalized Zernike Moment
3.2.2. Introduction of a Zernike Moments Descriptor
3.2.3. Feature Selection
3.4. Related Machine Learning Models
3.5. PCVM Algorithm
3.6. Initial Parameter Selection and Training
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Puig, O.; Caspary, F.; Rigaut, G.; Rutz, B.; Bouveret, E.; Bragado-Nilsson, E.; Wilm, M.; Seraphin, B. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods 2001, 24, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.; Zhou, J.; Burgess, R.; Elledge, S.J.; Olson, E.N. PICK1: A perinuclear binding protein and substrate for protein kinase C isolated by the yeast two-hybrid system. J. Cell Biol. 1995, 128, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Koegl, M.; Uetz, P. Improving yeast two-hybrid screening systems. Brief. Funct. Genom. 2007, 6, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Snyder, M. Protein chip technology. Curr. Opin. Chem. Biol. 2003, 7, 55–63. [Google Scholar] [CrossRef]
- Pazos, F.; Valencia, A. Similarity of phylogenetic trees as indicator of protein-protein interaction. Protein Eng. Des. Sel. 2001, 14, 609–614. [Google Scholar] [CrossRef]
- Wang, B.; Chen, P.; Huang, D.S.; Li, J.J.; Lok, T.M.; Lyu, M.R. Predicting protein interaction sites from residue spatial sequence profile and evolution rate. FEBS Lett. 2006, 580, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Maleki, M.; Hall, M.; Rueda, L. Using structural domains to predict obligate and non-obligate protein-protein interactions. CIBCB 2012, 252–261. [Google Scholar] [CrossRef]
- Huang, C.; Morcos, F.; Kanaan, S.P.; Wuchty, S.; Chen, D.Z.; Izaguirre, J.A. Predicting protein–protein interactions from protein domains using a set cover approach. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Yu, H.; Greenbaum, D.; Kluger, Y.; Krogan, N.J.; Chung, S.; Emili, A.; Snyder, M.; Greenblatt, J.F.; Gerstein, M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 2003, 302, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Cai, L. Predicting protein–protein interaction based on protein secondary structure information using Bayesian classifier. J. Inn. Mongolia Univ. Sci. Technol. 2010, 1, 021. (In Chinese) [Google Scholar]
- Cai, L.; Pei, Z.; Qin, S.; Zhao, X. Prediction of protein–protein interactions in Saccharomyces cerevisiae Based on Protein Secondary Structure. iCBEB 2012, 413–416. [Google Scholar] [CrossRef]
- You, Z.H.; Yu, J.Z.; Zhu, L.; Li, S.; Wen, Z.K. A MapReduce based parallel SVM for large-scale predicting protein–protein interactions. Neurocomputing 2014, 145, 37–43. [Google Scholar] [CrossRef]
- You, Z.H.; Zheng, Y.; Han, K.; Huang, D.S.; Zhou, X. A semi-supervised learning approach to predict synthetic genetic interactions by combining functional and topological properties of functional gene network. BMC Bioinform. 2010, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Hu, Q.; Guo, M.; Wang, G. HAlign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 2015, 31, 2475. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.R.; Gough, D.A. Whole-proteome interaction mining. Bioinformatics 2003, 19, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Roe, D.; Faulon, J.L. Predicting protein–protein interactions using signature products. Bioinformatics 2005, 21, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, H.S. Sequence-based prediction of protein–protein interactions by means of codon usage. Genome Biol. 2008, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein–protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; You, Z.H.; Xin, G.; Leon, W.; Wang, L. Using Weighted Sparse Representation Model Combined with Discrete Cosine Transformation to Predict Protein-Protein Interactions from Protein Sequence. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A. An ensemble of support vector machines for predicting the membrane protein type directly from the amino acid sequence. Amino Acids 2008, 35, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A. An ensemble of K-local hyperplanes for predicting protein-protein interactions. Bioinformatics 2006, 22, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L. Fusion of classifiers for predicting protein–protein interactions. Neurocomputing 2005, 68, 289–296. [Google Scholar] [CrossRef]
- Nanni, L.; Brahnam, S.; Lumini, A. High performance set of PseAAC and sequence based descriptors for protein classification. J. Theor. Biol. 2010, 266, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A. A genetic approach for building different alphabets for peptide and protein classification. BMC Bioinform. 2008, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Li, J.; Gao, X.; He, Z.; Zhu, L.; Lei, Y.K.; Ji, Z. Detecting protein-protein interactions with a novel matrix-based protein sequence representation and support vector machines. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Chan, K.C.C.; Hu, P. Predicting protein–protein interactions from primary protein sequences using a novel multi-scale local feature representation scheme and the random forest. PLoS ONE 2015, 10, e0125811. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; You, Z.H.; Chen, X.; Li, J.Q.; Yan, X.; Zhang, W.; Huang, Y.A. An ensemble approach for large-scale identification of protein- protein interactions using the alignments of multiple sequences. Oncotarget 2016, 8, 5149–5159. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Le, Y.; Zh, L.; Xi, J.; Wang, B. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinform. 2013, 14, S10. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; You, Z.H.; Ming, Z.; Li, J.; Chen, X.; Huang, Y.A. Detection of Interactions between Proteins through Rotation Forest and Local Phase Quantization Descriptors. Int. J. Mol. Sci. 2016, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.K.; You, Z.H.; Ji, Z.; Zhu, L.; Huang, D.S. Assessing and predicting protein interactions by combining manifold embedding with multiple information integration. BMC Bioinform. 2012, 13, S3. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L. Letters: Hyperplanes for predicting protein-protein interactions. Neurocomputing 2005, 69, 257–263. [Google Scholar] [CrossRef]
- You, Z.H.; Li, S.; Gao, X.; Luo, X.; Ji, Z. Large-scale protein-protein interactions detection by integrating big biosensing data with computational model. BioMed Res. Int. 2014, 2014, 598129. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; You, Z.H.; Li, X.; Chen, X.; Hu, P.; Li, S.; Luo, X. Construction of Reliable Protein–Protein Interaction Networks Using Weighted Sparse Representation Based Classifier with Pseudo Substitution Matrix Representation Features. Neurocomputing 2016, 218, 131–138. [Google Scholar] [CrossRef]
- An, J.Y.; You, Z.H.; Chen, X.; Huang, D.S.; Yan, G.Y. Robust and accurate prediction of protein self-interactions from amino acids sequence using evolutionary information. Mol. BioSyst. 2016, 12, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.B.; Hu, S.C.; Wang, H.; Zou, Q.; Ji, Z.L. PaGeFinder: Quantitative identification of spatiotemporal pattern genes. Bioinformatics 2012, 28, 1544–1545. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Li, X.B.; Jiang, W.R.; Lin, Z.Y.; Li, G.L.; Chen, K. Survey of MapReduce frame operation in bioinformatics. Brief. Bioinform. 2014, 15, 637. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2016, 17, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Guo, M.; Wang, C.; Liu, X.; Zou, Q. An overview of SNP interactions in genome-wide association studies. Brief. Funct. Genom. 2015, 14, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Huang, J.; Ling, C.X. Using AUC and accuracy in evaluating learning algorithms. Knowl. Data Eng. Trans. 2005, 17, 299–310. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2007, 2, 389–396. [Google Scholar] [CrossRef]
- Quan, Z.; Li, J.; Li, S.; Zeng, X.; Wang, G. Similarity computation strategies in the microRNA-disease network: A survey. Brief. Funct. Genom. 2016, 15, 55. [Google Scholar]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Donaldson, I.; Wolting, C.; Ouellette, B.F.F.; Pawson, T.; Hogue, C.W.V. BIND—The Biomolecular Interaction Network Database. Nucleic Acids Res. 2001, 29, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; de la Paz, M.L.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, J.G.; Henikoff, S. Using substitution probabilities to improve position-specific scoring matrices. Bioinformatics 1996, 12, 135–143. [Google Scholar] [CrossRef]
- Paliwal, K.K.; Sharma, A.; Lyons, J.; Dehzangi, A. A Tri-Gram Based Feature Extraction Technique Using Linear Probabilities of Position Specific Scoring Matrix for Protein Fold Recognition. J. Theor. Biol. 2014, 13, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Y.; You, Z.H.; Zhang, X.F.; Yong, Z. Prediction of Protein-Protein Interactions with Clustered Amino Acids and Weighted Sparse Representation. Int. J. Mol. Sci. 2015, 16, 10855–10869. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M. A model of evolutionary change in proteins. Atlas Protein Seq. Struct. 1977, 5, 345–352. [Google Scholar]
- Bhagwat, M.; Aravind, L. PSI-BLAST tutorial. Methods Mol. Biol. 2007, 395, 177–186. [Google Scholar] [PubMed]
- Xiao, R.Q.; Guo, Y.Z.; Zeng, Y.H.; Tan, H.F.; Tan, H.F.; Pu, X.M.; Li, M.L. Using position specific scoring matrix and auto covariance to predict protein subnuclear localization. J. Biomed. Sci. Eng. 2009, 2, 51–56. [Google Scholar] [CrossRef]
- An, J.Y.; Meng, F.R.; You, Z.H.; Fang, Y.H.; Zhao, Y.J.; Ming, Z. Using the Relevance Vector Machine Model Combined with Local Phase Quantization to Predict Protein-Protein Interactions from Protein Sequences. BioMed Res. Int. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Kim, Y.S. A region-based shape descriptor using Zernike moments. Signal Process. Image Commun. 2000, 16, 95–102. [Google Scholar] [CrossRef]
- Liao, S.X.; Pawlak, M. On the accuracy of Zernike moments for image analysis. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 1358–1364. [Google Scholar] [CrossRef]
- Li, S.; Lee, M.C.; Pun, C.M. Complex Zernike moments features for shape-based image retrieval. IEEE Trans. Syst. Man Cybern. Part A Syst. Hum. 2009, 39, 227–237. [Google Scholar] [CrossRef]
- Georgiou, D.N.; Karakasidis, T.E.; Megaritis, A.C. A short survey on genetic sequences, chou’s pseudo amino acid composition and its combination with fuzzy set theory. Open Bioinform. J. 2013, 7, 41–48. [Google Scholar] [CrossRef]
- Liu, T.; Qin, Y.; Wang, Y.; Wang, C. Prediction of Protein Structural Class Based on Gapped-Dipeptides and a Recursive Feature Selection Approach. Int. J. Mol. Sci. 2015, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, S. Protein Sub-Nuclear Localization Based on Effective Fusion Representations and Dimension Reduction Algorithm LDA. Int. J. Mol. Sci. 2015, 16, 30343–30361. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, D.N.; Karakasidis, T.E.; Nieto, J.J.; Torres, A. A study of entropy/clarity of genetic sequences using metric spaces and fuzzy sets. J. Theor. Biol. 2010, 267, 95. [Google Scholar] [CrossRef] [PubMed]
- Hse, H.; Newton, A.R. Sketched symbol recognition using Zernike moments. In Proceedings of the 17th International Conference on Pattern Recognition, Cambridge, UK, 23–26 August 2004; Volume 1, pp. 367–370. [Google Scholar]
- Noll, R.J. Zernike polynomials and atmospheric turbulence. JOsA 1976, 66, 207–211. [Google Scholar] [CrossRef]
- Wang, J.Y.; Silva, D.E. Wave-front interpretation with Zernike polynomials. Appl. Opt. 1980, 19, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Schwiegerling, J.; Greivenkamp, J.E.; Miller, J.M. Representation of videokeratoscopic height data with Zernike polynomials. JOsA 1995, 12, 2105–2113. [Google Scholar] [CrossRef]
- Chong, C.W.; Raveendran, P.; Mukundan, R. A comparative analysis of algorithms for fast computation of Zernike moments. Pattern Recognit. 2003, 36, 731–742. [Google Scholar] [CrossRef]
- Singh, C.; Walia, E.; Upneja, R. Accurate calculation of Zernike moments. Inf. Sci. 2013, 233, 255–275. [Google Scholar] [CrossRef]
- Hwang, S.K.; Billinghurst, M.; Kim, W.Y. Local Descriptor by Zernike Moments for Real-Time Keypoint Matching. Image Signal Process. 2008, 2, 781–785. [Google Scholar]
- Liao, S.X.; Pawlak, M. A study of Zernike moment computing. Asian Conf. Comput. Vis. 2006, 98, 394–401. [Google Scholar]
- Khotanzad, A.; Hong, Y.H. Invariant Image Recognition by Zernike Moments. IEEE Trans. Pattern Anal. Mach. Intell. 1990, 12, 489–497. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, H.K. Invariant image watermark using Zernike moments. IEEE Trans.Circuits Syst. Video Technol. 2003, 13, 766–775. [Google Scholar]
- Zou, Q.; Zeng, J.C.; Cao, L.J.; Ji, R.R. A Novel Features Ranking Metric with Application to Scalable Visual and Bioinformatics Data Classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Burges, C.J.C. A Tutorial on Support Vector Machines for Pattern Recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]
- Bishop, C.M.; Tipping, M.E.; Nh, C.C. Variational Relevance Vector Machines. Adv. Neural Inf. Process. Syst. 2000, 12, 299–334. [Google Scholar]
- Li, Y.; Campbell, C.; Tipping, M. Bayesian automatic relevance determination algorithms for classifying gene expression data. Bioinformatics 2002, 18, 1332–1339. [Google Scholar]
- Wei, L.Y.; Tang, J.J.; Zou, Q. Local-DPP: An Improved DNA-binding Protein Prediction Method by Exploring Local Evolutionary Information. Inf. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Chen, H.; Tino, P.; Yao, X. Probabilistic classification vector machines. IEEE Trans. Neural Netw. 2009, 20, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tino, P.; Xin, Y. Efficient Probabilistic Classification Vector Machine With Incremental Basis Function Selection. IEEE Trans. Neural Netw. Learn. Syst. 2014, 25, 356–369. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Testing Set | Acc (%) | Sen (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 96.38 | 97.21 | 95.57 | 93.02 |
| 2 | 94.05 | 95.23 | 92.77 | 88.81 |
| 3 | 93.07 | 96.73 | 90.27 | 87.06 |
| 4 | 94.46 | 94.20 | 94.71 | 89.53 |
| 5 | 94.42 | 92.26 | 96.26 | 89.46 |
| Average | 94.48 ± 1.2 | 95.13 ± 2.0 | 93.92 ± 2.4 | 89.58 ± 2.2 |
| Testing Set | Acc (%) | Sen (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 89.54 | 92.11 | 86.82 | 81.24 |
| 2 | 92.11 | 92.68 | 91.41 | 85.46 |
| 3 | 91.08 | 91.16 | 91.16 | 83.75 |
| 4 | 91.42 | 92.25 | 90.34 | 84.31 |
| 5 | 92.12 | 92.04 | 93.23 | 85.42 |
| Average | 91.25 ± 1.1 | 92.05 ± 0.6 | 90.06 ± 2.4 | 84.04 ± 1.7 |
| Model | Testing Set | Acc (%) | Sen (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|---|
| Probabilistic Classification Vector Machines (PCVM) | 1 | 96.38 | 97.21 | 95.57 | 93.02 |
| 2 | 94.05 | 95.23 | 92.77 | 88.81 | |
| 3 | 93.07 | 96.73 | 90.27 | 87.06 | |
| 4 | 94.46 | 94.20 | 94.71 | 89.53 | |
| 5 | 94.42 | 92.26 | 96.26 | 89.46 | |
| Average | 94.48 ± 1.2 | 95.13 ± 2.0 | 93.92 ± 2.4 | 89.58 ± 2.2 | |
| Support Vector Machin (SVM) | 1 | 89.23 | 87.75 | 90.27 | 80.76 |
| 2 | 90.48 | 88.73 | 91.49 | 82.74 | |
| 3 | 87.62 | 87.37 | 88.07 | 78.30 | |
| 4 | 89.63 | 88.05 | 90.97 | 81.40 | |
| 5 | 89.60 | 85.79 | 93.23 | 81.32 | |
| Average | 89.31 ± 1.7 | 87.54 ± 1.1 | 90.81 ± 1.9 | 80.91 ± 1.62 |
| Model | Testing Set | Acc (%) | Sen (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|---|
| Guo [20] | Auto Covariance (ACC) | 89.33 ± 2.67 | 89.93 ± 3.68 | 88.87 ± 6.16 | N/A |
| auto covariance (AC) | 87.36 ± 1.38 | 87.30 ± 4.68 | 87.82 ± 4.33 | N/A | |
| Yang [23] | Cod1 | 75.08 ± 1.13 | 75.81 ± 1.20 | 74.75 ± 1.23 | N/A |
| Cod2 | 80.04 ± 1.06 | 76.77 ± 0.69 | 82.17 ± 1.35 | N/A | |
| Cod3 | 80.41 ± 0.47 | 78.14 ± 0.90 | 81.66 ± 0.99 | N/A | |
| Cod4 | 86.15 ± 1.17 | 81.03 ± 1.74 | 90.24 ± 1.34 | N/A | |
| You [24] | Principal Component Analysis-Ensemble Extreme Learning Machines (PCA-EELM) | 87.00 ± 0.29 | 86.15 ± 0.43 | 87.59 ± 0.32 | 77.36 ± 0.44 |
| Wong [30] | Rotation Forest (RF) + Property Response-Local Phase Quantization (PR-LPQ) | 93.92 ± 0.36 | 91.10 ± 0.31 | 96.45 ± 0.45 | 88.56 ± 0.63 |
| Proposed Method | PCVM | 94.48 ± 1.20 | 95.13 ± 2.00 | 93.92 ± 2.40 | 89.58 ± 2.20 |
| Model | Acc (%) | Sen (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|
| Nanni [23] | 83.00 | 86.00 | 85.10 | N/A |
| Nanni [32] | 84.00 | 86.00 | 84.00 | N/A |
| Nanni and Lumini [25] | 86.60 | 86.70 | 85.00 | N/A |
| Z-H You [29] | 87.50 | 88.95 | 86.15 | 78.13 |
| L Nanni [24] | 84.00 | 84.00 | 84.00 | N/A |
| Proposed Method | 91.25 | 92.05 | 90.06 | 84.04 |
| N | Moments | No. | N | Moments | No. |
|---|---|---|---|---|---|
| 1 | 1 | 7 | 4 | ||
| 2 | 1 | 8 | 4 | ||
| 3 | 2 | 9 | 5 | ||
| 4 | 2 | 10 | 5 | ||
| 5 | 3 | 11 | 6 | ||
| 6 | 3 | 12 | 6 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; You, Z.; Li, X.; Chen, X.; Jiang, T.; Zhang, J. PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences. Int. J. Mol. Sci. 2017, 18, 1029. https://doi.org/10.3390/ijms18051029
Wang Y, You Z, Li X, Chen X, Jiang T, Zhang J. PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences. International Journal of Molecular Sciences. 2017; 18(5):1029. https://doi.org/10.3390/ijms18051029
Chicago/Turabian StyleWang, Yanbin, Zhuhong You, Xiao Li, Xing Chen, Tonghai Jiang, and Jingting Zhang. 2017. "PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences" International Journal of Molecular Sciences 18, no. 5: 1029. https://doi.org/10.3390/ijms18051029