
Protective Effect of Tyrosol and S-Adenosylmethionine against Ethanol-Induced Oxidative Stress of Hepg2 Cells Involves Sirtuin 1, P53 and Erk1/2 Signaling

 ,
,
Abstract
:
1. Introduction
2. Results
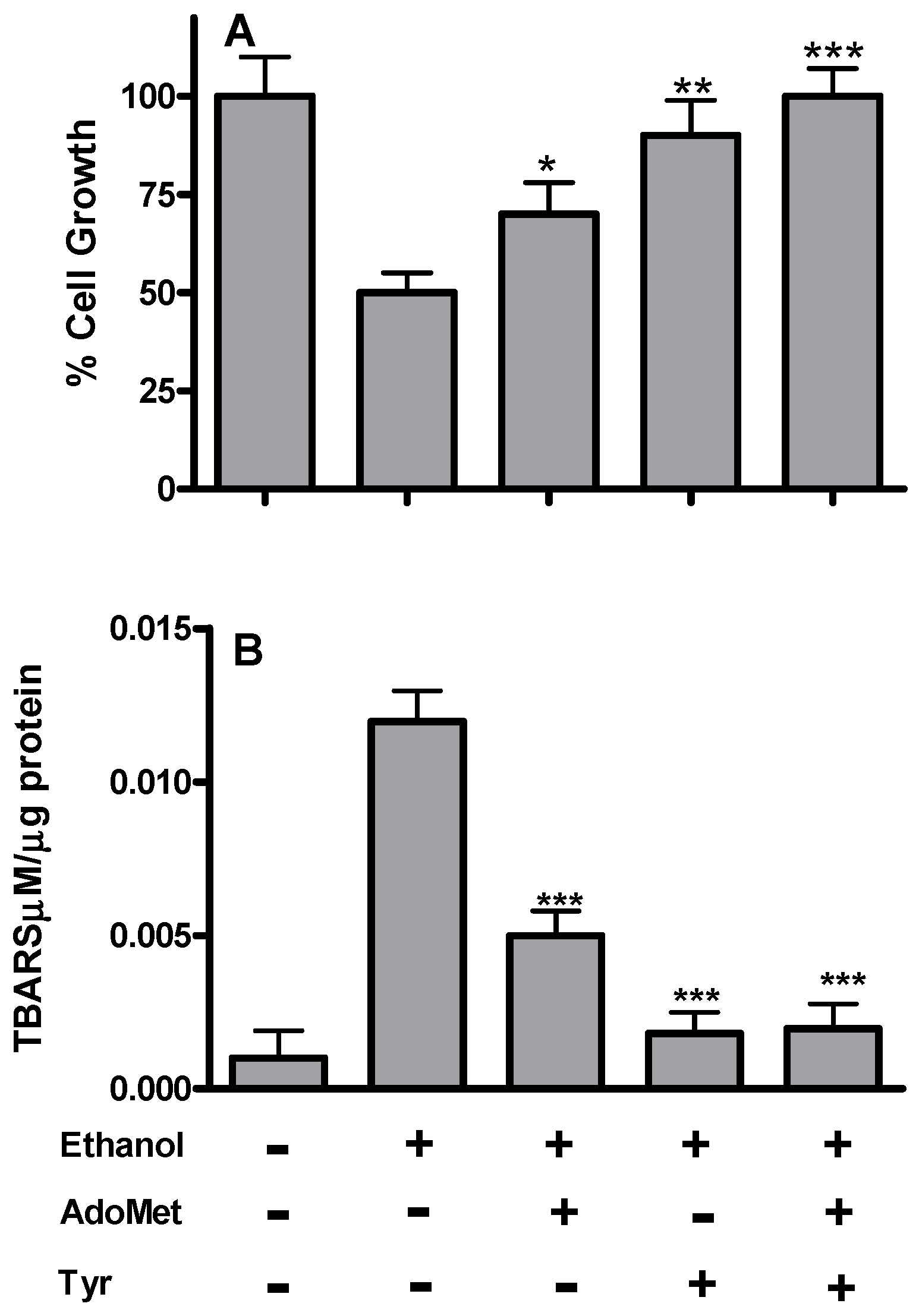
2.1. Tyrosol and AdoMet Affect Lipid Peroxidation and Fat Accumulation in HepG2 Cells Exposed to Acute Ethanol Treatment
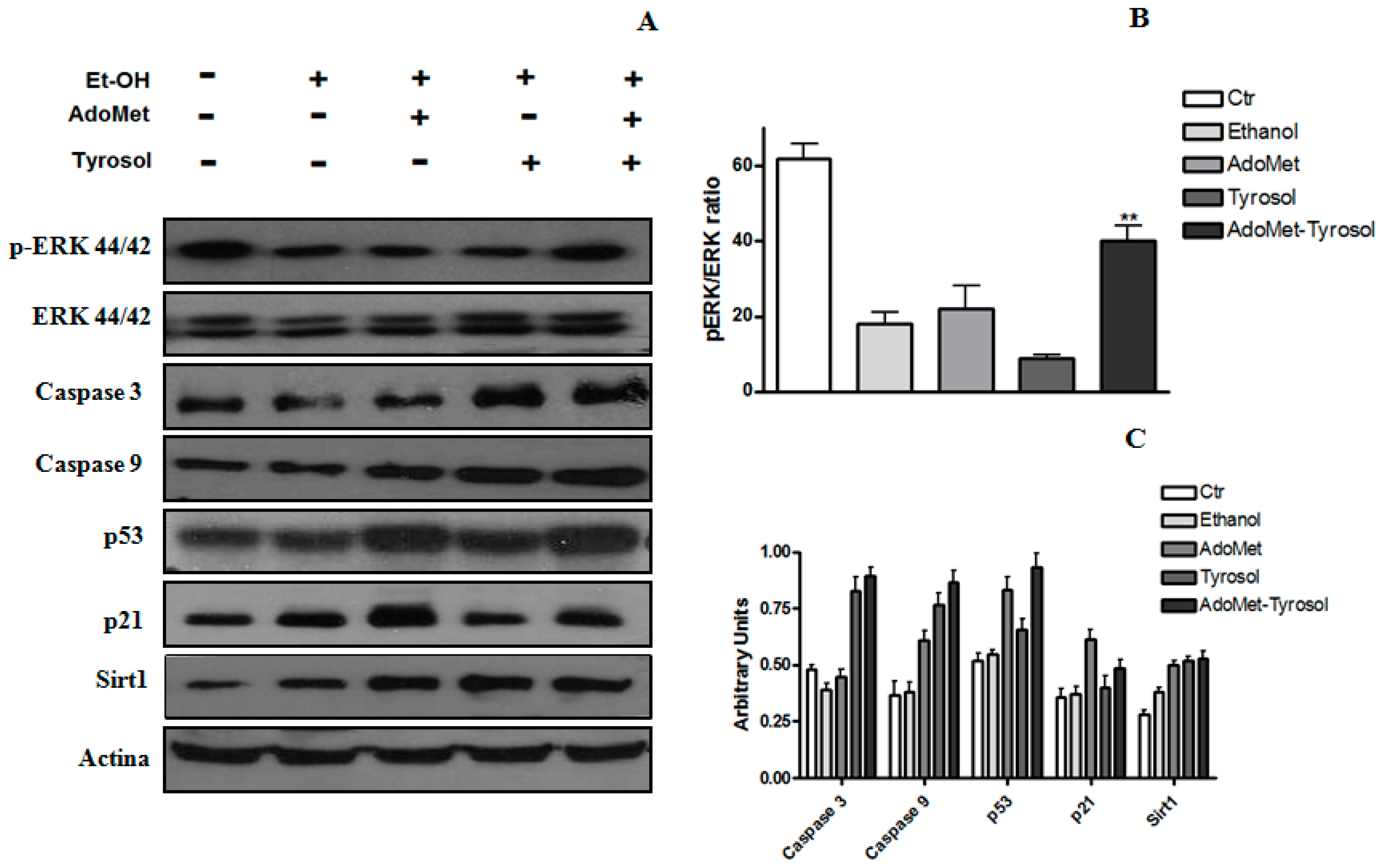
2.2. Tyrosol and AdoMet Affect SIRT1 Expression and Antagonize Ethanol-Induced SIRT1 Nucleo-Cytoplasmic Shuttling
2.3. AdoMet/Tyrosol Combination Activates Proliferative Pathways
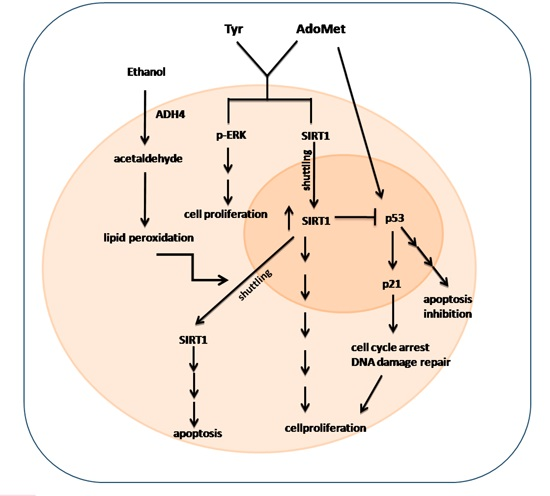
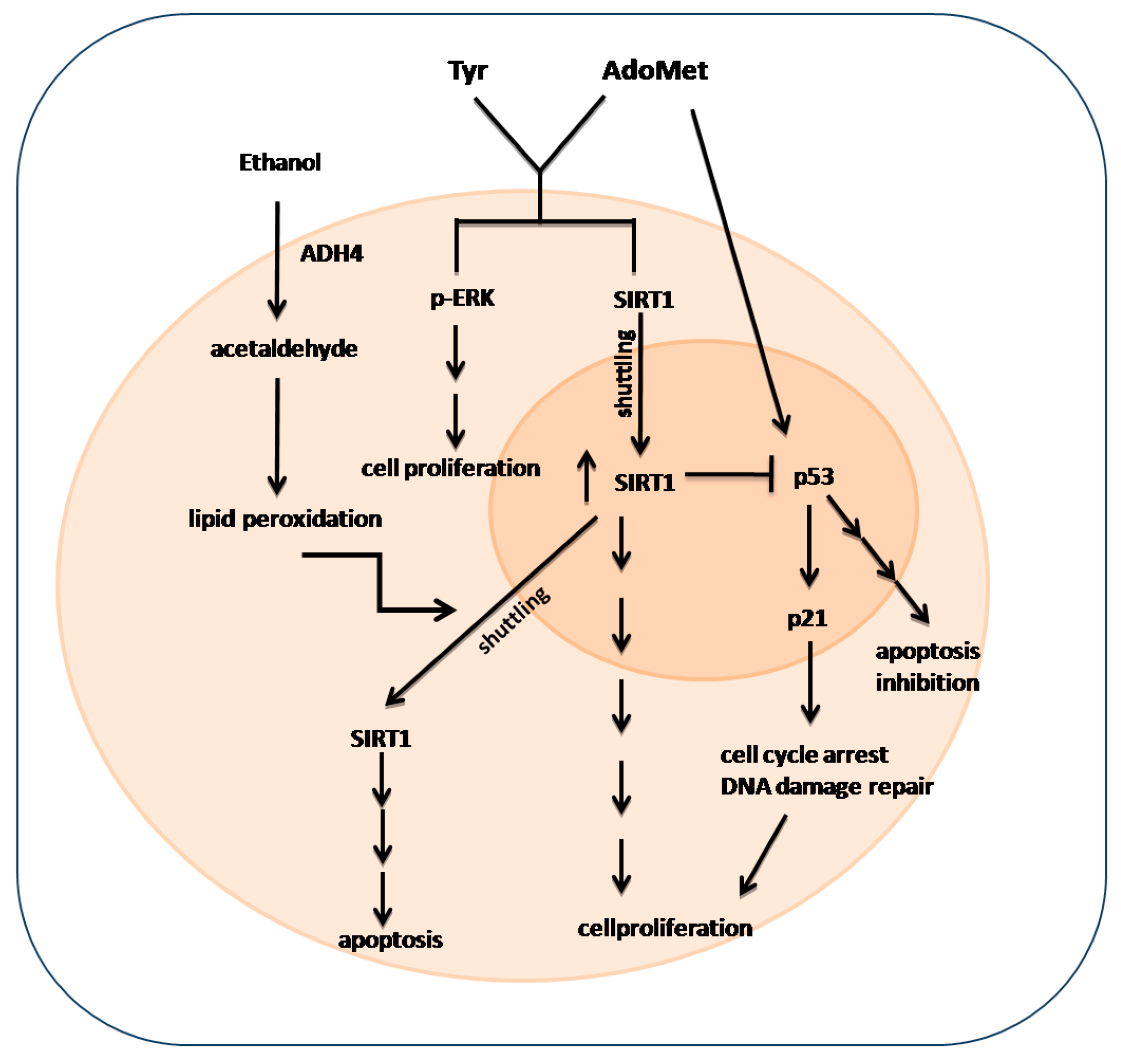
3. Discussion
4. Material and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Viability
4.4. Oil Red O Staining
4.5. Western Blot Analysis
4.6. Mitochondrial Superoxide Anion Levels
4.7. Thiobarbituric Acid-Reactive Species (TBARS) Levels
4.8. Laboratory Parameters
4.9. Immunostaining and Confocal Microscopy
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AdoMet | S-Adenosylmethionine |
| MAPK | mitogen-activated protein kinase |
| ERK1/2 | extracellular signal-regulated kinases 1 and 2 |
| Tyr | Tyrosol |
| ROS | reactive oxygen species |
| Hcy | homocysteine |
| ADH | alcohol dehydrogenase |
| BSA | bovine serum albumin |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| PBS | phosphate-buffered saline |
| FBS | fetal bovine serum |
| SDS | sodium dodecyl sulphate |
| TBARS | thiobarbituric acid-reactive species |
| HE | hydroethidine |
| AST | aspartate transaminase |
| ALT | alanine transaminase |
| G-GT | γ-glutamyltransferase |
| TG | triacylglycerol |
| CHO | total cholesterol |
| MSA | mithocondrial superoxide anions |
| ORO | Oil Red O |
| SIRT1 | Sirtuin 1 |
| pERK | phosphorylated ERK |
| HRP | horseradish peroxidase |
| MFIs | mean fluorescence intensity |
References
- Seth, D.; Haber, P.S.; Syn, W.K.; Diehl, A.M.; Day, C.P. Pathogenesis of alcohol-induced liver disease: Classical concepts and recent advances. J. Gastroenterol. Hepatol. 2011, 26, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Lim, R.W. Epigenetic effects of ethanol on the liver and gastrointestinal system. Alcohol Res. 2013, 35, 47–55. [Google Scholar] [PubMed]
- Zakhari, S. Alcohol metabolism and epigenetic changes. Alcohol Res. 2013, 35, 6–16. [Google Scholar] [PubMed]
- Osna, N.A. Epigenetic regulation in alcoholic liver disease. World J. Gastroenterol. 2011, 17, 2456–2464. [Google Scholar]
- Park, P.H.; Miller, R.; Shukla, S.D. Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochem. Biophys. Res. Commun. 2003, 306, 501–504. [Google Scholar] [CrossRef]
- Polavarapu, R.; Spitz, D.R.; Sim, J.E.; Follansbee, M.H.; Oberley, L.W.; Rahemtulla, A.; Nanji, A.A. Increased lipid peroxidation and impaired antioxidant enzyme function is associated with pathological liver injury in experimental alcoholic liver disease in rats fed diets high in corn oil and fish oil. Hepatology 1998, 27, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldheyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Farfán Labonne, B.E.; Gutiérrez, M.; Gómez-Quiroz, L.E.; Konigsberg Fainstein, M.; Bucio, L.; Souza, V.; Flores, O.; Ortíz, V.; Hernández, E.; Kershenobich, D.; et al. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol. Toxicol. 2009, 25, 599–609. [Google Scholar]
- Garro, A.J.; McBeth, D.L.; Lima, V.; Lieber, C.S. Ethanol consumption inhibits fetal DNA methylation in mice: Implications for the fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1991, 15, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Barak, A.J.; Beckenhauer, H.C.; Tuma, D.J. Methionine synthase a possible prime site of the ethanolic lesion in liver. Alcohol 2002, 26, 65–67. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Introduction-serial review: Alcohol, oxidative stress and cell injury. Free Radic. Biol. Med. 2001, 31, 1524–1526. [Google Scholar] [CrossRef]
- Choudury, M.; Park, P.H.; Jackson, D.; Shukla, S.D. Evidence for the role of oxidative stress in the acetylation of histone H3 by ethanol in rat hepatocytes. Alcohol 2010, 44, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Leo, M.A.; Wang, X.; Decarli, L.M. Effect of chronic alcohol consumption on hepatic SIRT1 and PGC-1α in rats. Biochem. Biophys. Res. Commun. 2008, 370, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Choi, S.W.; Kim, Y.I.; Bagley, P.J.; Seitz, H.K.; Russell, R.M.; Selhub, J.; Mason, J.B. Effect of chronic alcohol consumption on total plasma homocysteine level in rats. Alcohol. Clin. Exp. Res. 2000, 24, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K. Alcoholic liver disease and methionine metabolism. Semin. Liver Dis. 2009, 29, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Thompson, K.J.; McKillop, I.H.; Clemens, M.G.; Schrum, L.W. S-adenosyl-l-methionine attenuates oxidative stress and hepatic stellate cell activation in an ethanol-LPS-induced fibrotic rat model. Shock 2008, 30, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Del Pilar Cabrales-Romero, M.; Márquez-Rosado, L.; Fattel-Fazenda, S.; Trejo-Solís, C.; Arce-Popoca, E.; Alemán-Lazarini, L.; Villa-Treviño, S. S-adenosyl-methionine decreases ethanol-induced apoptosis in primary hepatocyte cultures by a c-Jun N-terminal kinase activity-independent mechanism. World J. Gastroenterol. 2006, 12, 1895–1904. [Google Scholar] [CrossRef]
- Kalaiselvan, I.; Dicson, S.M.; Kasi, P.D. Olive oil and its phenolic constituent Tyrosol attenuates dioxin-induced toxicity in peripheral blood mononuclear cells via an antioxidant-dependent mechanism. Nat. Prod. Res. 2015, 29, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Kalaiselvan, I.; Dicson, S.M.; Kasi, P.D. Protective effect of hydroxytyrosol and Tyrosol against oxidative stress in kidney cells. Toxicol. Ind. Health 2009, 25, 301–310. [Google Scholar]
- Sarna, L.K.; Sid, V.; Wang, P.; Siow, Y.L.; House, J.D.; Karmin, O. Tyrosol attenuates high fat diet-induced hepatic oxidative stress: Potential involvement of cystathionine β-synthase and cystathionine γ-lyase. Lipids 2015. [Google Scholar] [CrossRef] [PubMed]
- Manna, C.; Napoli, D.; Cacciapuoti, G.; Porcelli, M.; Zappia, V. Olive oil phenolic compounds inhibit homocysteine-induced endothelial cell adhesion regardless of their different antioxidant activity. J. Agric. Food Chem. 2009, 57, 3478–3482. [Google Scholar] [CrossRef] [PubMed]
- Au, A.Y.; Hasenwinkel, J.M.; Frondoza, C.G. Hepatoprotective effects of S-adenosylmethionine and silybin on canine hepatocytes in vitro. J. Anim. Physiol. Anim. Nutr. 2013, 97, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.G.; Bradford, B.U.; Iimuro, Y.; Frankenberg, M.V.; Knecht, K.T.; Connor, H.D.; Adachi, Y.; Wall, C.; Arteel, G.E.; Raleigh, J.A.; et al. Mechanisms of alcohol-induced hepatotoxicity: Studies in rats. Front. Biosci. 1999, 4, e42–e46. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcohol and the liver: Metabolism of alcohol and its role in hepatic and extrahepatic diseases. Mt. Sinai J. Med. 2000, 67, 84–94. [Google Scholar] [PubMed]
- Lieber, C.S. Alcoholic liver injury: Pathogenesis and therapy in 2001. Pathol. Biol. 2001, 49, 738–752. [Google Scholar] [CrossRef]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, F.; Kinne, R.K. Ethanol treatment of hepatocellular carcinoma: High potentials of low concentrations. Cancer Biol. Ther. 2004, 3, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Pochareddy, S.; Edenberg, H.J. Chronic alcohol exposure alters gene expression in HepG2 cells. Alcohol. Clin. Exp. Res. 2012, 36, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Pochareddy, S.; Edenberg, H.J. Identification of a FOXA-dependent enhancer of human alcohol dehydrogenase 4 (ADH4). Gene 2010, 460, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dahlhof, C.; Worsch, S.; Sailer, M.; Hummel, B.A.; Fiamoncini, J.; Uebel, K.; Obeid, R.; Scherling, C.; Geisel, J.; Bader, B.L.; et al. Methyl-donor supplementation in obese mice prevents the progression of NAFLD, activates AMPK and decreases acyl-carnitine levels. Mol. Metab. 2014, 3, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Hurley, T.D.; Edenberg, H.J.; Li, T.-K. Pharmacogenomics of Alcoholism. In Pharmacogenomics: The Search for Individualized Therapies; Licinio, J., Wong, M.-L., Eds.; Wiley-VCH: Weinheim, Germany; pp. 417–441.
- Sampey, B.P.; Stewart, B.J.; Petersen, D.R. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J. Biol. Chem. 2007, 282, 1925–1937. [Google Scholar] [CrossRef] [PubMed]
- Agoglia, A.E.; Sharko, A.C.; Psilos, K.E.; Holstein, S.E.; Reid, G.T.; Hodge, C.W. Alcohol alters the activation of ERK1/2, a functional regulator of binge alcohol drinking in adult C57BL/6J Mice. Alcohol. Clin. Exp. Res. 2015, 39, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Fusco, S.; Colavitti, R.; Borrello, S.; Maggiano, N.; Cravero, A.A.; Farré, S.M.; Galeotti, T.; Koch, O.R. Abrogation of hepatocyte apoptosis and early appearance of liver dysplasia in ethanol-fed p53-deficient mice. Biochem. Biophys. Res. Commun. 2004, 325, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yan, T.; Ge, X.; Sun, C.; Shi, X.; Zhai, Q. Cytoplasm-localized SIRT1 enhances apoptosis. J. Cell. Physiol. 2007, 213, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar] [PubMed]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Hu, M.; Rogers, C.Q.; Shen, Z.; You, M. Role of SIRT1-FoxO1 signaling in dietary saturated fat-dependent upregulation of liver adiponectin receptor 2 in ethanol-administered mice. Antioxid. Redox Signal. 2011, 15, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, Y. p53, Oxidative Stress, and Aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, T.; Song, Z. Involvement and mechanism of DGAT2 upregulation in the pathogenesis of alcoholic fatty liver disease. J. Lipid Res. 2010, 51, 3158–3165. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 261–270. [Google Scholar] [CrossRef] [PubMed]
- De Maria, S.; Scognamiglio, I.; Lombardi, A.; Amodio, N.; Caraglia, M.; Cartenì, M.; Ravagnan, G.; Stiuso, P. Polydatin, a natural precursor of resveratrol, induces cell cycle arrest and differentiation of human colorectal Caco-2 cell. J. Transl. Med. 2013, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiuso, P.; Scognamiglio, I.; Murolo, M.; Ferranti, P.; de Simone, C.; Rizzo, M.R.; Tuccillo, C.; Caraglia, M.; Loguercio, C.; Federico, A. Serum oxidative stress markers and lipidomic profile to detect NASH patients responsive to an antioxidant treatment: A pilot study. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Monterrey, I.; Campiglia, P.; Scognamiglio, I.; Vanacore, D.; Dicitore, A.; Lombardi, A.; Caraglia, M.; Novellino, E.; Stiuso, P. DTNQ-Pro, a mimetic dipeptide, sensitizes human colon cancer cells to 5-fluorouracil treatment. J. Amino Acids 2013, 2013. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HepG2 | Et-HepG2 | Et-HepG2 + AdoMet | Et-HepG2 + Tyr | Et-HepG2 + AdoMet-Tyr | |
|---|---|---|---|---|---|
| AST/GOT | 0.10 ± 0.01 | 0.14 ± 0.02 | 0.12 ± 0.015 | 0.14 ± 0.02 | 0.11 ± 0.01 |
| ALT/GOT | 0.027 ± 0.003 | 0.031 ± 0.002 | 0.034 ± 0.003 | 0.050 ± 0.001 | 0.029 ± 0.002 |
| CHO | 0.34 ± 0.03 | 0.77 ± 0.015 | 0.37 ± 0.03 | 0.54 ± 0.04 | 0.34 ± 0.05 |
| TG | 0.44 ± 0.05 | 1.0 ± 0.21 | 0.88 ± 0.06 | 1.2 ± 0.1 | 0.70 ± 0.06 |
| Albumin g/L | 0.06 ± 0.03 | 0.35 ± 0.08 | 0.13 ± 0.009 | 0.30 ± 0.01 | 0.07 ± 0.003 |
| Ferritin µg/L | 0.90 ± 0.1 | 2.4 ± 0.3 | 1.4 ± 0.4 | 2.3 ± 0.2 | 1.1 ± 0.3 |
| Homocysteine µmol/L | 0.25 ± 0.06 | 0.56 ± 0.05 | 0.26 ± 0.03 | 0.54 ± 0.04 | 0.26 ± 0.037 |
| MSAs | ORO A520nm/Cells | |
|---|---|---|
| HepG2 | 9.3 ± 0.6 | 3.3 × 10−5 ± 0.1 × 10−5 |
| Et-HepG2 | 9.6 ± 0.57 | 6.8 × 10−5 ± 0.25 × 10−5 |
| Et-HepG2 + AdoMet | 11.8 ± 0.6 | 5.0 × 10−5 ± 0.34 × 10−5 |
| Et-HepG2 + Tyr | 9.0 ± 0.8 | 6.0 × 10−5 ± 0.29 × 10−5 |
| Et-HepG2 + AdoMet/Tyr | 8.5 ± 0.5 | 5.3 × 10−5 ± 0.37 × 10−5 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiuso, P.; Bagarolo, M.L.; Ilisso, C.P.; Vanacore, D.; Martino, E.; Caraglia, M.; Porcelli, M.; Cacciapuoti, G. Protective Effect of Tyrosol and S-Adenosylmethionine against Ethanol-Induced Oxidative Stress of Hepg2 Cells Involves Sirtuin 1, P53 and Erk1/2 Signaling. Int. J. Mol. Sci. 2016, 17, 622. https://doi.org/10.3390/ijms17050622
Stiuso P, Bagarolo ML, Ilisso CP, Vanacore D, Martino E, Caraglia M, Porcelli M, Cacciapuoti G. Protective Effect of Tyrosol and S-Adenosylmethionine against Ethanol-Induced Oxidative Stress of Hepg2 Cells Involves Sirtuin 1, P53 and Erk1/2 Signaling. International Journal of Molecular Sciences. 2016; 17(5):622. https://doi.org/10.3390/ijms17050622
Chicago/Turabian StyleStiuso, Paola, Maria Libera Bagarolo, Concetta Paola Ilisso, Daniela Vanacore, Elisa Martino, Michele Caraglia, Marina Porcelli, and Giovanna Cacciapuoti. 2016. "Protective Effect of Tyrosol and S-Adenosylmethionine against Ethanol-Induced Oxidative Stress of Hepg2 Cells Involves Sirtuin 1, P53 and Erk1/2 Signaling" International Journal of Molecular Sciences 17, no. 5: 622. https://doi.org/10.3390/ijms17050622