
Comparison of Mutation Profiles in the Duchenne Muscular Dystrophy Gene among Populations: Implications for Potential Molecular Therapies

 ,
, 

Abstract
: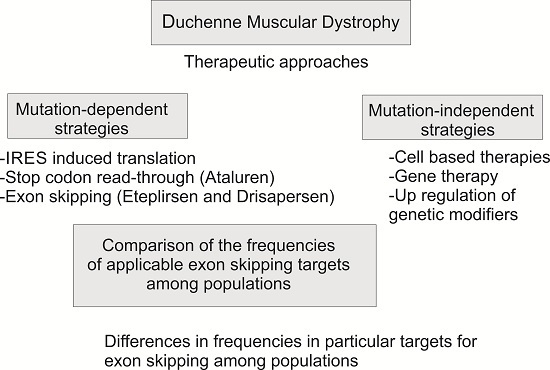
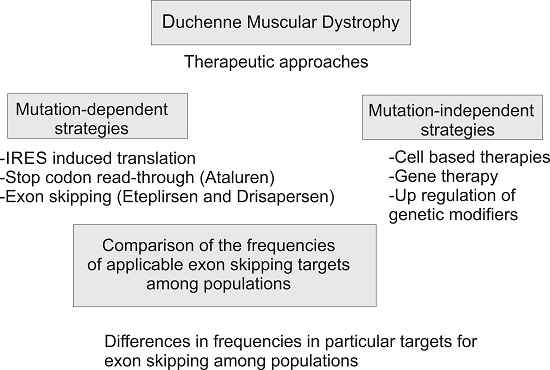
1. Introduction

2. Results and Discussion
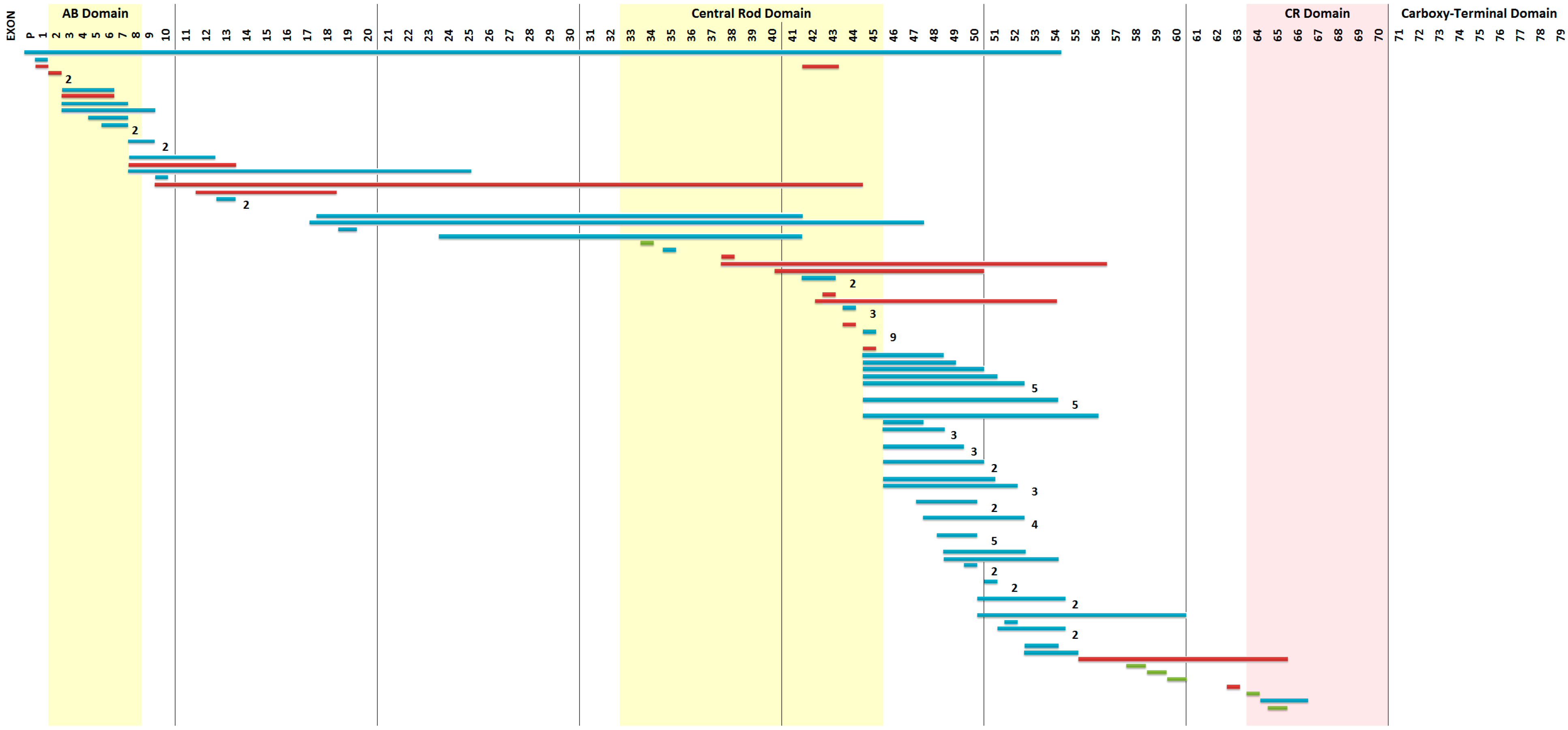
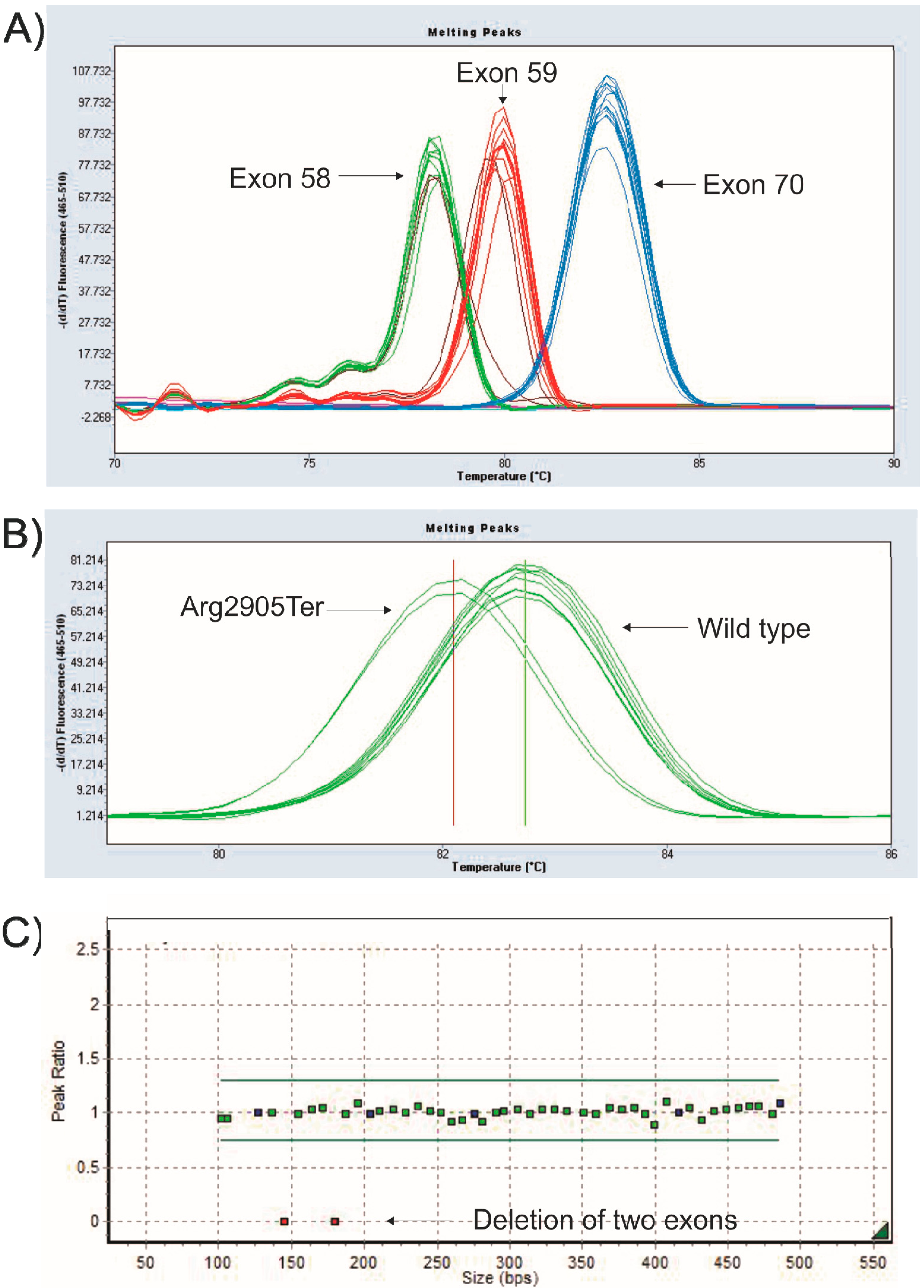
2.1. Mutation Detection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon | Change | Stop Codon | Reported in LEIDEN Database |
|---|---|---|---|
| 30 | c.4120G > T p.(Glu1374Ter) | UAG | Novel |
| 34 | c.4693C > T p.(Gln1565Ter) | UAG | 4 times |
| 59 | c.8713C > T p.(Arg2905Ter) | UGA | 20 times |
| 64 | c.9337C > T p.(Arg3113Ter) | UGA | 14 times |
| 65 | c.9380C > G p.(Ser3127Ter) | UGA | 8 times |
| 70 | c.10171C > T p.(Arg3391Ter) | UGA | 29 times |
| Involved Exon(s)/Change | Prediction |
|---|---|
| ex01ex54del→c.(?_-244)_8027+?del | No mRNA produced |
| ex65ex66del→c.9362-?_9649+?del | IN-FRAME duplication |
| ex10ex44dup→c.961-?_6438+?dup | IN-FRAME duplication |
| ex38ex56dup→c.5326-?_8390+?dup | OUT-OF-FRAME duplication |
| ex41ex50dup→c.5740-?_7309+?dup | OUT-OF-FRAME duplication |
| ex43ex54dup→c.6118-?_8027+?dup | OUT-OF-FRAME duplication |
| ex56ex65dup→c.8218-?_9563+?dup | OUT-OF-FRAME duplication |
| ex63dup→c.9225-?_9286+?dup | OUT-OF-FRAME duplication |
| ex 30 c.4120G > T p.(Glu1374Ter) | Stop codon |
2.2. Genotype-Phenotype Correlation
2.3. Theoretical Applicability of Exon Skipping and Stop Codon Read-Through among Populations
| Country | Total of Del/Dup | Exon | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 44 | 45 | 46 | 51 | 53 | |||||||
| Del/Dup | % | Del/Dup | % | Del/Dup | % | Del/Dup | % | Del/Dup | % | ||
| Australia | 159 | 7 | 4.4 | 10 | 6.29 | 5 | 3.14 | 16 | 10.06 | 17 | 10.69 |
| Belgium | 39 | 0 | 0 | 2 | 5.13 | 0 | 0 | 4 | 10.26 | 1 | 2.56 |
| Bulgaria | 23 | 1 | 4.35 | 2 | 8.7 | 1 | 4.35 | 4 | 17.39 | 1 | 4.35 |
| China | 491 | 18 | 3.67 | 39 | 7.94 | 14 | 2.85 | 67 | 13.65 | 58 | 11.81 |
| Denmark | 123 | 8 | 6.5 | 12 | 9.76 | 7 | 5.69 | 14 | 11.38 | 9 | 7.32 |
| France | 1829 | 93 | 5.08 | 132 | 7.22 | 65 | 3.55 | 174 | 9.51 | 148 | 8.09 |
| Germany | 95 | 11 | 11.58 | 3 | 3.16 | 9 | 9.47 | 9 | 9.47 | 5 | 5.26 |
| Greece | 178 | 5 | 2.81 | 8 | 4.49 | 4 | 2.25 | 36 | 20.22 | 19 | 10.67 |
| Hungary | 110 | 2 | 1.82 | 6 | 5.45 | 2 | 1.82 | 13 | 11.82 | 7 | 6.36 |
| Italy | 480 | 34 | 7.08 | 27 | 5.63 | 28 | 5.83 | 40 | 8.33 | 37 | 7.71 |
| Netherlands | 581 | 45 | 7.75 | 47 | 8.09 | 33 | 5.68 | 61 | 10.5 | 42 | 7.23 |
| Portugal | 50 | 1 | 2 | 3 | 6 | 1 | 2 | 4 | 8 | 2 | 4 |
| Romania | 62 | 2 | 3.23 | 6 | 9.68 | 0 | 0 | 12 | 19.35 | 8 | 12.9 |
| Serbia/Montenegro | 71 | 1 | 1.41 | 11 | 15.49 | 1 | 1.41 | 8 | 11.27 | 4 | 5.63 |
| Mexico (this study) | 105 | 18 | 17.14 | 12 | 11.43 | 10 | 9.52 | 11 | 10.48 | 11 | 10.48 |
| p-value | - | p < 0.0001 | p > 0.05 | p = 0.035 | p > 0.05 | p > 0.05 | |||||
| Population | Best Target Exon | p-Value | Frequency (%) | Second Best Target | Frequency (%) |
|---|---|---|---|---|---|
| Australia | N/A | p = 0.11 | - | N/A | - |
| Belgium | Exon 51 | p < 0.01 * | 10.26 | Exon 45 | 5.13 |
| Bulgaria | Exon 51 | p < 0.01 | 17.39 | Exon 45 | 8.7 |
| China | Exon 51 | p = 0.02 | 13.65 | Exon 53 | 7.94 |
| Denmark | N/A | p = 0.67 | - | N/A | - |
| France | N/A | p = 0.49 | - | N/A | - |
| Germany | N/A | p = 0.15 | - | N/A | - |
| Greece | Exon 51 | p < 0.01 | 20.22 | Exon 53 | 10.67 |
| Hungary | Exon 51 | p < 0.01 | 11.82 | Exon 53 | 6.36 |
| Italy | N/A | p = 0.96 | - | N/A | - |
| Netherlands | N/A | p = 0.77 | - | N/A | - |
| Portugal | Exon 51 | p < 0.01 * | 8 | Exon 45 | 6 |
| Romania | Exon 51 | p < 0.01 | 19.35 | Exon 53 | 12.9 |
| Serbia/ | Exon 45 | p < 0.01 | 11.27 | Exon 51 | 11.27 |
| Mexico (this study) | N/A | p = 0.53 | - | N/A | - |
3. Experimental Section
3.1. Data and Sample Collection
3.2. DNA Extraction
3.3. Mutation Detection Using PM-MLPA
3.4. Mutation Detection by High Resolution Melting

3.5. DNA Sequencing
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lopez-Hernandez, L.B.; Ayala-Madrigal, M.L.; van Heusden, D.; Estrada-Mena, F.J.; Canto, P.; Sandoval-Ramirez, L.; Gomez-Diaz, B.; Coral-Vazquez, R.M. Improvements in the diagnosis of dystrophinopathies: What have we learnt in these last 20 years? Rev. Neurol. 2011, 52, 239–249. [Google Scholar] [PubMed]
- Bunyan, D.J.; Skinner, A.C.; Ashton, E.J.; Sillibourne, J.; Brown, T.; Collins, A.L.; Cross, N.C.; Harvey, J.F.; Robinson, D.O. Simultaneous mlpa-based multiplex point mutation and deletion analysis of the dystrophin gene. Mol. Biotechnol. 2007, 35, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar]
- Moxley, R.T., 3rd; Pandya, S.; Ciafaloni, E.; Fox, D.J.; Campbell, K. Change in natural history of duchenne muscular dystrophy with long-term corticosteroid treatment: Implications for management. J. Child Neurol. 2010, 25, 1116–1129. [Google Scholar]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Deutekom, J.C.; Fokkema, I.F.; van Ommen, G.J.; Den Dunnen, J.T. Entries in the leiden duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 2008, 265, 191–225. [Google Scholar] [PubMed]
- Seto, J.T.; Bengtsson, N.E.; Chamberlain, J.S. Therapy of genetic disorders-novel therapies for duchenne muscular dystrophy. Curr. Pediatr. Rep. 2014, 2, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar]
- Aartsma-Rus, A.; Muntoni, F. 194th enmc international workshop. 3rd enmc workshop on exon skipping: Towards clinical application of antisense-mediated exon skipping for duchenne muscular dystrophy 8–10 december 2012, naarden, the netherlands. Neuromuscul. Disord. 2013, 23, 934–944. [Google Scholar]
- Wein, N.; Vulin, A.; Falzarano, M.S.; Szigyarto, C.A.; Maiti, B.; Findlay, A.; Heller, K.N.; Uhlen, M.; Bakthavachalu, B.; Messina, S.; et al. Translation from a dmd exon 5 ires results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat. Med. 2014, 20, 992–1000. [Google Scholar]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of pro051 in duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer avi-4658 in duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar]
- Hoffman, E.P.; Connor, E.M. Orphan drug development in muscular dystrophy: Update on two large clinical trials of dystrophin rescue therapies. Discov. Med. 2013, 16, 233–239. [Google Scholar] [PubMed]
- Aartsma-Rus, A.; van Ommen, G.J.; Kaplan, J.C. Innovating therapies for muscle diseases. Handb. Clin. Neurol. 2013, 113, 1497–1501. [Google Scholar] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation duchenne muscular dystrophy. PLoS One 2013, 8, e81302. [Google Scholar]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar]
- Rubi-Castellanos, R.; Martinez-Cortes, G.; Munoz-Valle, J.F.; Gonzalez-Martin, A.; Cerda-Flores, R.M.; Anaya-Palafox, M.; Rangel-Villalobos, H. Pre-hispanic mesoamerican demography approximates the present-day ancestry of mestizos throughout the territory of mexico. Am. J. Phys. Anthropol. 2009, 139, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.J. Ataluren: First global approval. Drugs 2014, 74, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Vlcek, V.; Balabanov, P.; Salmonson, T.; Bakchine, S.; Markey, G.; Weise, M.; Schlosser-Weber, G.; Brohmann, H.; Yerro, C.P.; et al. European medicines agency review of ataluren for the treatment of ambulant patients aged 5 years and older with duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul. Disord. 2015, 25, 5–13. [Google Scholar]
- Veltrop, M.; Aartsma-Rus, A. Antisense-mediated exon skipping: Taking advantage of a trick from mother nature to treat rare genetic diseases. Exp. Cell Res. 2014, 325, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.; Yan, M.; Huang, S.; Chen, T.J.; Zhong, N. Similarity of dmd gene deletion and duplication in the chinese patients compared to global populations. Behav. Brain Funct. 2008, 4. [Google Scholar] [CrossRef]
- Nakamura, H.; Kimura, E.; Mori-Yoshimura, M.; Komaki, H.; Matsuda, Y.; Goto, K.; Hayashi, Y.K.; Nishino, I.; Takeda, S.; Kawai, M. Characteristics of japanese duchenne and becker muscular dystrophy patients in a novel japanese national registry of muscular dystrophy (remudy). Orphanet J. Rare Dis. 2013, 8. [Google Scholar] [CrossRef]
- Tran, V.K.; Ta, V.T.; Vu, D.C.; Nguyen, S.T.; Do, H.N.; Ta, M.H.; Tran, T.H.; Matsuo, M. Exon deletion patterns of the dystrophin gene in 82 vietnamese duchenne/becker muscular dystrophy patients. J. Neurogenet. 2013, 27, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.Q.; Sasongko, T.H.; Sulong, S.; Bunyan, D.; Salmi, A.R.; Zilfalil, B.A.; Matsuo, M.; Zabidi-Hussin, Z.A. Mutation spectrum of dystrophin gene in malaysian patients with duchenne/becker muscular dystrophy. J. Neurogenet. 2013, 27, 11–15. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; den Dunnen, J.T. Copy number variation in the genome; the human dmd gene as an example. Cytogenet. Genome Res. 2006, 115, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Duno, M.; Palle, A.L.; Krag, T.; Vissing, J. Deletion of exon 16 of the dystrophin gene is not associated with disease. Hum. Mutat. 2007, 28, 205. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.S.; Takeshima, Y.; Adachi, K.; Van Tran, K.; Nguyen, H.T.; Low, P.S.; Matsuo, M. Comparative study on deletions of the dystrophin gene in three asian populations. J. Hum. Genet. 2002, 47, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.J.; Mahmood, S.; Ali, G.; Bibi, N.; Waheed, I.; Rafiq, M.A.; Ansar, M.; Ahmad, W. Intragenic deletions in the dystrophin gene in 211 pakistani duchenne muscular dystrophy patients. Pediatr. Int. 2008, 50, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Rafferty, K.; Straub, V.; Monges, S.; Moresco, A.; Dawkins, H.; Roy, A.; Chamova, T.; Guergueltcheva, V.; Korngut, L.; et al. The treat-nmd duchenne muscular dystrophy registries: Conception, design, and utilization by industry and academia. Hum. Mutat. 2013, 34, 1449–1457. [Google Scholar]
- Lopez-Hernandez, L.B.; Gomez-Diaz, B.; Escobar-Cedillo, R.E.; Gama-Moreno, O.; Camacho-Molina, A.; Soto-Valdes, D.M.; Anaya-Segura, M.A.; Luna-Padron, E.; Zuniga-Guzman, C.; Lopez-Hernandez, J.A.; et al. Duchenne muscular dystrophy in a developing country: Challenges in management and genetic counseling. Genet. Couns. 2014, 25, 129–141. [Google Scholar]
- Vazquez-Cardenas, N.A.; Ibarra-Hernandez, F.; Lopez-Hernandez, L.B.; Escobar-Cedillo, R.E.; Ruano-Calderon, L.A.; Gomez-Diaz, B.; Garcia-Calderon, N.; Carriedo-Davila, M.F.; Rojas-Hurtado, L.G.; Luna-Padron, E.; et al. Diagnosis and treatment with steroids for patients with duchenne muscular dystrophy: Experience and recommendations for mexico. Administracion del patrimonio de la beneficencia publica. Asociacion de distrofia muscular de occidente. Rev. Neurol. 2013, 57, 455–462. [Google Scholar]
- Gustincich, S.; Manfioletti, G.; del Sal, G.; Schneider, C.; Carninci, P. A fast method for high-quality genomic DNA extraction from whole human blood. Biotechniques 1991, 11, 298–300, 302. [Google Scholar] [PubMed]
- Lopez-Hernandez, L.B.; Gomez-Diaz, B.; Bahena-Martinez, E.; Neri-Gomez, T.; Camacho-Molina, A.; Ruano-Calderon, L.A.; Garcia, S.; Coral-Vazquez, R.M. A novel noncontiguous duplication in the dmd gene escapes the ‘reading-frame rule’. J. Genet. 2014, 93, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Almomani, R.; van der Stoep, N.; Bakker, E.; den Dunnen, J.T.; Breuning, M.H.; Ginjaar, I.B. Rapid and cost effective detection of small mutations in the dmd gene by high resolution melting curve analysis. Neuromuscul. Disord. 2009, 19, 383–390. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Hernández, L.B.; Gómez-Díaz, B.; Luna-Angulo, A.B.; Anaya-Segura, M.; Bunyan, D.J.; Zúñiga-Guzman, C.; Escobar-Cedillo, R.E.; Roque-Ramírez, B.; Ruano-Calderón, L.A.; Rangel-Villalobos, H.; et al. Comparison of Mutation Profiles in the Duchenne Muscular Dystrophy Gene among Populations: Implications for Potential Molecular Therapies. Int. J. Mol. Sci. 2015, 16, 5334-5346. https://doi.org/10.3390/ijms16035334
López-Hernández LB, Gómez-Díaz B, Luna-Angulo AB, Anaya-Segura M, Bunyan DJ, Zúñiga-Guzman C, Escobar-Cedillo RE, Roque-Ramírez B, Ruano-Calderón LA, Rangel-Villalobos H, et al. Comparison of Mutation Profiles in the Duchenne Muscular Dystrophy Gene among Populations: Implications for Potential Molecular Therapies. International Journal of Molecular Sciences. 2015; 16(3):5334-5346. https://doi.org/10.3390/ijms16035334
Chicago/Turabian StyleLópez-Hernández, Luz Berenice, Benjamín Gómez-Díaz, Alexandra Berenice Luna-Angulo, Mónica Anaya-Segura, David John Bunyan, Carolina Zúñiga-Guzman, Rosa Elena Escobar-Cedillo, Bladimir Roque-Ramírez, Luis Angel Ruano-Calderón, Héctor Rangel-Villalobos, and et al. 2015. "Comparison of Mutation Profiles in the Duchenne Muscular Dystrophy Gene among Populations: Implications for Potential Molecular Therapies" International Journal of Molecular Sciences 16, no. 3: 5334-5346. https://doi.org/10.3390/ijms16035334