
Limitations and Extensions of the Lock-and-Key Principle: Differences between Gas State, Solution and Solid State Structures

Abstract
:
1. Introduction
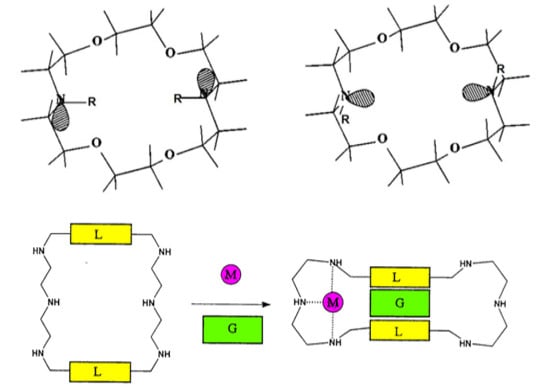
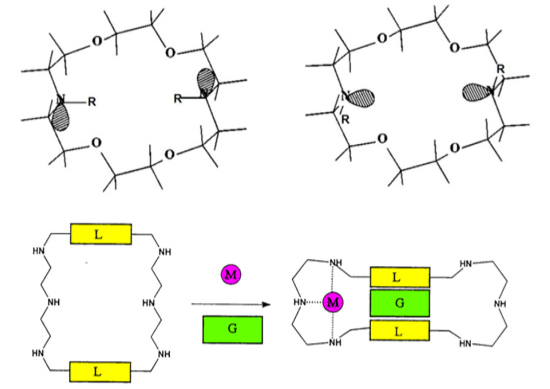
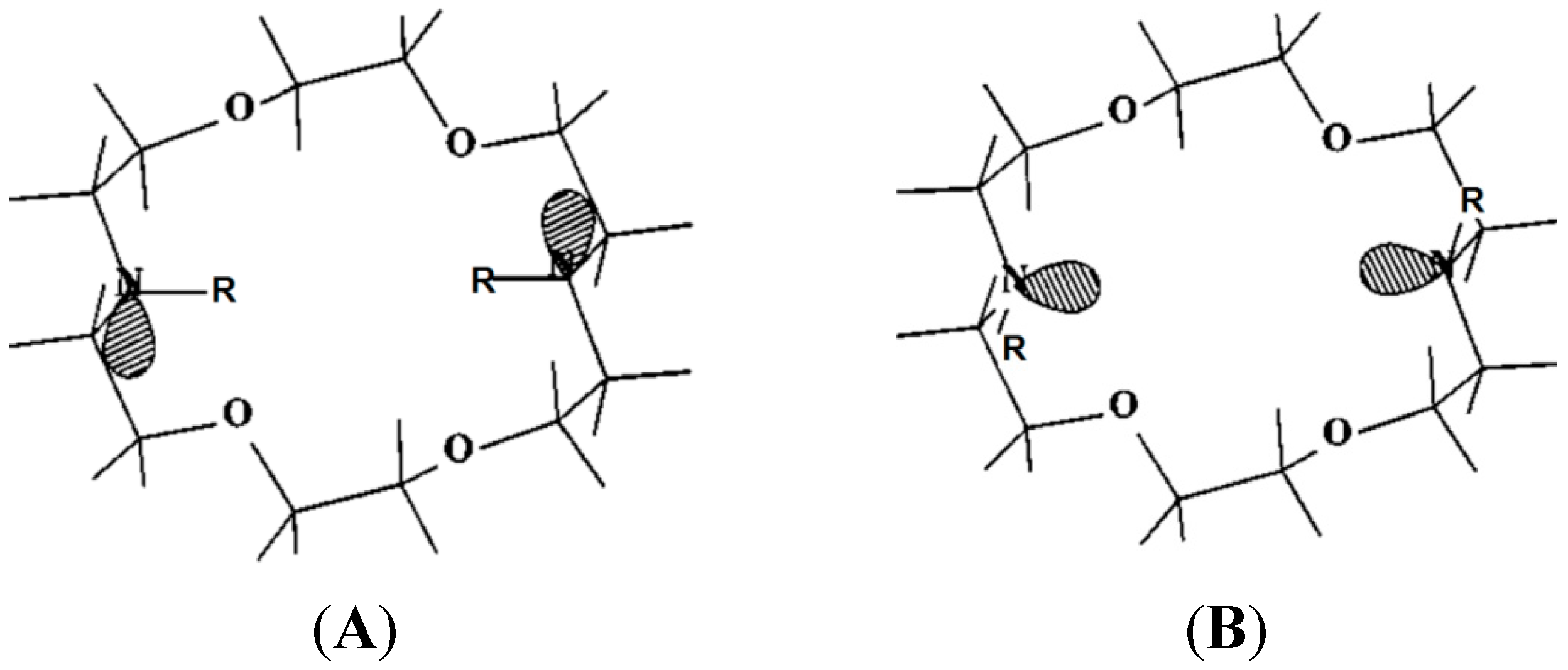
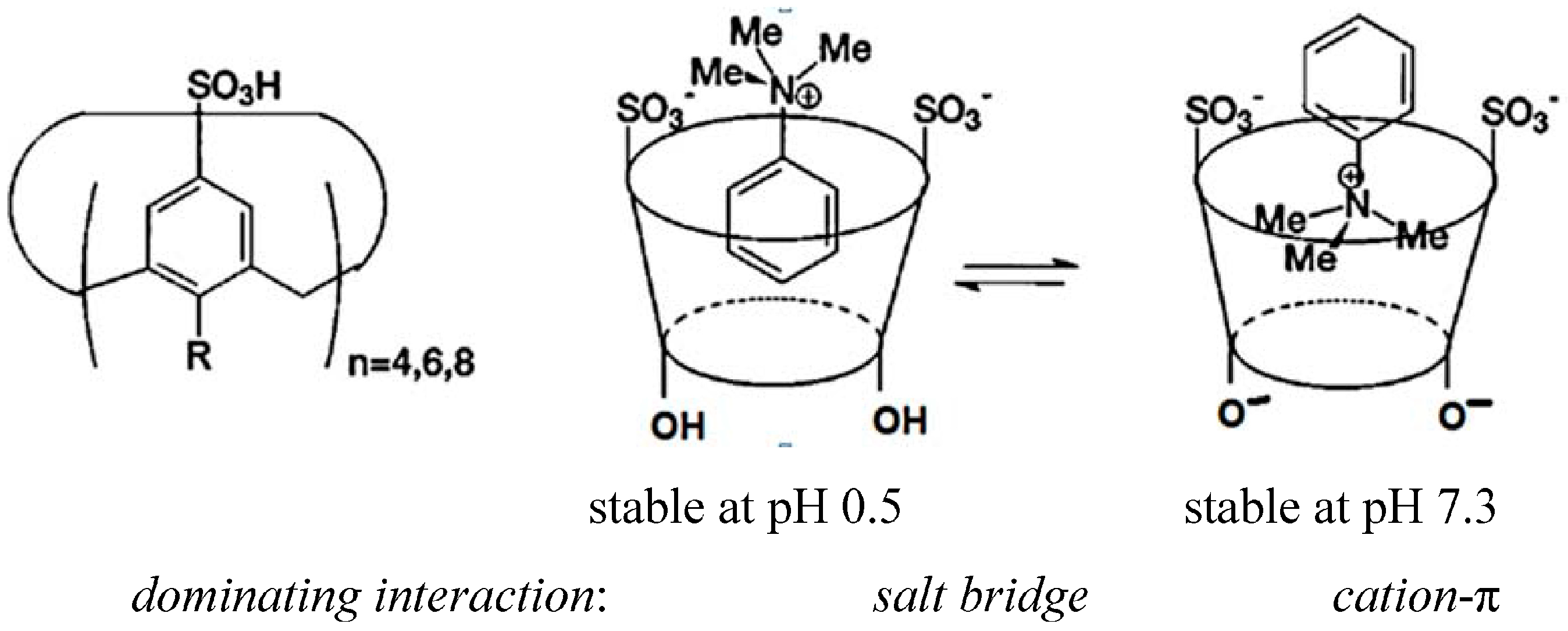
2. Dependence on the Binding Mechanism/Medium, pH and Stereoelectronic Effects


| KCl in H2O | KCl in MeOH | NaCl in H2O | NaCl in MeOH | |
|---|---|---|---|---|
| 18C6 ΔG | 11.6 | 34.5 | 4.6 | 25.0 |
| 18C5 ΔG | 7.5 | 15.9 | 4.5 | 14.0 |
| ΔΔG | 4.1 | 18.6 | 0.1 | 11.0 |



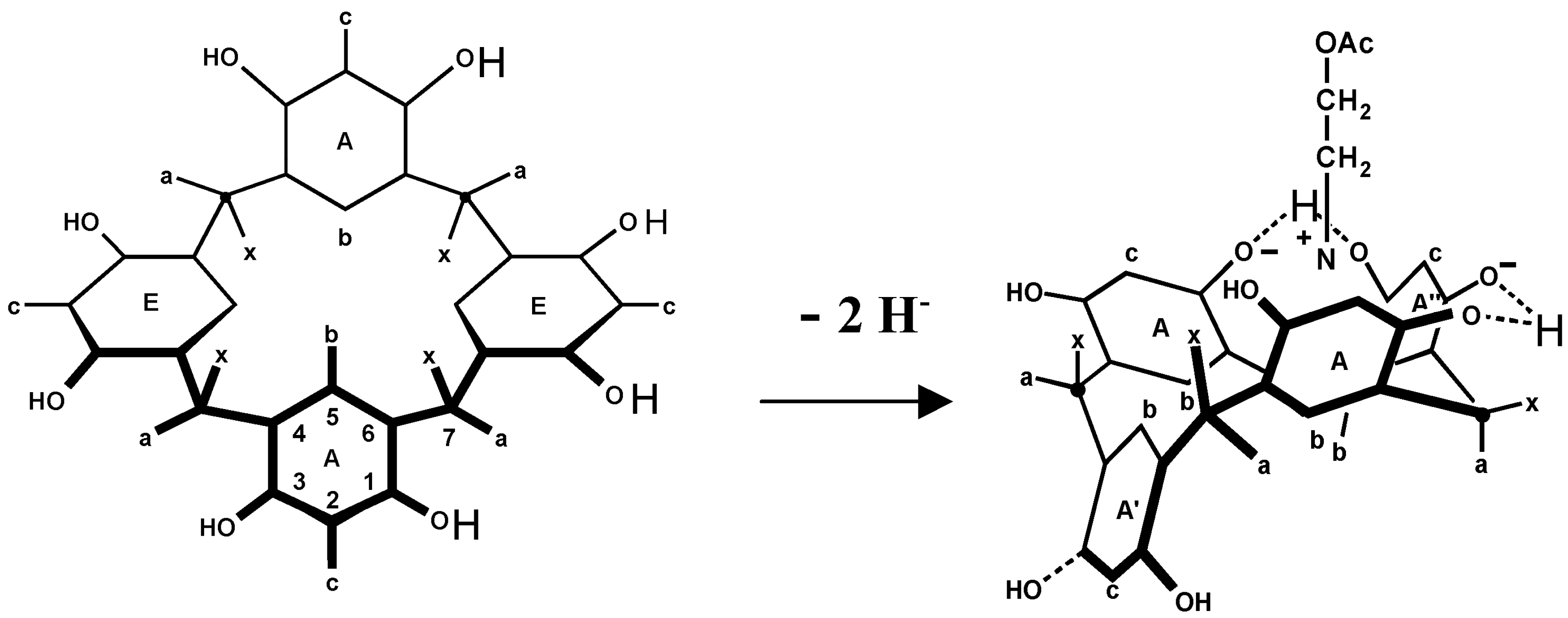
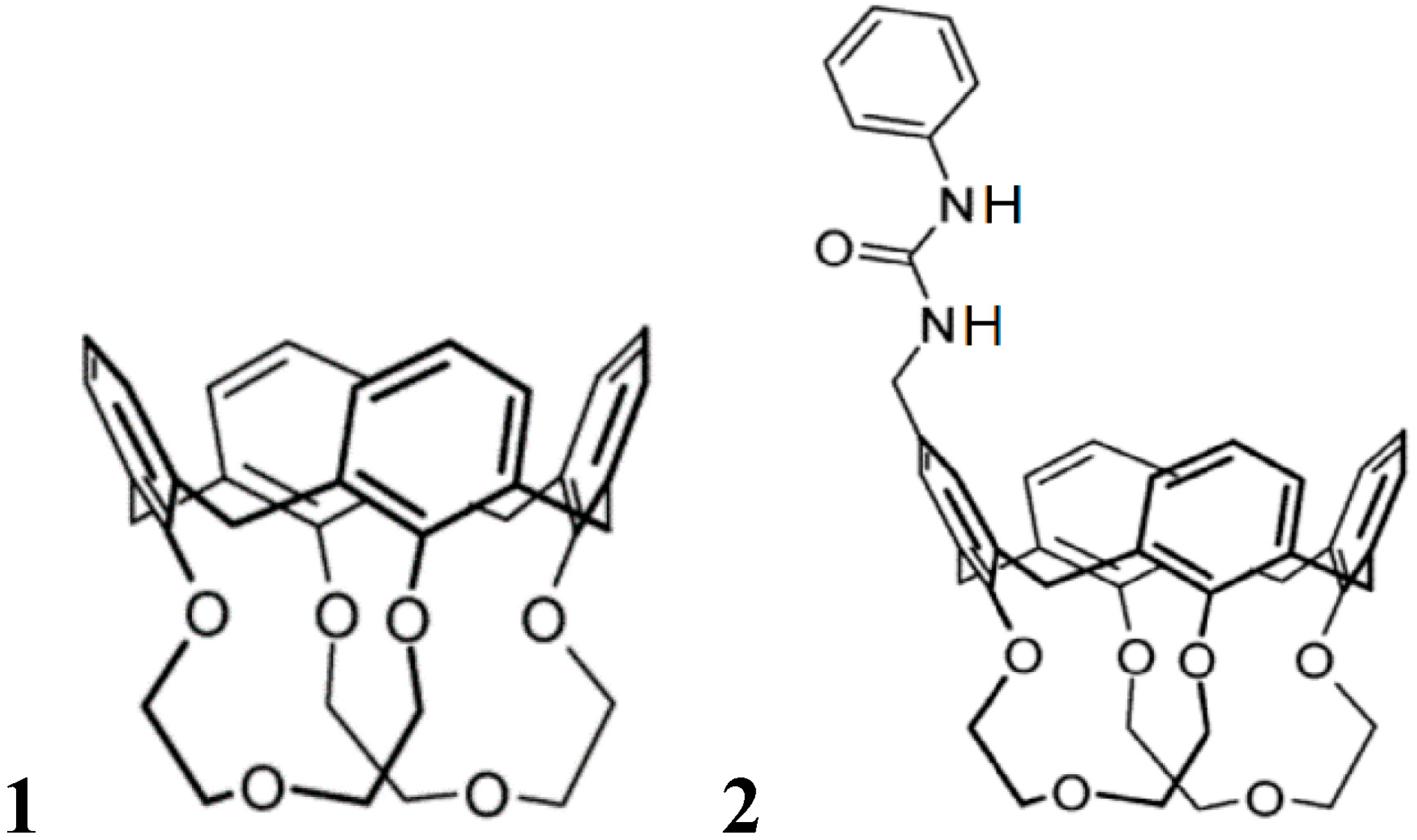
3. Induced Fit



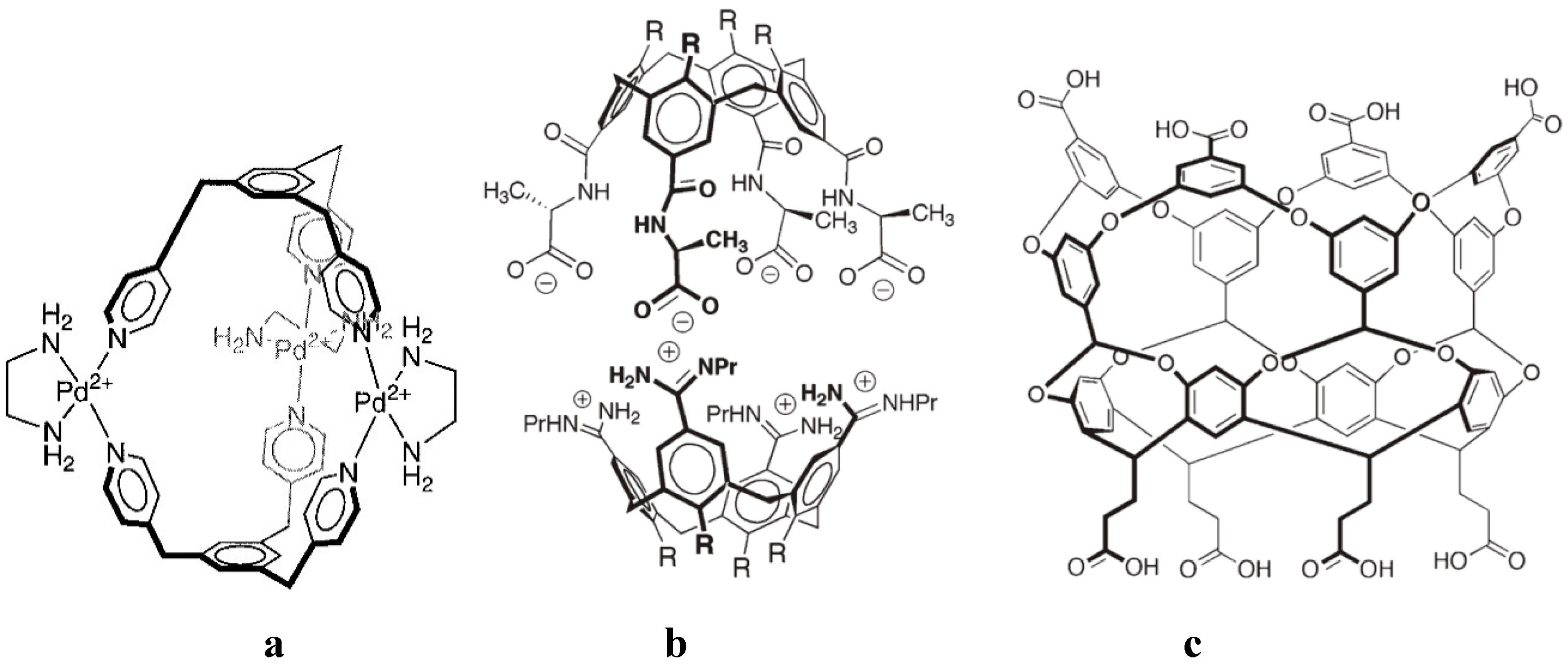
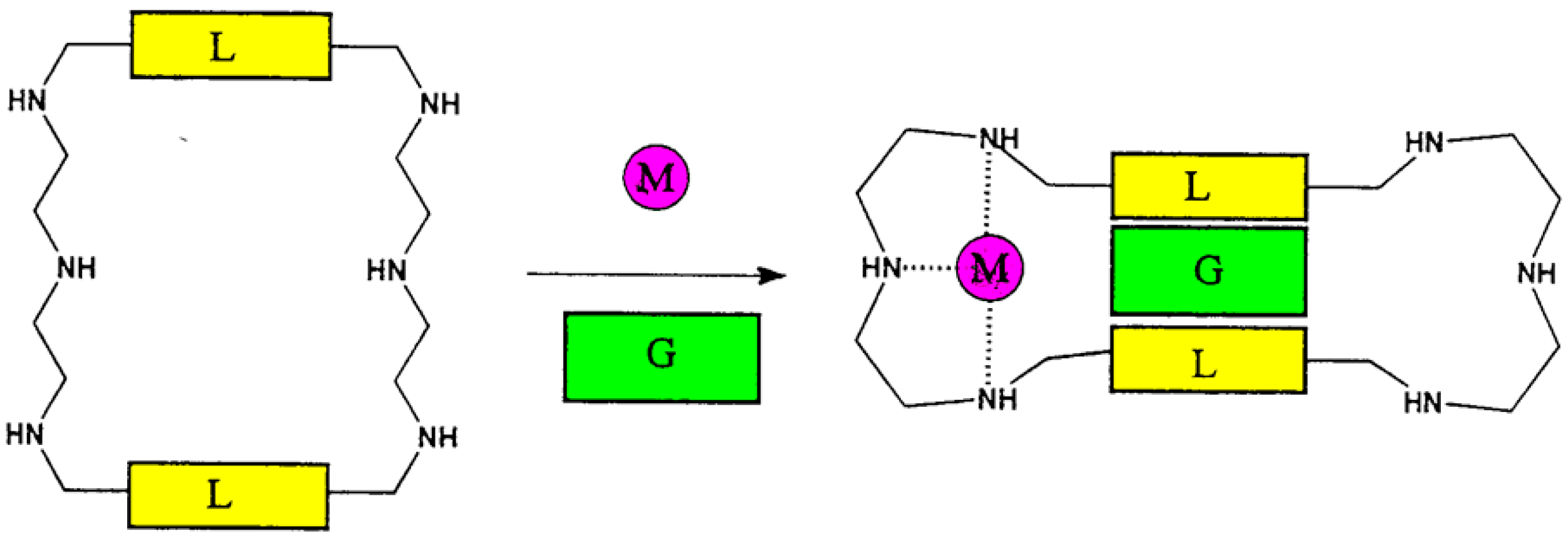
4. Allosteric Effects

| X | TsO | Cl | OAc | TFA |
|---|---|---|---|---|
| Host 1 | 33 | 80 | 250 | 360 |
| Host 2 | 700 | 8800 | 5000 | 13,000 |

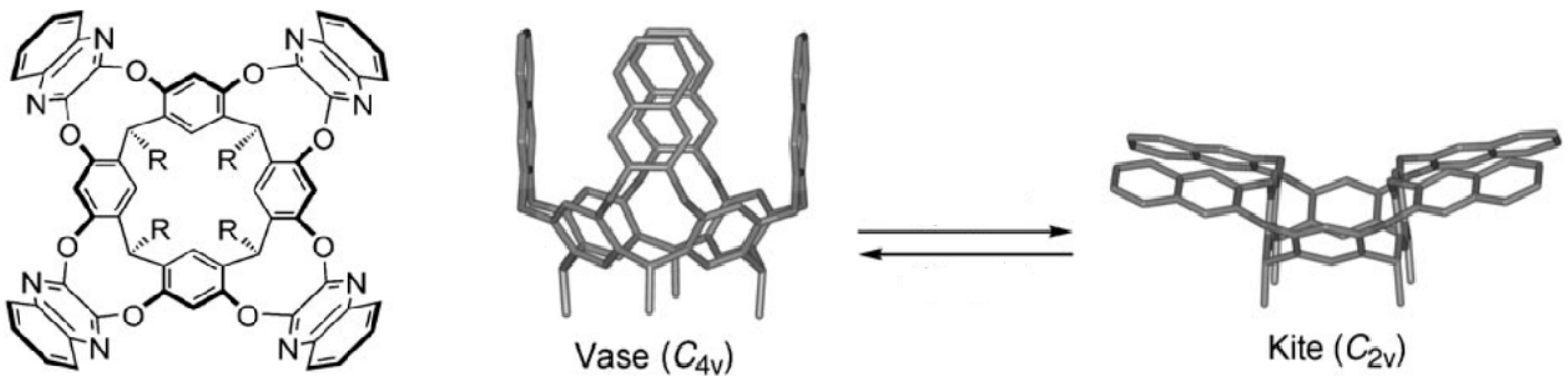
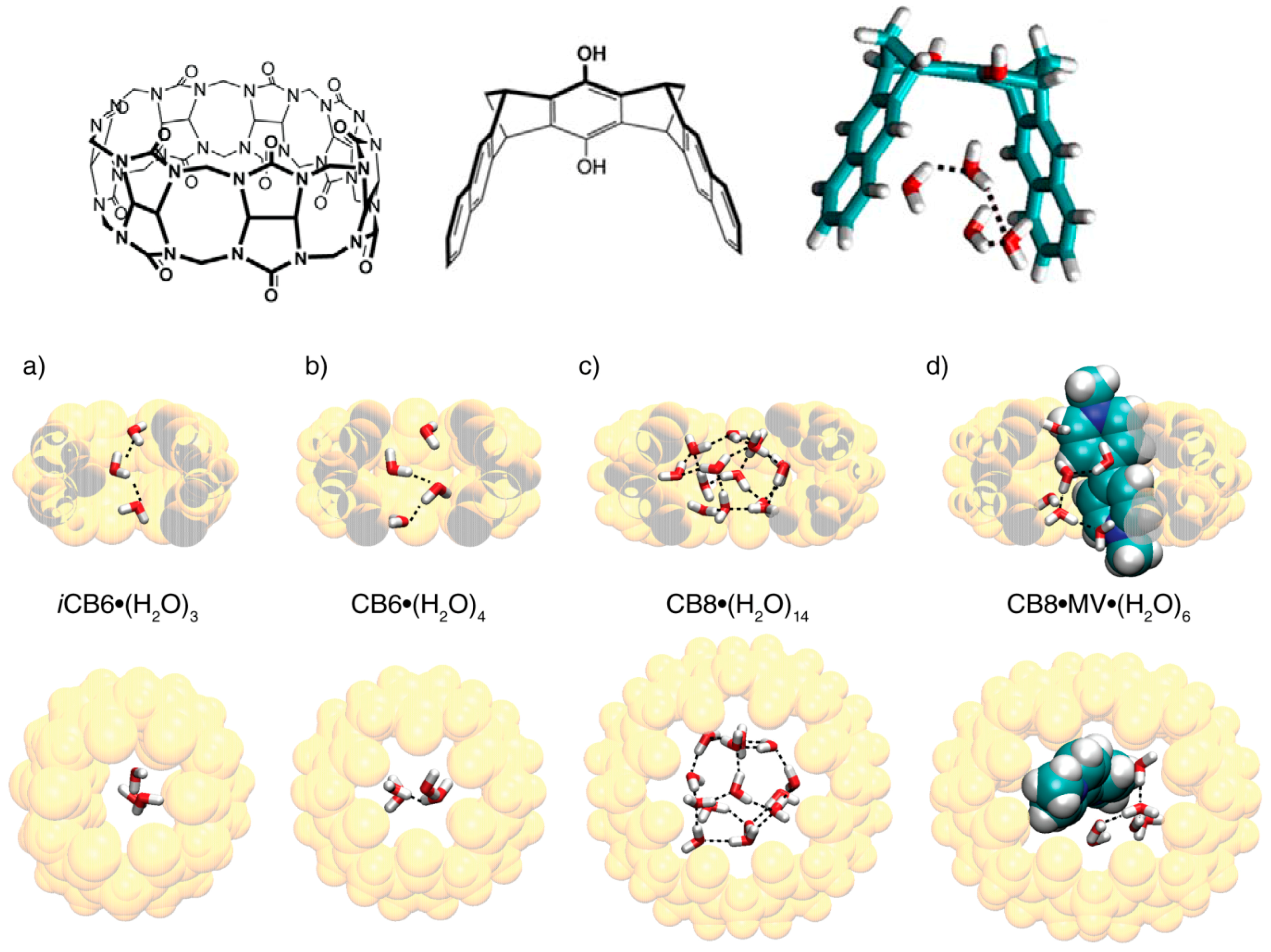
5. Hydrophobic Interactions beyond the Lock-and-Key Picture

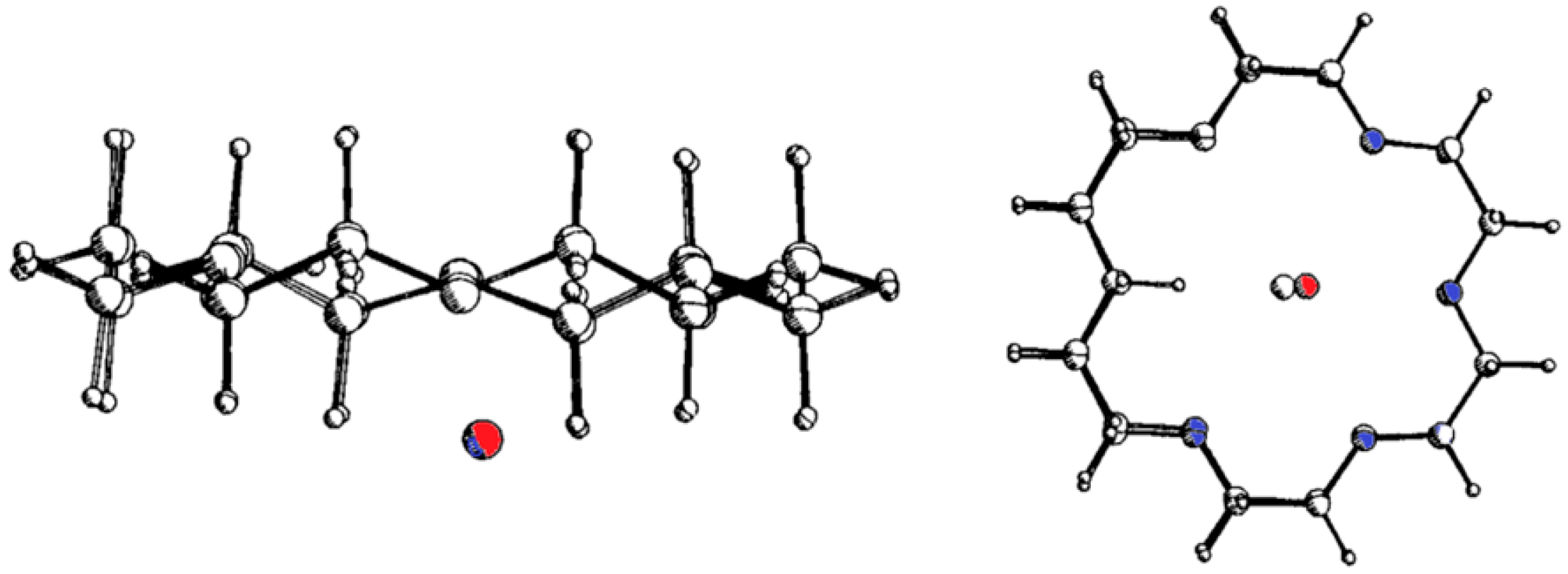
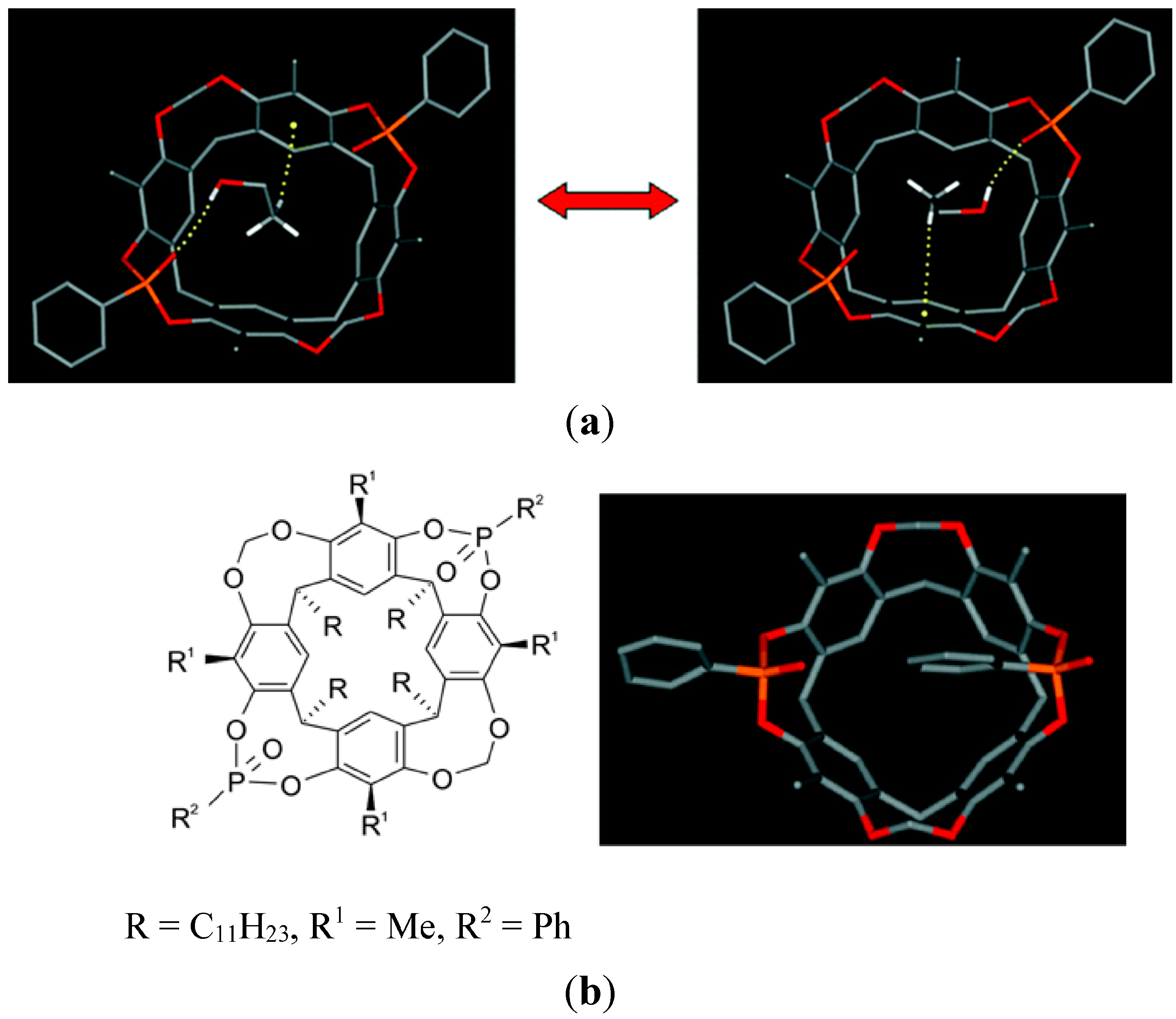
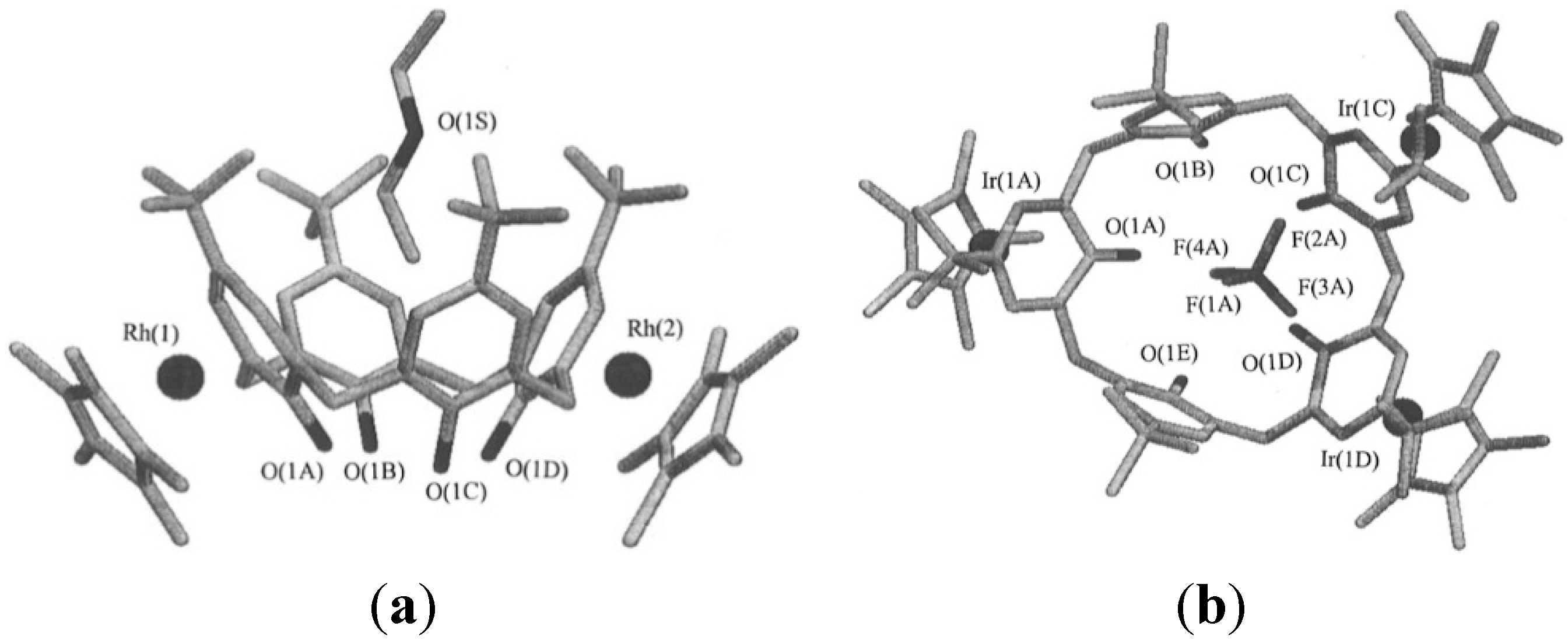
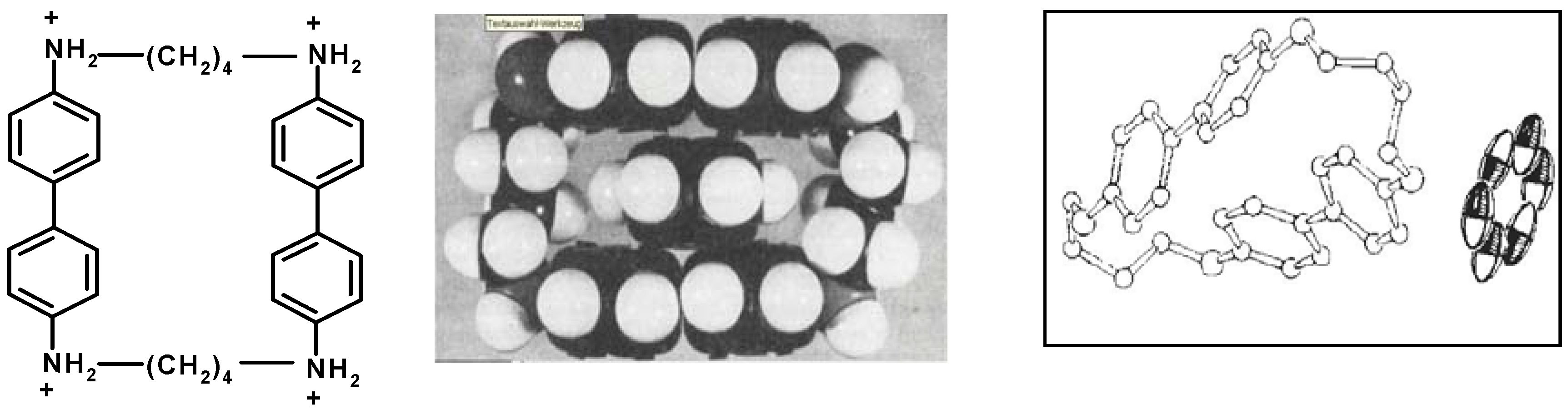
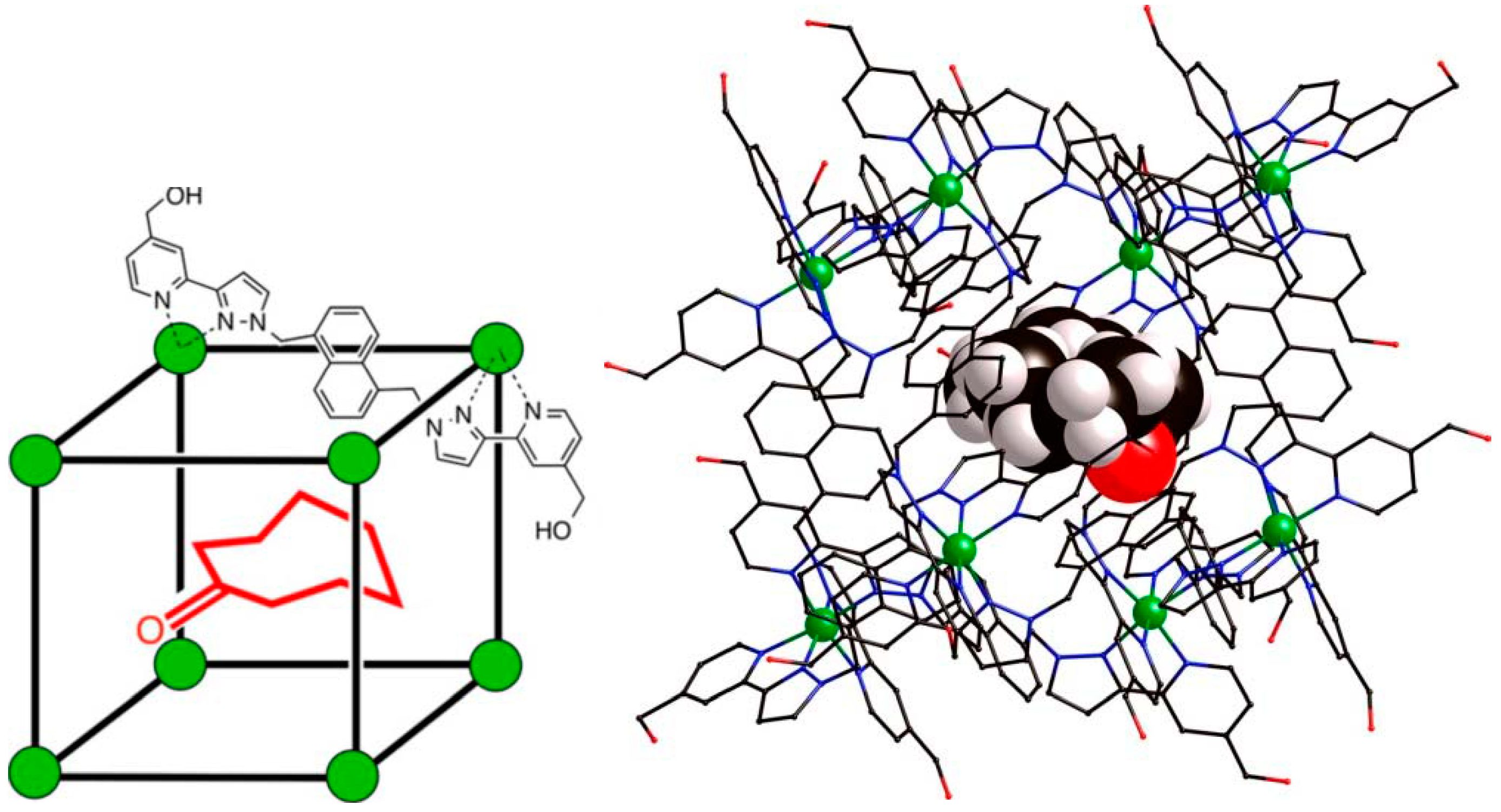
6. Host and Guest Complexes in the Solid State

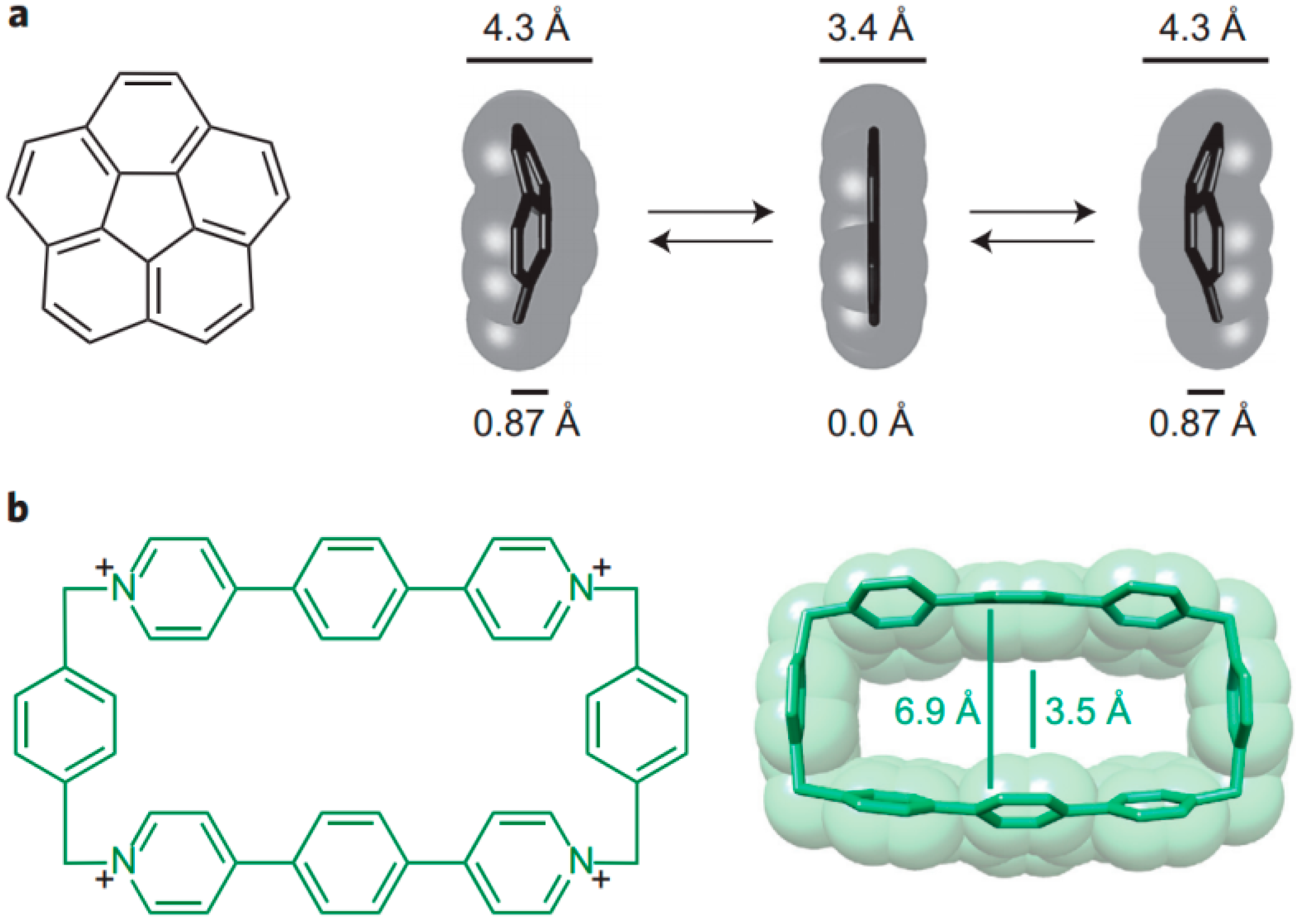
7. Intra- and Extra Cavity Complexation in Macrocyclic Receptors/Differences between Solid State, Gas State and Solution Structure






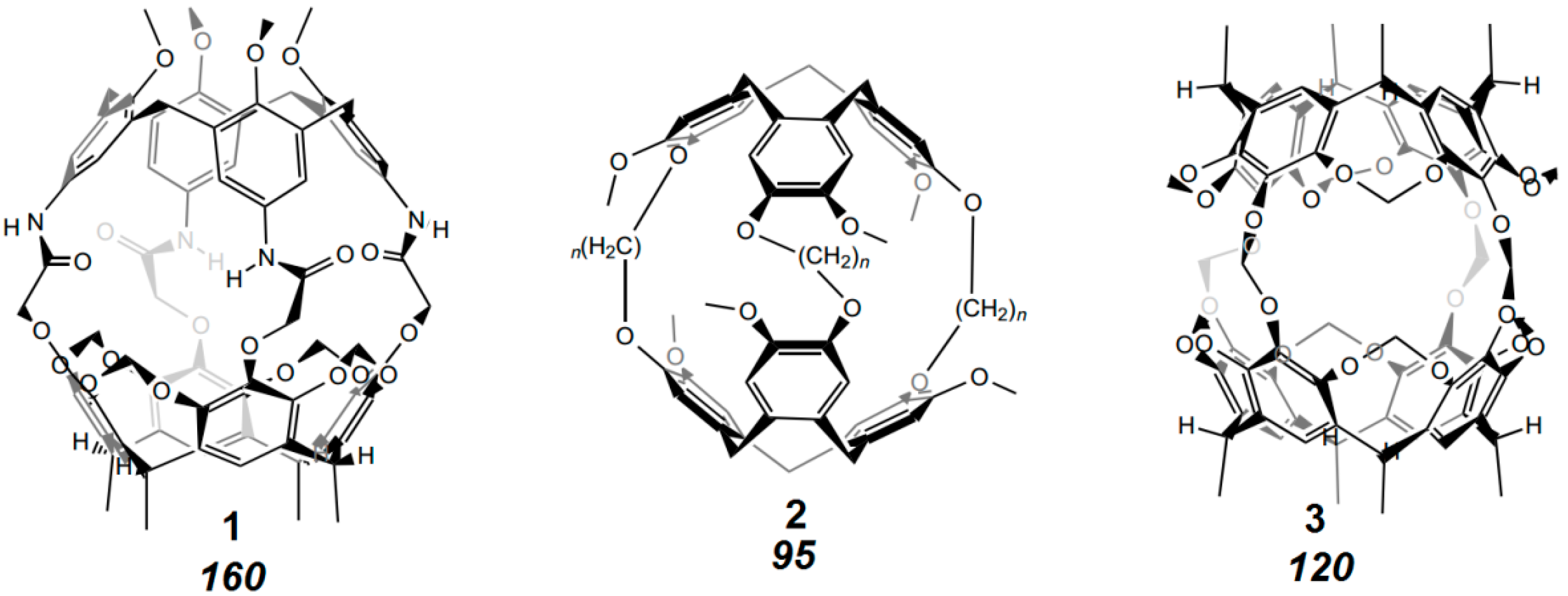
8. Cavity Filling Factors—Conflict with the Lock-and-Key Principle?


9. Problems with Identification of Weak Interactions in Crystals
10. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Conflicts of Interest
References
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme Influence of configuration on the action of enzymes. Ber. Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Cram, D.J.; Cram, J.M. Container Molecules and Their Guests; Royal Society of Chemistry: Cambridge, UK, 1997. [Google Scholar]
- Mazik, M. Recent developments in the molecular recognition of carbohydrates by artificial receptors. RSC Adv. 2012, 2, 2630–2642. [Google Scholar] [CrossRef]
- Walker, D.B.; Joshi, G; Davis, A.P. Progress in biomimetic carbohydrate recognition. Cell. Mol. Life Sci. 2009, 66, 3177–3191. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.-J. Binding mechanisms in supramolecular complexes. Angew. Chem. 2009, 48, 3924–3977. [Google Scholar] [CrossRef]
- Solovev, V.P.; Strakhova, N.N.; Raevsky, O.A.; Rüdiger, V.; Schneider, H.-J. Supramolecular chemistry 60. Solvent effects on crown ether complexations. J. Org. Chem. 1996, 61, 5221–5526. [Google Scholar] [CrossRef]
- Raevsky, O.A.; Solovev, V.P.; Solotnov, A.F.; Schneider, H.-J.; Rüdiger, V. Conformation of 18-crown-5 and its influence on complexation with alkali and ammonium cations: Why 18-crown-5 binds more than 1000 times weaker than 18C6. J. Org. Chem. 1996, 61, 8113–8116. [Google Scholar] [CrossRef] [PubMed]
- Solovev, V.P.; Strakhova, N.N.; Kazachenko, V.P.; Solotnov, A.F.; Baulin, V.E.; Raevsky, O.A.; Rüdiger, V.; Eblinger, F.; Schneider, H.-J. Steric and stereoelectronic effects in aza crown ether complexes. Eur. J. Org. Chem. 1998, 1379–1389. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Rüdiger, V.; Raevsky, O.A. The incremental description of host-guest complexes—Free-energy increments derived from hydrogen-bonds applied to crown-ethers and cryptands. J. Org. Chem. 1993, 58, 3648–3653. [Google Scholar] [CrossRef]
- Lhotak, P.; Shinkai, S. Cation-π interactions in calix[n]arene and related systems. J. Phys. Org. Chem. 1997, 10, 273–285. [Google Scholar] [CrossRef]
- Klärner, F.-G.; Schrader, T. Aromatic interactions by molecular tweezers and clips in chemical and biological systems. Acc. Chem. Res. 2013, 46, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Cable, M.L.; Kirby, J.P.; Gray, H.B.; Ponce, A. Enhancement of anion binding in lanthanide optical sensors. Acc. Chem. Res. 2013, 46, 2576–2584. [Google Scholar] [CrossRef] [PubMed]
- Roncucci, L.; Pirondini, L.; Paderni, G.; Massera, C.; Dalcanale, E.; Azov, V.A.; Diederich, F. Conformational behavior of pyrazine-bridged and mixed-bridged cavitands: A general model for solvent effects on thermal “Vase-Kite” switching. Chem. Eur. J. 2006, 12, 4775–4784. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Deutman, A.B.C.; Smits, J.M.M.; de Gelder, R.; Elemans, J.A.A.W.; Nolte, R.J.M.; Rowan, A.E. Strong induced-fit binding of viologen and pyridine derivatives in adjustable porphyrin cavities. Chem. Eur. J. 2014, 20, 11574–11583. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.-J.; Güttes, D.; Schneider, U. Host guest chemistry 15 host guest complexes with water-soluble macrocyclic polyphenolates including induced fit and simple elements of a proton pump. J. Am. Chem. Soc. 1988, 110, 6449–6454. [Google Scholar] [CrossRef]
- Menand, M.; Leroy, A.; Marrot, J.; Luhmer, M.; Jabin, I. Induced-fit encapsulation by a 1,3,5-alternate calix[6]arene. Angew. Chem. Int. Ed. 2009, 48, 5509–5512. [Google Scholar] [CrossRef]
- Coquiere, D.; le Gac, S.; Darbost, U.; Seneque, O.; Jabin, I.; Reinaud, O. Biomimetic and self-assembled calix[6]arene-based receptors for neutral molecules. Org. Biomol. Chem. 2009, 7, 2485–2500. [Google Scholar] [CrossRef] [PubMed]
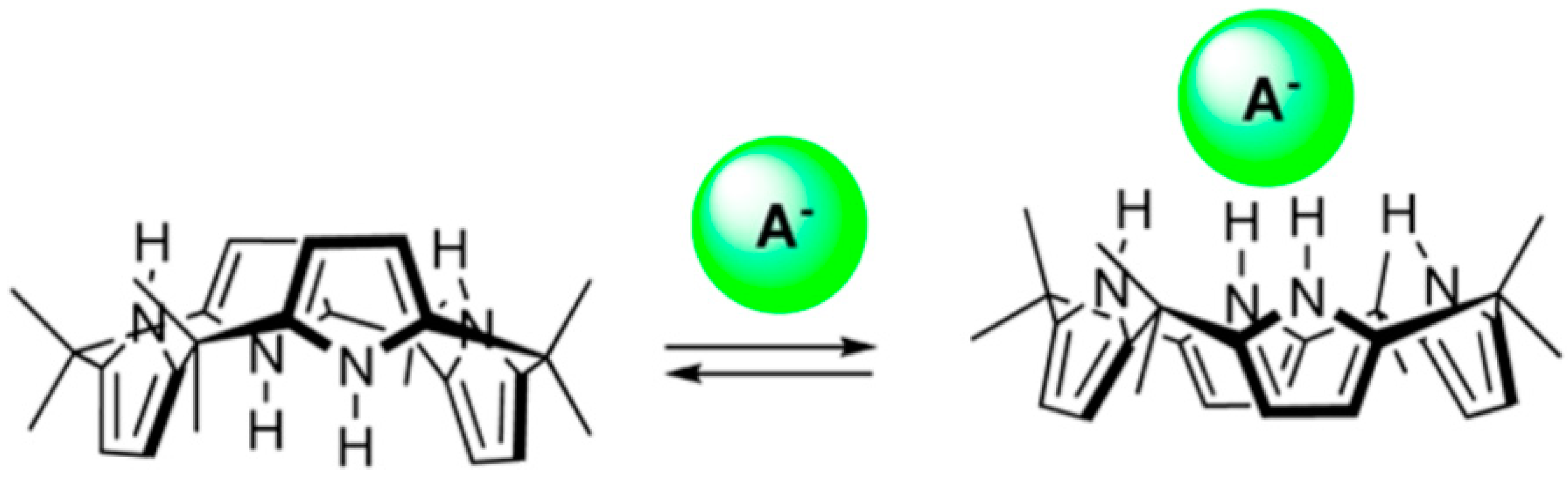
- Kim, D.S.; Sessler, J.L. Calix[4] pyrroles: Versatile molecular containers with ion transport, recognition, and molecular switching functions. Chem. Soc. Rev. 2015, 44, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Sessler, J.L. Calix[4] pyrrole-based ion pair receptors. Acc. Chem. Res. 2014, 47, 2525–2536. [Google Scholar] [CrossRef] [PubMed]
- Biros, A.; Shannon, M.; Rebek, J., Jr. Structure and binding properties of water-soluble cavitands and capsules. Chem. Soc. Rev. 2007, 36, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Corbellini, F.; Knegtel, R.M.A.; Grootenhuis, P.D.J.; Crego-Calama, M.; Reinhoudt, D.N. Water-soluble molecular capsules: Self-assembly and binding properties. Chem. Eur. J. 2005, 11, 298–307. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Klosterman, J.K.; Fujita, M. Functional molecular flasks: New properties and reactions within discrete, self-assembled hosts. Angew. Chem. Int. Ed. 2009, 48, 3418–3438. [Google Scholar] [CrossRef]
- Fujita, M.; Nagao, S.; Ogura, K. Guest-induced organization of a 3-dimensional palladium(ii) cage-like complex—A prototype for induced-fit molecular recognition. J. Am. Chem. Soc. 1995, 117, 1649–1650. [Google Scholar] [CrossRef]
- Gibb, C.L.D.; Gibb, B.C. Well-defined, organic nanoenvironments in water: The hydrophobic effect drives a capsular assembly. J. Am. Chem. Soc. 2004, 126, 11408–11409. [Google Scholar] [CrossRef] [PubMed]
- Kovbasyuk, L.; Krämer, R. Allosteric supramolecular receptors and catalysts. Chem. Rev. 2004, 104, 3161–3187. [Google Scholar] [CrossRef] [PubMed]
- Kremer, C.; Luetzen, A. Artificial allosteric receptors. Chem. Eur. J. 2013, 19, 6162–6196. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, S.; Takeuchi, M. Molecular design of synthetic receptors with dynamic, imprinting, and allosteric functions. Biosens. Biolelectron. 2004, 20, 1250–1259. [Google Scholar] [CrossRef]
- Takeuchi, M.; Ikeda, S.; Sugasaki, A.; Shinkai, S. Molecular design of artificial molecular and ion recognition systems with allosteric guest responses. Acc. Chem. Res. 2001, 34, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Arduini, A.; Giorgi, G.; Pochini, A.; Secchi, A.; Ugozzoli, F. Anion allosteric effect in the recognition of tetramethylammonium salts by calix[4]arene cone conformers. J. Org. Chem. 2001, 66, 8302–8308. [Google Scholar] [CrossRef] [PubMed]
- Baldes, R.; Schneider, H.-J. Complexes from polyazacyclophanes, fluorescence indicators, and metal cations—An example of allosterism through ring contraction. Angew. Chem. Int. Ed. 1995, 34, 321–323. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Ruf, D. A synthetic allosteric system with high cooperativity between polar and hydrophobia binding sites. Angew. Chem. Int. Ed. 1990, 29, 1159. [Google Scholar] [CrossRef]
- Biedermann, F.; Nau, W.; Schneider, H.-J. The hydrophobic effect revisited-studies with supramolecular complexes imply high-energy water as a noncovalent driving force. Angew. Chem. Int. Ed. 2014, 53, 2–16. [Google Scholar] [CrossRef]
- Snyder, P.W.; Lockett, M.R.; Moustakas, D.T.; Whiteside, G.M. Is it the shape of the cavity, or the shape of the water in the cavity? Eur. Phys. J. Spec. Top. 2014, 223, 853–891. [Google Scholar] [CrossRef]
- Baron, R.; Setny, P.; McCammon, J.A. Water in cavity-ligand recognition. J. Am. Chem. Soc. 2010, 132, 12091–12097. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Rogers, K.E.; de Oliveira, C.A.F.; Baron, R.; Cheng, L.T.; Dzubiella, J.; Li, B.; McCammon, J.A. Variational implicit-solvent modeling of host-guest binding: A case study on cucurbit[7]urill. J. Chem. Theory Comput. 2013, 9, 4195–4204. [Google Scholar] [CrossRef] [PubMed]
- Kühne, T.D.; Khaliullin, R.Z. Electronic signature of the instantaneous asymmetry in the first coordination shell of liquid water. Nat. Commun. 2013, 4, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.-J.; Tianjun, L. Additivity and quantification of dispersive interactions-from cyclopropyl to nitro groups: Measurements on porphyrin derivatives. Angew. Chem. Int. Ed. 2002, 41, 1368–1370. [Google Scholar] [CrossRef]
- Lockett, M.R.; Lange, H.; Breiten, B.; Heroux, A.; Sherman, W.; Rappoport, D.; Yau, P.O.; Snyder, P.W.; Whitesides, G.M. The binding of benzoarylsulfonamide ligands to human carbonic anhydrase is insensitive to formal fluorination of the ligand. Angew. Chem. Int. Ed. 2013, 52, 7714–7717. [Google Scholar] [CrossRef]
- Breiten, B.; Lockett, M.R.; Sherman, W.; Fujita, S.; Al-Sayah, M.; Lange, H.; Bowers, C.M.; Heroux, A.; Krilov, G.; Whitesides, G.M. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 2013, 135, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- Nangia, A. Conformational polymorphism in organic crystals. Acc. Chem. Res. 2008, 41, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef] [PubMed]
- Aitipamula, S.; Chow, P.S.; Tan, R.B.H. Polymorphism in cocrystals: A review and assessment of its significance. Cryst. Eng. Commun. 2014, 16, 3451–3465. [Google Scholar] [CrossRef]
- De Zorzi, R.; Brancatelli, G.; Melegari, M.; Pinalli, R.; Dalcanale, E.; Geremia, S. Selectivity assessment in host-guest complexes from single-crystal X-ray diffraction data: The cavitand-alcohol case. Cryst. Eng. Commun. 2014, 16, 10987–10996. [Google Scholar] [CrossRef]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 1990, 495, 461–466. [Google Scholar] [CrossRef]
- Nassimbeni, L.R.; Su, H. How to monitor guest exchange in host-guest systems. Cryst. Eng. Commun. 2013, 15, 7396–7401. [Google Scholar] [CrossRef]
- Ebata, T.; Hontama, N.; Inokuchi, Y.; Haino, T.; Aprà, E.; Xantheas, S.S. Encapsulation of Arn complexes by calix[4]arene: Endo- vs. exo-complexes. Phys. Chem. Chem. Phys. 2010, 12, 4569. [Google Scholar] [CrossRef] [PubMed]
- Cabral, B.J.C.; Coutinho, K.; Canuto, S. Dynamics of endo- vs. exo-complexation and electronic absorption of calix[4]arene-Ar-2. Chem. Phys. Lett. 2014, 612, 266–272. [Google Scholar] [CrossRef]
- Kaneko, S.; Inokuchi, Y.; Ebata, T.; Aprà, E.; Xantheas, S.S. Laser spectroscopic and theoretical studies of encapsulation complexes of calix[4]arene. J. Phys. Chem. A 2011, 115, 10846–10853. [Google Scholar] [CrossRef] [PubMed]
- Pirondini, L.; Dalcanale, E. Molecular recognition at the gas–solid interface: A powerful tool for chemical sensing. Chem. Soc. Rev. 2007, 36, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Andreetti, D.; Pochini, A.; Ungaro, R. Molecular inclusion in functionalized macrocycles 6. The crystal and molecular-structures of the calix[4]arene from para-(1,1,3,3-tetramethylbutyl)phenol and its 1:1 complex with toluene. J. Chem. Soc. Perkin Trans. 1983, 2, 1773–1779. [Google Scholar] [CrossRef]
- Mutihac, L.; Buschmann, H.-J.; Mutihac, R.C.; Schollmeyer, E. Complexation and separation of amines, amino acids, and peptides by functionalized calix[n]arenes. J. Incl. Phenom. Mol. Recognit. Chem. 2005, 51, 1–10. [Google Scholar] [CrossRef]
- General Review: Calixarenes 2001; Asfari, Z.; Böhmer, V.; Harrowfield, J.; Vicens, J. (Eds.) Kluwer: Dordrecht, The Netherlands, 2001.
- Brouwer, E.B.; Udachin, K.A.; Enright, G.D.; Ripmeester, J.A. Amine guest size and hydrogen-bonding influence the structures of p-tert-butylcalix[4]arene inclusions. Chem. Commun. 2000, 1905–1906. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, K.D.; Yeo, H.M.; Ko, S.W.; Ryu, B.J.; Nam, K.C. The molecular recognition of amines with calix[6]arene: Conclusive X-ray and NMR evidence for endo and exo complex formation between calix[6]arene and amines. Supramol. Chem. 2007, 19, 167–173. [Google Scholar] [CrossRef]
- Andreetti, G. D.; Ugozzoli, F.; Nakamoto, Y.; Ishida, S.-I. Molecular inclusion in calixarenes 18. Crystal and molecular-structure of the p-tert-butylcalix[7]arene 1:3 pyridine complex/clathrate. J. Incl. Phenom. Mol. Recognit. Chem. 1991, 10, 241–253. [Google Scholar] [CrossRef]
- Czugler, M.; Tisza, S.; Speier, G. Versatility in inclusion hosts—Unusual conformation in the crystal-structure of the para-tert-butylcalix[8]aren—Pyridine (1:8) clathrate. J. Incl. Phenom. Mol. Recognit. Chem. 1991, 11, 323–331. [Google Scholar] [CrossRef]
- Staffilani, M.; Hancock, K.S.B.; Steed, J.W.; Holman, K.T.; Atwood, J.L.; Juneja, R.K.; Burkhalter, R.S. Anion binding within the cavity of π-metalated calixarenes. J. Am. Chem. Soc. 1997, 119, 6324–6335. [Google Scholar] [CrossRef]
- Cotton, F.A.; Dikarev, E.V.; Murillo, C.A.; Petrukhina, M.A. Dinuclear Ti-IV and Ti-III complexes supported by calix[4]arene ligands. Binding alkali-metal cations inside and outside the cavity of calix[4]arenes. Inorg. Chim. Acta 2002, 332, 41–46. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, S.Y.; Seo, J.; Park, C.S.; Go, J.N.; Sim, W.; Lee, S.S. Calix[4]bis(thiacrown): Assembly of an endocyclic disilver(I) complex and exocyclic 3D copper(I) coordination polymers. Inorg. Chem. 2007, 46, 6221–6223. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.T.; Metrangolo, P.; Pappalardo, S.; Parisi, M.F.; Pilati, T.; Resnati, G. Interactions at the outside faces of calix[4]arenes: Two-dimensional infinite network formation with perfluoroarenes. Chem. Eur. J. 2000, 6, 3495–3500. [Google Scholar] [CrossRef] [PubMed]
- Stetter, H.; Roos, E.E. Zur kenntnis der makrocyclischen ringsysteme 2. Uber die bis-[n,n'-alkylenbenzidine. Chem. Ber. 1955, 88, 1390–1395. [Google Scholar]
- Hilgenfeld, R.; Saenger, W. stetters complexes are no intramolecular inclusion-compounds. Angew. Chem. Int. Ed. 1982, 21, 787–788. [Google Scholar] [CrossRef]
- Wald, P.; Schneider, H.-J. Reinvestigation of supramolecular complexes with cyclophanes of the Stetter and Koga type: Agreement and disagreement with solid-state structures. Eur. J. Org. Chem. 2009, 3450–3453. [Google Scholar] [CrossRef]
- Ciampolini, M.; Dapporto, P.; Nardi, N. Structure and properties of some lanthanoid (III) perchlorates with the cryptand 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo [8.8.8] hexacosane. J. Chem. Soc. Dalton Trans. 1979, 974–977. [Google Scholar] [CrossRef]
- Shestakova, A.K.; Chertkov, V.A.; Schneider, H.-J. Structures and equilibria involving the (222) cryptand and europium ions. Tetrahedron Lett. 2000, 41, 6753–6756. [Google Scholar] [CrossRef]
- Fiehn, T.; Goddard, R.; Seidel, R.W.; Kubik, S. A cyclopeptide-derived molecular cage for sulfate ions that closes with a click. Chem. Eur. J. 2010, 16, 7241–7255. [Google Scholar] [CrossRef] [PubMed]
- Lahiani-Skiba, M.; Boulet, Y.; Youm, I.; Bounoure, F.; Verite, P.; Arnaud, P.; Skiba, M. Interaction between hydrophilic drug and α-cyclodextrins: Physico-chemical aspects. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 211–217. [Google Scholar] [CrossRef]
- Connors, K.A. Prediction of binding constants of α-cyclodextrin complexes. J. Pharm. Sci. 2000, 85, 796–802. [Google Scholar] [CrossRef]
- Uccello-Barretta, G.; Balzano, F.; Pertici, F.; Jicsinszky, L.; Sicoli, G.; Schurig, V. External vs. internal interactions in the enantiodiscrimination of fluorinated alpha-amino acid derivatives by heptakis[2,3-di-O-acetyl-6-O-(tert-butyldimethylsilyl)]-β-cyclodextrin, a powerful chiral solvating agent for NMR spectroscopy. Eur. J. Org. Chem. 2008, 1855–1863. [Google Scholar] [CrossRef]
- Sicoli, G.; Pertici, F.; Jiang, Z.; Jicsinszky, L.; Schurig, V. Gas-chromatographic approach to probe the absence of molecular inclusion in enantioseparations by carbohydrates. Investigation of linear dextrins (“acyclodextrins”) as novel chiral stationary phases. Chirality 2007, 19, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Schurig, V.; Juza, M. Enantiomer separation by gas chromatography on chiral stationary phases. Adv. Chromatogr. 2014, 52, 117–168. [Google Scholar] [PubMed]
- Gleiter, R.; Hopf, H. Modern Cyclophane Chemistry; Wiley/VCH: Weinheim, Germany, 2004. [Google Scholar]
- Diederich, F. Cyclophanes; RSC: Cambridge, UK, 1991. [Google Scholar]
- Diederich, F. Complexation of neutral molecules by cyclophane hosts. Angew. Chem. Int. Ed. 1988, 100, 372–396. [Google Scholar]Top. Curr. Chem. Cyclophanes 1994, 172; 1983, 115; 1983, 113.
- König, B. Carbon rich cyclophanes with unusual properties—An update. Top. Curr. Chem. 1998, 196, 91–136. [Google Scholar]
- Hopf, H. Acetylenic cyclophanes: Emerging carbon-rich compounds for molecular construction and practical applications. Tetrahedron 2008, 64, 11504–11516. [Google Scholar] [CrossRef]
- JJeppesen, O.; Nielsen, M.B.; Becher, J. Tetrathiafulvalene cyclophanes and cage molecules. Chem. Rev. 2004, 104, 5115–5131. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Yamanaka, M. Self-assembled capsules based on tetrafunctionalized calix[4]resorcinarene cavitands. Chem. Soc. Rev. 2015, 44, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P. Supramolecular capsules derived from calixpyrrole Scaffolds. Israel J. Chem. 2011, 51, 710–724. [Google Scholar] [CrossRef]
- Garel, L.; Lozach, B.; Dutasta, J.-P.; Collet, A. Remarkable effect of the receptor size in the binding of acetylcholine and related ammonium ions to water-soluble cryptophanes. J. Am. Chem. Soc. 1993, 115, 11652. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Blatter, T.; Simova, S. Host guest chemistry. 26. NMR and fluorescence studies of cyclodextrin complexes with guest molecules containing both phenyl and naphthyl units. J. Am. Chem. Soc. 1991, 113, 1996. [Google Scholar] [CrossRef]
- Garel, L.; Dutasta, J.; Collet, A. Complexation of methane and chlorofluorocarbons by cryptophane—A in organic solution. Angew. Chem. Int. Ed. 1993, 32, 1169–1171. [Google Scholar] [CrossRef]
- Collet, A. Comprehensive Supramolecular Chemistry Vol. 2; Atwood, J.L., Davies, J.E., MacNicol, D.D., Vögtle, F., Eds.; Elsevier: Oxford, UK, 1996; pp. 325–365. [Google Scholar]
- Mecozzi, S.; Rebek, J., Jr. The 55% solution: A formula for molecular recognition in the liquid state. Chem. Eur. J. 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- Ajami, D.; Rebek, J., Jr. More chemistry in small spaces. Acc. Chem. Res. 2013, 46, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Custelcean, R. Anion encapsulation and dynamics in self-assembled coordination cages. Chem. Soc. Rev. 2014, 43, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Assaf, K.I.; Nau, W.M. Cucurbiturils as fluorophilic receptors. Supramol. Chem. 2014, 26, 657–669. [Google Scholar] [CrossRef]
- Jelfs, K.E.; Cooper, A.I. Molecular simulations to understand and to design porous organic molecules. Curr. Opin. Solid State Mater. Sci. 2013, 17, 19–30. [Google Scholar] [CrossRef]
- Kubik, S. Molecular cages and capsules with functionalized inner surfaces. Top. Curr. Chem. 2012, 319, 1–34. [Google Scholar] [PubMed]
- Scarso, A.; Pellizzaro, L.; de Lucchi, O.; Linden, A.; Fabris, F. Gas hosting in enantiopure self-assembled oximes. Angew. Chem. Int. Ed. 2007, 46, 4972–4975. [Google Scholar] [CrossRef]
- Rebek, J., Jr. Contortions of encapsulated alkyl groups. Chem. Commun. 2007, 2777–2278. [Google Scholar] [CrossRef]
- Pirondini, L.; Bonifazi, D.; Cantadori, B.; Braiuca, P.; Campagnolo, M.; de Zorzi, R.; Geremia, S.; Diederich, F.; Dalcanale, E. Inclusion of methano[60]fullerene derivatives in cavitand-based coordination cages. Tetrahedron 2006, 62, 2008–2015. [Google Scholar] [CrossRef]
- Zürcher, M.; Gottschalk, T.; Meyer, S.; Bur, D.; Diederich, F. Exploring the flap pocket of the antimalarial target plasmepsin II: The “55% rule” applied to enzymes. Chem. Med. Chem. 2007, 3, 237–240. [Google Scholar] [CrossRef]
- Gottschalk, T.; Jarowski, P.D.; Diederich, F. Reversibly controllable guest binding in precisely defined cavities: Selectivity, induced fit, and switching in novel resorcin[4]arene-based container molecules. Tetrahedron 2006, 62, 2008–2015. [Google Scholar] [CrossRef]
- Ruan, Y.; Wang, B.; Erb, J.M.; Chen, S.; Hadad, C.M.; Badjić, J.D. On the role of guests in enforcing the mechanism of action of gated baskets. Org. Biomol. Chem. 2013, 11, 7667–7675. [Google Scholar] [CrossRef] [PubMed]
- Dale, E.J.; Vermeulen, N.A.; Thomas, A.A.; Barnes, J.C.; Jurícek, M.; Blackburn, A.K.; Strutt, N.L.; Sarjeant, A.A.; Stern, C.L.; Denmark, S.E.; et al. ExCage. J. Am. Chem. Soc. 2014, 136, 10669–10682. [Google Scholar] [CrossRef] [PubMed]
- Gibb, C.L.D.; Gibb, B.C. Binding of cyclic carboxylates to octa-acid deep-cavity cavitand. J. Comput. Aided Mol. Des. 2014, 28, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.I.; El-Ayle, G.; Boutin, C.; Léonce, E.; Berthault, P.; Holman, K.T. Rim-functionalized cryptophane-111 derivatives via heterocapping, and their xenon complexes. Chem. Commun. 2014, 50, 15905–15908. [Google Scholar] [CrossRef]
- Turega, S.; Cullen, W.; Whitehead, M.; Hunter, C.A.; Ward, M.D. Mapping the internal recognition surface of an octanuclear coordination cage using guest libraries. J. Am. Chem. Soc. 2014, 136, 8475–8483. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D. X-Ray Analysis and the Structure of Organic Molecules Wiley, 2nd ed.; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Bürgi, H.B.; Dunitz, J.D. Structure Correlation; VCH: Weinheim, Germany, 1994. [Google Scholar]
- Allen, F.H.; Motherwell, W.D.S. Applications of the cambridge structural database in organic chemistry and crystal chemistry. Acta Cryst. 2002, B58, 407–422. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. How molecules stick together in organic crystals: Weak intermolecular interactions. Chem. Soc. Rev. 2009, 38, 2622–2633. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Taylor, R. Organic fluorine hardly ever accepts hydrogen bonds. Chem. Biol. Chem. 2004, 5, 614–621. [Google Scholar] [CrossRef]
- Mehta, G.; Sen, S. Probing fluorine interactions in a polyhydroxylated environment: Conservation of a C–F···H–C recognition motif in presence of O–H···O hydrogen bonds. Eur. J. Org. Chem. 2010, 3387–3394. [Google Scholar] [CrossRef]
- Thakur, T.S.; Kirchner, M.T.; Blaeser, D.; Boese, R.; Desiraju, G.R. C–H···F–C hydrogen bonding in 1,2,3,5-tetrafluorobenzene and other fluoroaromatic compounds and the crystal structure of alloxan revisited. Cryst. Eng. Commun. 2010, 12, 2079–2085. [Google Scholar] [CrossRef]
- Thalladi, V.R.; Weiss, H.C.; Bläser; Nangia; Desiraju, G.R. C–H···F interactions in the crystal structures of some fluorobenzenes. J. Am. Chem. Soc. 1998, 120, 8702–8710. [Google Scholar] [CrossRef]
- Schneider, H.-J. Hydrogen bonds with fluorine. Studies in solution, in gas phase and by computations, conflicting conclusions from crystallographic analyses. Chem. Sci. 2012, 3, 1381–1394. [Google Scholar] [CrossRef]
- Ouvrard, C.; Berthelot, M.; Laurence, C. An enthalpic scale of hydrogen-bond basicity, part 1: Halogenoalkanes. J. Phys. Org. Chem. 2001, 14, 804–810. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-Q.; Sang, P.; Tao, Y.; Fu, Y.-X.; Zhang, K.-Q.; Xie, Y.-H.; Liu, S.-Q. Protein dynamics and motions in relation to their functions: several case studies and the underlying mechanisms. J. Biomol. Struct. Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef] [PubMed]
- Robles, V.M.; Durrenberger, M.; Heinisch, T.; Lledos, A.; Schirmer, T.; Ward, T.R.; Marechal, J.-D. Structural, kinetic, and docking studies of artificial imine reductases based on biotin-streptavidin technology: An induced lock-and-key hypothesis. J. Am. Chem. Soc. 2014, 136, 15676–15683. [Google Scholar] [CrossRef] [PubMed]
- Juricek, M.; Strutt, N.L.; Barnes, J.C.; Butterfield, A.M.; Dale, E.J.; Baldridge, K.K.; Stoddart, J.F.; Siegel, J.S. Induced-fit catalysis of corannulene bowl-to-bowl inversion. Nat. Chem. 2014, 6, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Zhang, J.Z.H.; He, X. Quantum calculation of protein NMR chemical shifts based on the automated fragmentation method. Adv. Exp. Med. Biol. 2015, 827, 49–70. [Google Scholar] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of H-1 and C-13 chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Packer, M.J.; Zonta, C. From structure to chemical shift and vice-versa. Prog. NMR Spectrosc. 2005, 47, 27–39. [Google Scholar] [CrossRef]
- Facelli, J.C. Chemical shift tensors: Theory and application to molecular structural problems. Prog. NMR Spectrosc. 2011, 58, 176–201. [Google Scholar] [CrossRef]
- Boomsma, W.; Tian, P.; Frellsen, J.; Ferkinghoff-Borg, J.; Hamelryck, T.; Lindorff-Larsen, K.; Vendruscolo, M. Equilibrium simulations of proteins using molecular fragment replacement and NMR chemical shifts. Proc. Natl. Acad. Sci. USA 2014, 111, 13852–13857. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.T.; Eghbalnia, H.R.; Nielsen, N.C. Chemical shift prediction for protein structure calculation and quality assessment using an optimally parameterized force field. Prog. NMR Spectrosc. 2012, 60, 1–28. [Google Scholar] [CrossRef]
- Mulder, F.A.A.; Filatov, M. NMR chemical shift data and ab initio shielding calculations: Emerging tools for protein structure determination. Chem. Soc. Rev. 2010, 39, 578–590. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, H.-J. Limitations and Extensions of the Lock-and-Key Principle: Differences between Gas State, Solution and Solid State Structures. Int. J. Mol. Sci. 2015, 16, 6694-6717. https://doi.org/10.3390/ijms16046694
Schneider H-J. Limitations and Extensions of the Lock-and-Key Principle: Differences between Gas State, Solution and Solid State Structures. International Journal of Molecular Sciences. 2015; 16(4):6694-6717. https://doi.org/10.3390/ijms16046694
Chicago/Turabian StyleSchneider, Hans-Jörg. 2015. "Limitations and Extensions of the Lock-and-Key Principle: Differences between Gas State, Solution and Solid State Structures" International Journal of Molecular Sciences 16, no. 4: 6694-6717. https://doi.org/10.3390/ijms16046694