
Development of an Advanced HPLC–MS/MS Method for the Determination of Carotenoids and Fat-Soluble Vitamins in Human Plasma

 ,
,  and
and
Abstract
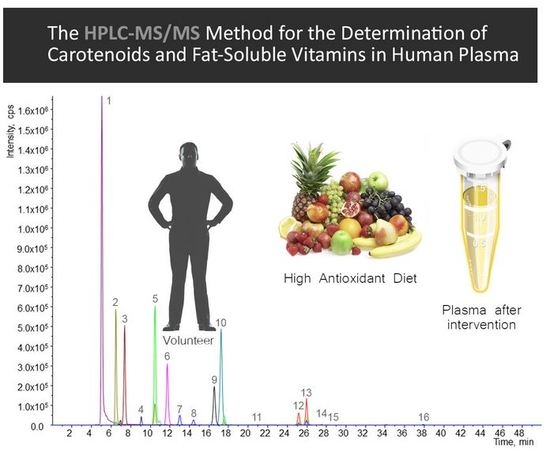
:
1. Introduction
2. Results
2.1. HPLC-MS/MS Method Development
2.1.1. Extraction of Carotenoids and Fat-Soluble Vitamins
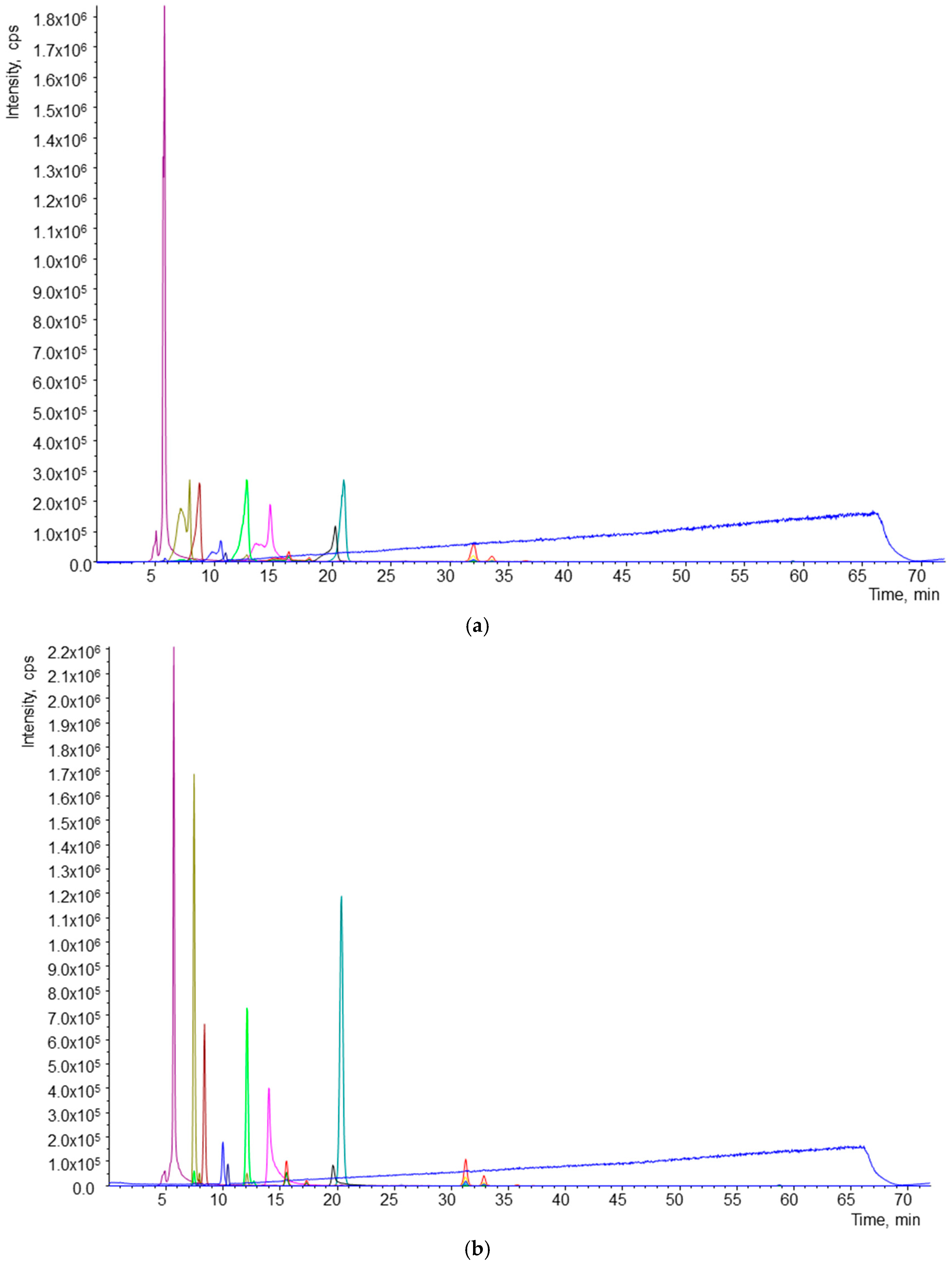
2.1.2. Optimization of Chromatographic and MS/MS Conditions
2.2. Validation of the Method
2.2.1. Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.2.2. Linearity
2.2.3. Recovery
2.2.4. Accuracy and Precision
2.2.5. Matrix Effect
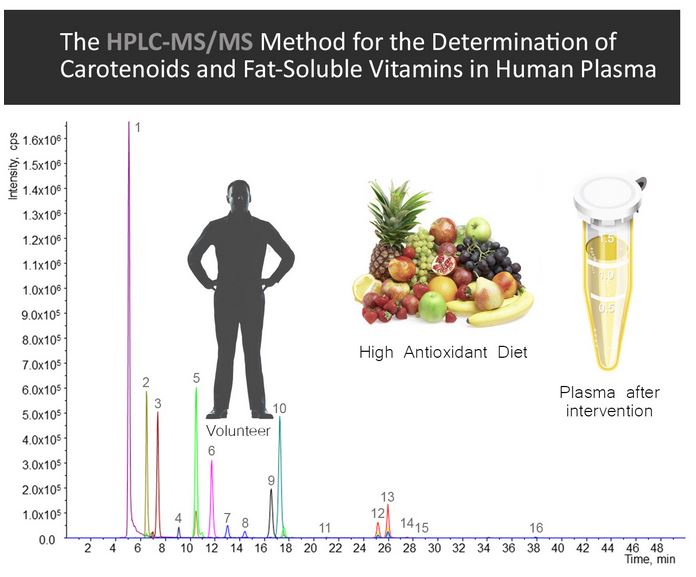
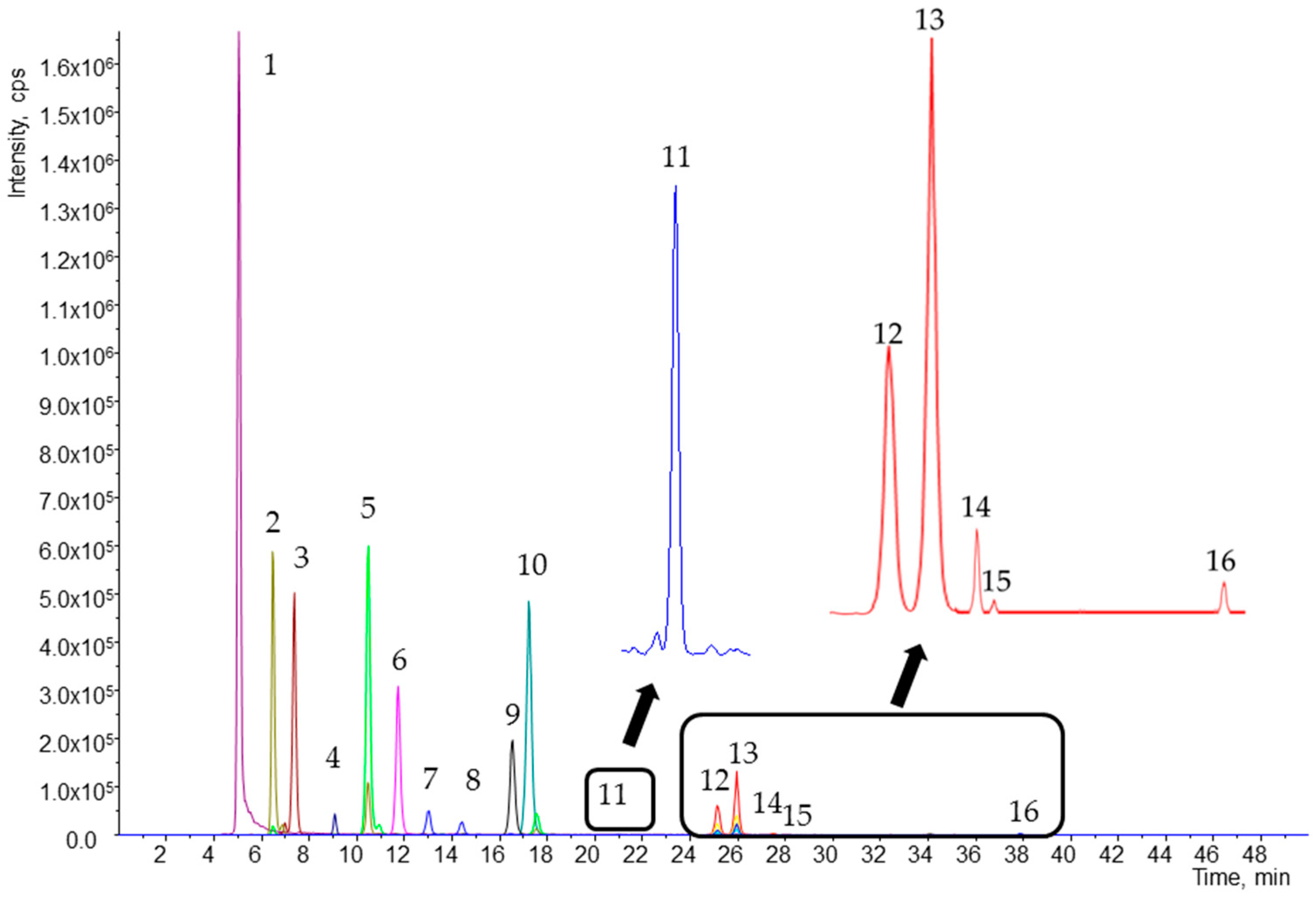
2.3. Quantification of Carotenoids and Fat-Soluble Vitamins in Human Plasma
3. Discussion
3.1. HPLC-MS/MS Method Development
3.1.1. Extraction of Carotenoids and Fat-Soluble Vitamins
3.1.2. Chromatographic and MS/MS Conditions
3.2. Method Validation
3.3. Quantification of Carotenoids and Fat-Soluble Vitamins in Human Plasma
4. Materials and Methods
4.1. Standards, Solvents and Reagents
Preparation of Standard Solutions
4.2. UHPLC-MS/MS Method Development
4.2.1. Instrumentation
4.2.2. Chromatographic Conditions
4.2.3. MS Conditions
4.2.4. Quality Parameters
4.3. Method Application to Real Samples: Human Dietary Intervention Study
Extraction of Carotenoids and Fat-Soluble Vitamins
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MeOH | Methanol |
| MTBE | Methyl-tert-butyl ether |
| BHT | Butylated hydroxy toluene |
| AMAC | Ammonium acetate |
| AA | Acetic acid |
| W | Water |
| CAD | Collision-activated dissociation |
| DP | Declustering potential |
| APCI | Atmospheric pressure chemical |
| EP | Entrance potential |
| CE | Collision energy |
| HPLC-MS/MS | High performance liquid chromatography coupled to mass spectrometry in tandem mode |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| MRM | Multiple reaction monitoring |
| ME | Matrix effect |
| MS | Mass spectrometry |
| CXP | Cell exit potential |
| RSD | Relative standard deviation |
| RT | Retention time |
References
- Maiani, G.; Periago Castón, M.J.; Catasta, G.; Toti, E.; Cambrodón, I.G.; Bysted, A.; Granado-Lorencio, F.; Olmedilla-Alonso, B.; Knuthsen, P.; Valoti, M.; et al. Carotenoids: Actual knowledge on food sources, intakes, stability and bioavailability and their protective role in humans. Mol. Nutr. Food Res. 2009, 53, S194–S218. [Google Scholar] [CrossRef] [PubMed]
- Granado, F.; Olmedilla, B.; Blanco, I. Nutritional and clinical relevance of lutein in human health. Br. J. Nutr. 2003, 90, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Balvers, M.G.J.; Brouwer-Brolsma, E.M.; Endenburg, S.; de Groot, L.C.P.G.M.; Kok, F.J.; Gunnewiek, J.K. Recommended intakes of vitamin D to optimise health, associated circulating 25-hydroxyvitamin D concentrations, and dosing regimens to treat deficiency: Workshop report and overview of current literature. J. Nutr. Sci. 2015, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Romney, S.L.; Palan, P.R.; Basu, J.; Mikhail, M. Nutrient antioxidants in the pathogenesis and prevention of cervical dysplasias and cancer. J. Cell Biochem. 1995, 59, S96–S103. [Google Scholar] [CrossRef]
- Perraud, A.; Nouaille, M.; Akil, H.; Petit, D.; Labrousse, F.; Jauberteau, M.; Mathonnet, M. Retinoid acid receptors in human colorectal cancer: An unexpected link with patient outcome. Exp. Ther. Med. 2011, 3, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Scientific Report of the 2015 Dietary Guidelines Advisory Committee. Available online: http://health.gov/dietaryguidelines/2015-scientific-report/pdfs/scientific-report-of-the-2015-dietary-guidelines-advisory-committee.pdf (accessed on 20 June 2016).
- Food Based Dietary Guidelines in the WHO European Region. Available online: http://www.euro.who.int/__data/assets/pdf_file/0017/150083/E79832.pdf (accessed on 20 June 2016).
- Stahl, W.; Sies, H. Bioactivity and protective effects of natural carotenoids. BBA Mol. Basis Dis. 2005, 1740, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Amaya, D.B. A Guide to Carotenoid Analysis in Foods; ILSI Press: Washington, DC, USA, 2001; pp. 1–22. [Google Scholar]
- Su, C.-C.; Chan, C.-M.; Chen, H.-M.; Wu, C.-C.; Hsiao, C.-Y.; Lee, P.-L.; Lin, V.C.-H.; Hung, C.-F. Lutein Inhibits the Migration of Retinal Pigment Epithelial Cells via Cytosolic and Mitochondrial Akt Pathways (Lutein Inhibits RPE Cells Migration). Int. J. Mol. Sci. 2014, 15, 13755–13767. [Google Scholar] [CrossRef] [PubMed]
- Gómez-García, M.R.; Ochoa-Alejo, N. Biochemistry and Molecular Biology of Carotenoid Biosynthesis in Chili Peppers (Capsicum spp.). Int. J. Mol. Sci. 2013, 14, 19025–19053. [Google Scholar] [CrossRef] [PubMed]
- Laddomada, B.; Durante, M.; Minervini, F.; Garbetta, A.; Cardinali, A.; D’Antuono, I.; Caretto, S.; Blanco, A.; Mita, G. Phytochemical Composition and Anti-Inflammatory Activity of Extracts from the Whole-Meal Flour of Italian Durum Wheat Cultivars. Int. J. Mol. Sci. 2015, 16, 3512–3527. [Google Scholar] [CrossRef] [PubMed]
- Nishino, A.; Ichihara, T.; Takaha, T.; Kuriki, T.; Nihei, H.; Kawamoto, K.; Yasui, H.; Maoka, T. Accumulation of Paprika Carotenoids in Human Plasma and Erythrocytes. J. Oleo Sci. 2015, 64, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Bernaldo de Quirós, A.; Costa, H.S. Analysis of carotenoids in vegetable and plasma samples: A review. J. Food Comp. Anal. 2006, 19, 97–111. [Google Scholar] [CrossRef]
- Bertram, J.S. Carotenoids and Gene Regulation. Nutr. Rev. 1999, 57, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Eldridge, A.L.; Peters, J.C. Dietary carotenoids and lung cancer: A review of recent research. Nutr. Rev. 1999, 57, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Palace, V.P.; Khaper, N.; Qin, Q.; Singal, P.K. Antioxidant potentials of vitamin A and carotenoids and their relevance to heart disease. Free Radic. Biol. Med. 1999, 26, 746–761. [Google Scholar] [CrossRef]
- Mostafa, W.Z.; Hegazy, R.A. Vitamin D and the skin: Focus on a complex relationship: A review. J. Adv. Res. 2015, 6, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E. Absorption of vitamin A and carotenoids by the enterocyte: Focus on transport proteins. Nutrients 2013, 5, 3563–3581. [Google Scholar] [CrossRef] [PubMed]
- Albarhani, A.A.; Collier, F.; Greaves, R.F.; Ponsonby, A.-L.; Allen, K.J.; Vuillermin, P.J.; Roche, P.; Clarke, M.W. Vitamins D and A can be successfully measured by LC–MS/MS in cord blood diluted plasma. Clin. Biochem. 2015, 48, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M. Development of a rapid, simple assay of plasma total carotenoids. BMC Res. Notes 2012, 5, 521. [Google Scholar] [CrossRef] [PubMed]
- Arathi, B.P.; Sowmya, P.R.R.; Vijay, K.; Dilshad, P.; Saikat, B.; Gopal, V.; Lakshminarayana, R. An Improved Method of UPLC-PDA-MS/MS Analysis of Lycopene Isomers. Food Anal. Methods 2015, 8, 1962–1969. [Google Scholar] [CrossRef]
- Lee, B.L.; New, A.L.; Ong, C.N. Simultaneous determination of tocotrienols, tocopherols, retinol, and major carotenoids in human plasma. Clin. Chem. 2003, 49, 2056–2066. [Google Scholar] [CrossRef] [PubMed]
- Colmán-Martínez, M.; Martínez-Huélamo, M.; Miralles, E.; Estruch, R.; Lamuela-Raventós, R.M. A new method to simultaneously quantify the antioxidants: Carotenes, xanthophylls, and vitamin A in human plasma. Oxid. Med. Cell Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kopec, R.E.; Schweiggert, R.M.; Riedl, K.M.; Carle, R.; Schwartz, S.J. Comparison of high-performance liquid chromatography/tandem mass spectrometry and high-performance liquid chromatography/photo-diode array detection for the quantitation of carotenoids, retinyl esters, α-tocopherol and phylloquinone in chylomicron-rich fr: Fat-soluble compounds from chylomicron fractions. Rapid Commun. Mass Spectrom. 2013, 27, 1393–1402. [Google Scholar] [PubMed]
- Van Meulebroek, L.; Vanden Bussche, J.; Steppe, K.; Vanhaecke, L. High-resolution Orbitrap mass spectrometry for the analysis of carotenoids in tomato fruit: Validation and comparative evaluation towards UV–VIS and tandem mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 2613–2626. [Google Scholar] [CrossRef] [PubMed]
- Vallverdú-Queralt, A.; Martínez-Huélamo, M.; Arranz-Martinez, S.; Miralles, E.; Lamuela-Raventós, R.M. Differences in the carotenoid content of ketchups and gazpachos through HPLC/ESI(Li+)–MS/MS correlated with their antioxidant capacity. J. Sci. Food Agric. 2012, 92, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.; Burri, B.J. Rapid Isocratic HPLC Method and Sample Extraction Procedures for Measuring Carotenoid, Retinoid, and Tocopherol Concentrations in Human Blood and Breast Milk for Intervention Studies. Chromatographia 2012, 75, 241–252. [Google Scholar] [CrossRef]
- Karppi, J.; Nurmia, T.; Olmedilla-Alonso, B.; Granado-Lorencio, F.; Nyyssönen, K. Simultaneous measurement of retinol, tocopherol and six carotenoids in human plasma by using an isocratic reversed-phase HPLC method. J. Chromatogr. B 2008, 867, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Arranz, S.; Martínez-Huélamo, M.; Vallverdu-Queralt, A.; Valderas-Martinez, P.; Illán, M.; Sacanella, E.; Escribano, E.; Estruch, R.; Lamuela-Raventos, R.M. Influence of olive oil on carotenoid absorption from tomato juice and effects on postprandial lipemia. Food Chem. 2015, 168, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Daykin, C.A.; Foxall, P.J.D.; Connor, S.C.; Lindon, J.C.; Nicholson, J.K. The comparison of plasma deproteinization methods for the detection of low-molecular-weight metabolites by 1H nuclear magnetic resonance spectroscopy. Anal. Biochem. 2002, 304, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Pajkovic, N.; Wang, Y.; Gu, C.; van Breemen, R.B. Quantitative Analysis of Lycopene Isomers in Human Plasma Using High-Performance Liquid Chromatography Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Bruce, S.J.; Tavazzi, I.; Parisod, V.; Rezzi, S.; Kochlar, S.; Guy, P.A. Investigation of human blood plasma sample preparation for performing metabolomics using ultrahigh performance liquid chromatography/mass spectrometry. Anal. Chem. 2009, 81, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Rowley, K.G.; Balazs, N.D.H. Carotenoids: Separation methods applicable to biological samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 731, 393–418. [Google Scholar] [CrossRef]
- Priego Capote, F.; Jiménez, J.R.; Granados, M.M.; de Castro, M.D.L. Identification and determination of fat-soluble vitamins and metabolites in human serum by liquid chromatography/triple quadrupole mass spectrometry with multiple reaction monitoring. Rapid Commun. Mass Spectrom. 2007, 21, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, A.; Uchikata, T.; Shinohara, M.; Nishiumi, S.; Yoshida, M.; Fukusaki, E.; Bamba, T. Highly sensitive and rapid profiling method for carotenoids and their epoxidized products using supercritical fluid chromatography coupled with electrospray ionization-triple quadrupole mass spectrometry. J. Biosci. Bioeng. 2012, 113, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Albahrani, A.A.; Greaves, R.F. Fat-soluble vitamins: Clinical indications and current challenges for chromatographic measurement. Clin. Biochem. Rev. 2016, 37, 27–47. [Google Scholar] [PubMed]
- Albahrani, A.A.; Rotarou, V.; Roche, P.J.; Greaves, R.F. A simultaneous quantitative method for vitamins A, D and E in human serum using liquid chromatography-tandem mass spectrometry. J. Steroid Biochem. Mol. Biol. 2016, 159, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Official Methods of Analysis of AOAC International. Available online: http://www.eoma.aoac.org/app_e.pdf (accessed on 15 April 2016).



{kind=link}
{kind=link}
{kind=link}
| Analyte | 0.4 g/L AMAC | 0.7 g/L AMAC | 1 g/L AMAC | 0.4 g/L AMAC + 0.1% AA | 0.7 g/L AMAC + 0.1% AA | 1 g/L AMAC + 0.1% AA |
|---|---|---|---|---|---|---|
| retinol | 2,200,000 | 1,400,000 | 1,700,000 | 1,600,000 | 2,400,000 | 1,800,000 |
| 25-hydroxycholecalciferol | 1,700,000 | 1,600,000 | 1,700,000 | 1,900,000 | 1,800,000 | 1,800,000 |
| retinol acetate | 660,000 | 640,000 | 600,000 | 720,000 | 630,000 | 700,000 |
| α-tocotrienol | 88,000 | 96,000 | 110,000 | 100,000 | 90,000 | 98,000 |
| cholecalciferol | 730,000 | 790,000 | 740,000 | 860,000 | 800,000 | 840,000 |
| astaxanthin | 400,000 | 480,000 | 530,000 | 370,000 | 500,000 | 500,000 |
| lutein | 100,000 | 140,000 | 130,000 | 140,000 | 140,000 | 140,000 |
| zeaxanthin | 1150 | 1883 | 1200 | 1800 | 1772 | 1925 |
| cantaxanthin | 86,000 | 190,000 | 150,000 | 170,000 | 200,000 | 180,000 |
| E-β-apo-8′-carotenal | 1,190,000 | 1,900,000 | 2,000,000 | 2,000,000 | 2,000,000 | 2,000,000 |
| cryptoxanthin | 6000 | 7950 | 6483 | 7500 | 8000 | 7317 |
| 13-Z-β-carotene | 108,000 | 100,000 | 89,000 | 107,000 | 110,000 | 100,000 |
| α-carotene | 40,000 | 40,000 | 32,000 | 40,000 | 43,000 | 37,000 |
| β-carotene | 5350 | 5000 | 4467 | 5000 | 5367 | 5242 |
| 9-Z-β-carotene | 1440 | 1400 | 1360 | 1100 | 1500 | 1500 |
| 5-Z-lycopene | 4000 | 3000 | 2942 | 3000 | 2800 | 2800 |
| Analyte | Rt (min) | DP (V) | EP (V) | CXP (V) | Quantification Transition | CE (eV) |
|---|---|---|---|---|---|---|
| retinol | 5.00 | 35 | 10 | 15 | 269 → 181 | 14 |
| 25-hydroxycholecalciferol | 6.45 | 58 | 10 | 15 | 383 → 365 | 17 |
| retinol acetate | 7.34 | 41 | 10 | 15 | 329 → 269 | 18 |
| α-tocotrienol | 9.03 | 181 | 10 | 15 | 411 → 165 | 57 |
| cholecalciferol | 10.45 | 60 | 10 | 15 | 385 → 367 | 24 |
| astaxanthin | 11.69 | 84 | 10 | 15 | 597 → 147 | 40 |
| lutein | 12.98 | 102 | 10 | 15 | 551 → 429 | 26 |
| zeaxanthin | 14.40 | 85 | 10 | 15 | 568 → 476 | 25 |
| cantaxanthin | 16.47 | 70 | 10 | 15 | 565 → 363 | 15 |
| E-β-apo-8′-carotenal | 17.16 | 70 | 10 | 15 | 417 → 325 | 14 |
| cryptoxanthin | 20.71 | 94 | 10 | 15 | 553 → 535 | 20 |
| 13-Z-β-carotene | 25.03 | 48 | 10 | 15 | 536 → 444 | 24 |
| α-carotene | 25.85 | 120 | 10 | 15 | 536 → 444 | 24 |
| β-carotene | 27.42 | 85 | 10 | 15 | 537 → 413 | 28 |
| 9-Z-β-carotene | 28.13 | 75 | 10 | 15 | 537 → 413 | 30 |
| 5-Z-lycopene | 37.65 | 87 | 10 | 15 | 537 → 413 | 23 |
| Analyte | LOD a (µg/mL) | LOQ b (µg /mL) | Rec. c (%) | ME d (%) | Conc. Range e (µg /mL) | (r2) f |
|---|---|---|---|---|---|---|
| retinol | 0.002 | 0.005 | 102.6 ± 8.9 | 108.5 ± 9.1 | 0.005–10 | 0.990 |
| 25-hydroxycholecalciferol | 0.003 | 0.011 | 92.0 ± 3.2 | 87.3 ± 1.4 | 0.011–5 | 0.995 |
| retinol acetate | 0.002 | 0.008 | 103.4 ± 1.5 | 95.1 ± 1.7 | 0.008–10 | 0.995 |
| α-tocotrienol | 0.113 | 0.376 | 99.0 ± 5.3 | 89.9 ± 3.4 | 0.376–5 | 0.992 |
| cholecalciferol | 0.005 | 0.018 | 102.9 ± 2.9 | 89.6 ± 1.3 | 0.018–10 | 0.998 |
| astaxanthin | 0.001 | 0.003 | 102.3 ± 2.6 | 100.1 ± 3.3 | 0.003–1 | 0.995 |
| lutein | 0.008 | 0.028 | 86.1 ± 1.4 | 91.0 ± 2.0 | 0.028–10 | 0.992 |
| zeaxanthin | 0.422 | 1.406 | 86.2 ± 2.2 | 86.9 ± 3.2 | 1.406–10 | 0.992 |
| cantaxanthin | 0.002 | 0.006 | 100.1 ± 2.5 | 103.5 ± 8.0 | 0.006–1 | 0.992 |
| E-β-apo-8′-carotenal | 0.003 | 0.010 | 103.6 ± 2.1 | 100.4 ± 1.4 | 0.010–10 | 0.994 |
| cryptoxanthin | 0.244 | 0.812 | 104.8 ± 3.2 | 94.0 ± 2.3 | 0.812–10 | 0.993 |
| 13-Z-β-carotene | 0.056 | 0.187 | 100.6 ± 2.0 | 87.8 ± 1.0 | 0.187–10 | 0.992 |
| α-carotene | 0.022 | 0.073 | 104.2 ± 5.4 | 102.8 ± 3.9 | 0.073–5 | 0.994 |
| β-carotene | 0.041 | 0.138 | 101.1 ± 2.2 | 95.7 ± 5.3 | 0.138–5 | 0.999 |
| 9-Z-β-carotene | 0.293 | 0.975 | 97.4 ± 6.7 | 91.5 ± 2.4 | 0.975–10 | 0.996 |
| 5-Z-lycopene | 0.189 | 0.631 | 104.6 ± 8.2 | 96.5 ± 3.3 | 0.631–10 | 0.998 |
| Concentration | LOW (n = 5) | MEDIUM (n = 5) | HIGH (n = 5) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Day 1 (RSD%) | Day 2 (RSD%) | Day 3 (RSD%) | Inter-Day (RSD%) | Day 1 (RSD%) | Day 2 (RSD%) | Day 3 (RSD%) | Inter-Day (RSD%) | Day 1 (RSD%) | Day 2 (RSD%) | Day 3 (RSD%) | Inter-Day (RSD%) |
| retinol | 11.5 | 8.4 | 3.6 | 3.8 | 6.0 | 6.0 | 6.7 | 6.7 | 9.5 | 9.2 | 6.8 | 3.4 |
| 25-hydroxycholecalciferol | 10.2 | 10.0 | 8.0 | 1.8 | 4.4 | 6.0 | 11.5 | 3.3 | 7.2 | 8.5 | 2.5 | 1.7 |
| retinol acetate | 8.6 | 7.0 | 5.7 | 0.7 | 9.2 | 6.4 | 6.8 | 1.3 | 8.3 | 11.1 | 3.6 | 0.7 |
| α-tocotrienol | 4.7 | 2.9 | 3.0 | 3.5 | 3.5 | 8.0 | 8.8 | 10.4 | 9.5 | 3.8 | 7.9 | 3.5 |
| cholecalciferol | 12.7 | 10.0 | 4.6 | 1.1 | 7.4 | 2.8 | 9.5 | 2.0 | 8.0 | 9.1 | 5.7 | 1.0 |
| astaxanthin | 9.9 | 1.0 | 7.5 | 2.7 | 9.5 | 4.7 | 8.8 | 8.8 | 6.2 | 7.0 | 10.3 | 4.0 |
| lutein | 12.1 | 9.7 | 4.5 | 4.1 | 5.8 | 5.2 | 9.6 | 2.5 | 6.2 | 11.8 | 5.9 | 2.0 |
| zeaxanthin | 7.3 | 1.9 | 2.2 | 5.5 | 9.0 | 9.3 | 7.5 | 4.8 | 8.5 | 10.8 | 5.1 | 0.7 |
| cantaxanthin | 11.5 | 3.3 | 7.0 | 6.2 | 11.1 | 2.9 | 9.7 | 6.2 | 8.5 | 11.0 | 9.9 | 2.9 |
| E-β-apo-8′-carotenal | 11.5 | 5.3 | 5.1 | 10.6 | 8.6 | 8.0 | 9.7 | 13.1 | 5.3 | 5.8 | 2.9 | 9.4 |
| cryptoxanthin | 5.2 | 7.6 | 8.2 | 4.4 | 10.4 | 10.1 | 8.3 | 1.5 | 6.9 | 14.0 | 4.2 | 0.3 |
| 13-Z-β-carotene | 10.8 | 3.1 | 4.6 | 3.1 | 4.4 | 10.6 | 10.0 | 9.1 | 12.3 | 13.6 | 13.1 | 4.2 |
| α-carotene | 5.5 | 9.8 | 9.4 | 5.9 | 0.7 | 5.3 | 10.2 | 5.0 | 7.1 | 12.1 | 8.1 | 5.7 |
| β-carotene | 8.9 | 12.2 | 5.7 | 2.4 | 8.1 | 3.7 | 2.5 | 3.4 | 10.5 | 2.9 | 7.8 | 1.8 |
| 9-Z-β-carotene | 7.0 | 10.0 | 5.1 | 1.3 | 3.3 | 4.0 | 14.0 | 4.9 | 8.0 | 9.7 | 3.8 | 4.9 |
| 5-Z-lycopene | 2.8 | 7.3 | 4.9 | 5.2 | 5.1 | 0.1 | 6.5 | 2.8 | 13.4 | 9.2 | 9.5 | 5.1 |
| Concentration | LOW (n = 5) | MEDIUM (n = 5) | HIGH (n = 5) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Day 1 (RSD%) | Day 2 (RSD%) | Day 3 (RSD%) | Inter-Day (RSD%) | Day 1 (RSD%) | Day 2 (RSD%) | Day 3 (RSD%) | Inter-Day (RSD%) | Day 1 (RSD%) | Day 2 (RSD%) | Day 3 (RSD%) | Inter-Day (RSD%) |
| retinol | 100.6 ± 11.4 | 97.7 ± 8.6 | 105.3 ± 3.6 | 101.2 ± 3.7 | 98.8 ± 6.0 | 104.4 ± 6.0 | 91.4 ± 6.7 | 98.2 ± 6.6 | 100.6 ± 9.5 | 97.8 ± 9.2 | 104.6 ± 6.8 | 101.0 ± 3.4 |
| 25-hydroxycholecalciferol | 99.2 ± 10.3 | 97.3 ± 10.1 | 100.7 ± 8.0 | 99.1 ± 1.7 | 101.6 ± 4.5 | 105.4 ± 5.9 | 98.8 ± 11.5 | 101.9 ± 3.3 | 99.2 ± 7.2 | 97.3 ± 8.5 | 100.7 ± 2.5 | 99.1 ± 1.7 |
| retinol acetate | 98.3 ± 8.5 | 97.1 ± 7.0 | 97.3 ± 5.6 | 97.5 ± 0.7 | 103.3 ± 9.2 | 106.0 ± 6.5 | 104.4 ± 7.0 | 104.6 ± 1.3 | 98.3 ± 8.3 | 97.0 ± 11.1 | 97.9 ± 3.6 | 97.7 ± 0.7 |
| α-tocotrienol | 100.8 ± 4.6 | 108.0 ± 2.8 | 104.0 ± 3.0 | 104.3 ± 3.5 | 96.4 ± 3.5 | 86.8 ± 1.7 | 87.7 ± 3.6 | 90.3 ± 5.9 | 100.6 ± 9.5 | 106.5 ± 3.8 | 104.0 ± 5.8 | 103.7 ± 2.9 |
| cholecalciferol | 97.9 ± 12.5 | 95.8 ± 9.8 | 96.8 ± 4.5 | 96.8 ± 1.0 | 103.9 ± 7.3 | 103.5 ± 11.0 | 101.6 ± 7.4 | 103.0 ± 1.2 | 97.9 ± 8.0 | 96.0 ± 9.1 | 97.6 ± 5.7 | 97.1 ± 1.0 |
| astaxanthin | 102.6 ± 9.8 | 105.3 ± 1.2 | 99.9 ± 7.5 | 102.6 ± 2.6 | 94.9 ± 9.7 | 92.1 ± 4.7 | 104.7 ± 5.6 | 97.2 ± 6.8 | 102.5 ± 6.2 | 103.7 ± 7.0 | 96.3 ± 10.3 | 100.8 ± 4.0 |
| lutein | 94.5 ± 8.2 | 97.0 ± 9.8 | 97.7 ± 4.5 | 96.4 ± 1.7 | 108.6 ± 5.8 | 105.2 ± 5.3 | 103.6 ± 9.5 | 105.8 ± 2.4 | 94.7 ± 6.2 | 97.5 ± 11.8 | 98.4 ± 5.9 | 96.9 ± 2.0 |
| zeaxanthin | 103.0 ± 7.2 | 103.8 ± 2.1 | 112.8 ± 1.7 | 106.5 ± 5.1 | 98.2 ± 9.0 | 97.2 ± 9.3 | 89.9 ± 7.4 | 95.1 ± 4.8 | 100.4 ± 8.5 | 100.5 ± 10.8 | 101.7 ± 5.1 | 100.9 ± 0.7 |
| cantaxanthin | 108.9 ± 11.4 | 104.8 ± 3.2 | 96.7 ± 7.1 | 103.5 ± 6.0 | 94.4 ± 10.9 | 92.6 ± 2.9 | 104.0 ± 9.7 | 97.0 ± 6.3 | 102.6 ± 8.5 | 108.0 ± 2.7 | 98.0 ± 9.9 | 102.9 ± 4.9 |
| E-β-apo-8′-carotenal | 111.3 ± 1.3 | 100.0 ± 5.1 | 96.6 ± 5.0 | 102.6 ± 7.5 | 95.2 ± 4.0 | 85.9 ± 4.1 | 103.3 ± 9.4 | 94.8 ± 9.2 | 109.0 ± 5.3 | 114.0 ± 2.3 | 97.0 ± 2.9 | 106.7 ± 8.2 |
| cryptoxanthin | 101.1 ± 5.2 | 94.7 ± 7.6 | 93.0 ± 8.2 | 96.2 ± 4.4 | 99.8 ± 10.6 | 101.6 ± 10.1 | 102.6 ± 8.4 | 101.3 ± 1.4 | 100.1 ± 6.9 | 99.8 ± 14.0 | 99.5 ± 4.2 | 99.8 ± 0.3 |
| 13-Z-β-carotene | 96.5 ± 10.8 | 102.3 ± 2.9 | 97.9 ± 4.8 | 98.9 ± 3.0 | 108.7 ± 4.3 | 91.0 ± 10.6 | 103.5 ± 10.0 | 101.1 ± 9.0 | 93.9 ± 12.3 | 109.0 ± 1.6 | 97.8 ± 13.1 | 100.2 ± 7.8 |
| α-carotene | 111.5 ± 0.6 | 105.0 ± 9.5 | 103.3 ± 9.2 | 106.6 ± 4.1 | 86.1 ± 0.7 | 93.9 ± 5.4 | 94.2 ± 10.3 | 91.4 ± 5.0 | 109.5 ± 2.6 | 104.1 ± 12.2 | 102.9 ± 8.5 | 105.3 ± 3.1 |
| β-carotene | 100.3 ± 9.0 | 96.8 ± 12.0 | 101.8 ± 5.7 | 99.6 ± 2.6 | 99.8 ± 7.9 | 102.6 ± 3.5 | 95.9 ± 2.5 | 99.4 ± 3.4 | 100.1 ± 10.5 | 97.6 ± 2.9 | 101.1 ± 7.7 | 99.6 ± 1.8 |
| 9-Z-β-carotene | 101.7 ± 7.0 | 99.2 ± 10.0 | 100.6 ± 5.1 | 100.5 ± 1.3 | 98.9 ± 3.0 | 107.2 ± 3.9 | 98.1 ± 14.3 | 101.4 ± 5.0 | 100.4 ± 8.0 | 92.1 ± 9.7 | 100.6 ± 3.8 | 97.7 ± 4.9 |
| 5-Z-lycopene | 97.3 ± 2.8 | 100.7 ± 7.3 | 90.7 ± 4.9 | 96.2 ± 5.2 | 103.5 ± 5.3 | 98.6 ± 0.1 | 103.5 ± 6.4 | 101.8 ± 2.8 | 96.8 ± 13.4 | 106.8 ± 9.2 | 99.5 ± 9.5 | 101.0 ± 5.1 |
| Analyte | Quantification Transition | In Plasma (nM) |
|---|---|---|
| retinol | 269 → 181 | 115.2 ± 10.5 |
| 25-hydroxycholecalciferol | 383 → 365 | 189.7 ± 32.5 |
| retinol acetate | 329 → 269 | <LOQ a |
| α-tocotrienol | 411 → 165 | n.d. b |
| cholecalciferol | 385 → 367 | n.d. b |
| astaxanthin | 597 → 147 | <LOQ a |
| lutein | 551 → 429 | 260.2 ± 138.9 |
| zeaxanthin | 568 → 476 | n.d. b |
| cantaxanthin | 565 → 363 | 28.32 ± 12.4 |
| E-β-apo-8′-carotenal | 417 → 325 | <LOQ a |
| cryptoxanthin | 553 → 535 | <LOQ a |
| 13-Z-β-carotene | 536 → 444 | n.d. b |
| α-carotene | 536 → 444 | 100.6 ± 18.6 |
| β-carotene | 537 → 413 | 2634.1 ± 1870.3 |
| 9-Z-β-carotene | 537 → 413 | n.d. b |
| 5-Z-lycopene | 537 → 413 | n.d. b |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrvolová, B.; Martínez-Huélamo, M.; Colmán-Martínez, M.; Hurtado-Barroso, S.; Lamuela-Raventós, R.M.; Kalina, J. Development of an Advanced HPLC–MS/MS Method for the Determination of Carotenoids and Fat-Soluble Vitamins in Human Plasma. Int. J. Mol. Sci. 2016, 17, 1719. https://doi.org/10.3390/ijms17101719
Hrvolová B, Martínez-Huélamo M, Colmán-Martínez M, Hurtado-Barroso S, Lamuela-Raventós RM, Kalina J. Development of an Advanced HPLC–MS/MS Method for the Determination of Carotenoids and Fat-Soluble Vitamins in Human Plasma. International Journal of Molecular Sciences. 2016; 17(10):1719. https://doi.org/10.3390/ijms17101719
Chicago/Turabian StyleHrvolová, Barbora, Miriam Martínez-Huélamo, Mariel Colmán-Martínez, Sara Hurtado-Barroso, Rosa Maria Lamuela-Raventós, and Jiří Kalina. 2016. "Development of an Advanced HPLC–MS/MS Method for the Determination of Carotenoids and Fat-Soluble Vitamins in Human Plasma" International Journal of Molecular Sciences 17, no. 10: 1719. https://doi.org/10.3390/ijms17101719