
The Role of Heme Oxygenase-1 Promoter Polymorphisms in Perinatal Disease

 ,
,
Abstract
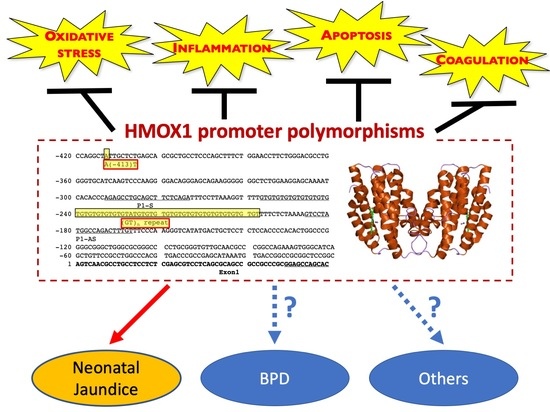
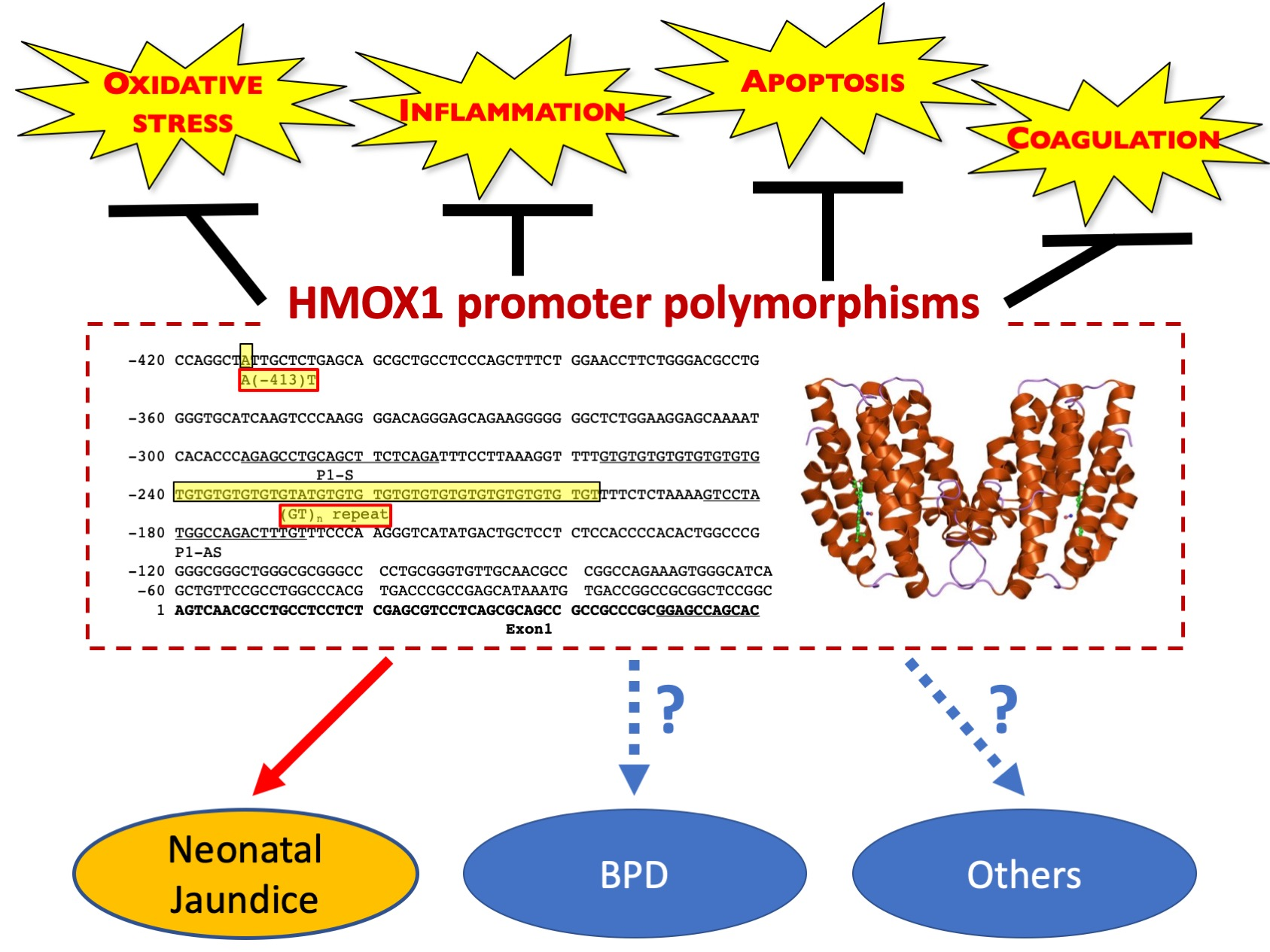
:
1. Introduction
2. Materials and Methods
3. Neonatal Jaundice
4. Bronchopulmonary Dysplasia (BPD)
5. Other Perinatal Morbidities
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poggi, C.; Giusti, B.; Vestri, A.; Pasquini, E.; Abbate, R.; Dani, C. Genetic polymorphisms of antioxidant enzymes in preterm infants. J. Mater. Fetal Neon. Med. Off. J. Eur. Assoc. Perin. Med. Federat. Asia Ocean. Perin. Soc. Int. Soc. Perin. Obstet 2012, 25 (Suppl. 4), 131–134. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [Green Version]
- Exner, M.; Minar, E.; Wagner, O.; Schillinger, M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Rad. Biol. Med. 2004, 37, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, L.E.; Perrella, M.A.; Mitsialis, S.A. The role of heme oxygenase-1 in pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2007, 36, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Taha, H.; Skrzypek, K.; Guevara, I.; Nigisch, A.; Mustafa, S.; Grochot-Przeczek, A.; Ferdek, P.; Was, H.; Kotlinowski, J.; Kozakowska, M.; et al. Role of heme oxygenase-1 in human endothelial cells: Lesson from the promoter allelic variants. Arterioscl. Thromb Vasc. Biol. 2010, 30, 1634–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozen, M.; Zhao, H.; Lewis, D.B.; Wong, R.J.; Stevenson, D.K. Heme oxygenase and the immune system in normal and pathological pregnancies. Front. Pharmacol. 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Wong, R.J.; Nguyen, X.; Kalish, F.; Mizobuchi, M.; Vreman, H.J.; Stevenson, D.K.; Contag, C.H. Expression and regulation of heme oxygenase isozymes in the developing mouse cortex. Pediatr. Res. 2006, 60, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Dennery, P.A.; Lee, C.S.; Ford, B.S.; Weng, Y.H.; Yang, G.; Rodgers, P.A. Developmental expression of heme oxygenase in the rat lung. Ped. Res. 2003, 53, 42–47. [Google Scholar] [CrossRef]
- Schulz, S.; Wong, R.J.; Jang, K.Y.; Kalish, F.; Chisholm, K.M.; Zhao, H.; Vreman, H.J.; Sylvester, K.G.; Stevenson, D.K. Heme oxygenase-1 deficiency promotes the development of necrotizing enterocolitis-like intestinal injury in a newborn mouse model. Am. J. Physiol. Gastrointest Liver Physiol. 2013, 304, G991–G1001. [Google Scholar] [CrossRef] [Green Version]
- Yamada, N.; Yamaya, M.; Okinaga, S.; Nakayama, K.; Sekizawa, K.; Shibahara, S.; Sasaki, H. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am. J. Hum. Genet. 2000, 66, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavrovsky, Y.; Schwartzman, M.L.; Abraham, N.G. Novel regulatory sites of the human heme oxygenase-1 promoter region. Biochem. Biophys. Res. Commun. 1993, 196, 336–341. [Google Scholar] [CrossRef]
- Ono, K.; Goto, Y.; Takagi, S.; Baba, S.; Tago, N.; Nonogi, H.; Iwai, N. A promoter variant of the heme oxygenase-1 gene may reduce the incidence of ischemic heart disease in Japanese. Atherosclerosis 2004, 173, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.A.; Yeh, Y.H.; Kuo, C.T.; Chen, Y.H.; Chang, G.J.; Tsai, F.C.; Chen, W.J. Microsatellite polymorphism in the heme oxygenase-1 gene promoter and the risk of atrial fibrillation in Taiwanese. PLoS ONE 2014, 9, e108773. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.W.; Yeung, V.T.; Collins, A.R.; Benzie, I.F. Redox-linked effects of green tea on DNA damage and repair, and influence of microsatellite polymorphism in HMOX-1: Results of a human intervention trial. Mutagenesis 2015, 30, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, K.; Yang, W.; Wallenstein, M.B.; Zhao, H.; Wong, R.J.; Stevenson, D.K.; Shaw, G.M. Heme oxygenase-1 promoter polymorphisms and risk of spina bifida. Birth Defects Res. A Clin. Mol. Teratol. 2015. [Google Scholar] [CrossRef]
- Katayama, Y.; Yokota, T.; Zhao, H.; Wong, R.J.; Stevenson, D.K.; Taniguchi-Ikeda, M.; Nakamura, H.; Iijima, K.; Morioka, I. Association of HMOX1 gene promoter polymorphisms with hyperbilirubinemia in the early neonatal period. Pediatr. Int. 2015. [Google Scholar] [CrossRef]
- Kaplan, M.; Renbaum, P.; Hammerman, C.; Vreman, H.J.; Wong, R.J.; Stevenson, D.K. Heme oxygenase-1 promoter polymorphisms and neonatal jaundice. Neonatology 2014, 106, 323–329. [Google Scholar] [CrossRef]
- Kuesap, J.; Hirayama, K.; Kikuchi, M.; Ruangweerayut, R.; Na-Bangchang, K. Study on association between genetic polymorphisms of haem oxygenase-1, tumour necrosis factor, cadmium exposure and malaria pathogenicity and severity. Malar J. 2010, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Walther, M.; De Caul, A.; Aka, P.; Njie, M.; Amambua-Ngwa, A.; Walther, B.; Predazzi, I.M.; Cunnington, A.; Deininger, S.; Takem, E.N.; et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. 2012, 8, e1002579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambo, M.R.; Trovoada, M.J.; Benchimol, C.; Quinhentos, V.; Goncalves, L.; Velosa, R.; Marques, M.I.; Sepulveda, N.; Clark, T.G.; Mustafa, S.; et al. Transforming growth factor beta 2 and heme oxygenase 1 genes are risk factors for the cerebral malaria syndrome in Angolan children. PLoS ONE 2010, 5, e11141. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Mannami, T.; Iwai, N. Association of a promoter variant of the haeme oxygenase-1 gene with hypertension in women. J. Hypertens 2003, 21, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G.; Aminuddin, F.; Akhabir, L.; He, J.Q.; Shumansky, K.; Connett, J.E.; Anthonisen, N.R.; Abboud, R.T.; Pare, P.D.; Sandford, A.J. Effect of heme oxygenase-1 polymorphisms on lung function and gene expression. BMC Med. Genet. 2011, 12, 117. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, V.R.; Luz, N.F.; Santos, N.J.; Borges, V.M.; Goncalves, M.S.; Andrade, B.B.; Barral-Netto, M. Association between the haptoglobin and heme oxygenase 1 genetic profiles and soluble CD163 in susceptibility to and severity of human malaria. Infect Immun. 2012, 80, 1445–1454. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, H.; Ohno, M.; Taguchi, J.; Togo, M.; Hashimoto, H.; Ogasawara, K.; Aizawa, T.; Ishizaka, N.; Nagai, R. Heme oxygenase-1 gene promoter polymorphism is associated with coronary artery disease in Japanese patients with coronary risk factors. Arterioscl. Thromb. Vasc. Biol. 2002, 22, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Lin, S.J.; Lin, M.W.; Tsai, H.L.; Kuo, S.S.; Chen, J.W.; Charng, M.J.; Wu, T.C.; Chen, L.C.; Ding, Y.A.; et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum. Genet. 2002, 111, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Schillinger, M.; Minar, E.; Mlekusch, W.; Schlerka, G.; Haumer, M.; Mannhalter, C.; Wagner, O. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with restenosis after percutaneous transluminal angioplasty. J. Endovasc. Ther. Off. J. Int. Soc. Endovasc. Spec. 2001, 8, 433–440. [Google Scholar] [CrossRef]
- Pechlaner, R.; Willeit, P.; Summerer, M.; Santer, P.; Egger, G.; Kronenberg, F.; Demetz, E.; Weiss, G.; Tsimikas, S.; Witztum, J.L.; et al. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with progressive atherosclerosis and incident cardiovascular disease. Arteriosc. Thromb. Vasc. Biol. 2015, 35, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.P.; Sun, C.; Dai, L.M.; Yang, L.F.; Zhang, Y.P. Relationship between COPD and polymorphisms of HOX-1 and mEPH in a Chinese population. Oncol. Rep. 2007, 17, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Vashist, Y.K.; Uzungolu, G.; Kutup, A.; Gebauer, F.; Koenig, A.; Deutsch, L.; Zehler, O.; Busch, P.; Kalinin, V.; Izbicki, J.R.; et al. Heme oxygenase-1 germ line GTn promoter polymorphism is an independent prognosticator of tumor recurrence and survival in pancreatic cancer. J. Surg. Oncol. 2011, 104, 305–311. [Google Scholar] [CrossRef]
- Lo, S.S.; Lin, S.C.; Wu, C.W.; Chen, J.H.; Yeh, W.I.; Chung, M.Y.; Lui, W.Y. Heme oxygenase-1 gene promoter polymorphism is associated with risk of gastric adenocarcinoma and lymphovascular tumor invasion. Ann. Surg. Oncol. 2007, 14, 2250–2256. [Google Scholar] [CrossRef]
- Chang, K.W.; Lee, T.C.; Yeh, W.I.; Chung, M.Y.; Liu, C.J.; Chi, L.Y.; Lin, S.C. Polymorphism in heme oxygenase-1 (HO-1) promoter is related to the risk of oral squamous cell carcinoma occurring on male areca chewers. Br. J. Cancer 2004, 91, 1551–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buis, C.I.; van der Steege, G.; Visser, D.S.; Nolte, I.M.; Hepkema, B.G.; Nijsten, M.; Slooff, M.J.; Porte, R.J. Heme oxygenase-1 genotype of the donor is associated with graft survival after liver transplantation. Am. J. Trans. Off. J. Am. Soc. Transpl. Am. Soc. Transpl. Surg. 2008, 8, 377–385. [Google Scholar] [CrossRef]
- Exner, M.; Bohmig, G.A.; Schillinger, M.; Regele, H.; Watschinger, B.; Horl, W.H.; Raith, M.; Mannhalter, C.; Wagner, O.F. Donor heme oxygenase-1 genotype is associated with renal allograft function. Transplantation 2004, 77, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Baan, C.; Peeters, A.; Lemos, F.; Uitterlinden, A.; Doxiadis, I.; Claas, F.; Ijzermans, J.; Roodnat, J.; Weimar, W. Fundamental role for HO-1 in the self-protection of renal allografts. Am. J. Transpl. Off. J. Am. Soc. Transpl. Am. Soc. Transpl. Surg. 2004, 4, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Akaba, K.; Sasaki, A.; Sato, M.; Harano, T.; Shibahara, S.; Kurachi, H.; Yoshida, T.; Hayasaka, K. Neonatal hyperbilirubinemia in Japanese neonates: Analysis of the heme oxygenase-1 gene and fetal hemoglobin composition in cord blood. Pediatr. Res. 2003, 54, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozkaya, O.G.; Kumral, A.; Yesilirmak, D.C.; Ulgenalp, A.; Duman, N.; Ercal, D.; Ozkan, H. Prolonged unconjugated hyperbilirubinaemia associated with the haem oxygenase-1 gene promoter polymorphism. Acta Paediatr. 2010, 99, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.K.; Sethi, A.; Basu, S.; Raman, R.; Kumar, A. Heme oxygenase-1 gene variants and hyperbilirubinemia risk in North Indian newborns. Eur. J. Pediatr. 2013, 172, 1627–1632. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.N.; Li, H.; Zha, W.; Wang, X.; Liu, Y.; Sun, J.; Peng, Q.; Li, S.; Chen, Y.; et al. Association of UGT1A1 variants and hyperbilirubinemia in breast-fed full-term Chinese infants. PLoS ONE 2014, 9, e104251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, S.N.; Li, H.; Zha, W.; Peng, Q.; Li, S.; Chen, Y.; Jin, L. Quantitative trait analysis of polymorphisms in two bilirubin metabolism enzymes to physiologic bilirubin levels in Chinese newborns. J. Pediatr. 2014, 165, 1154–1160.e1. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Q.; Zheng, L.; Lin, M.; Zheng, X.B.; Lin, F.; Yang, L.Y. Multiple Genetic Modifiers of Bilirubin Metabolism Involvement in Significant Neonatal Hyperbilirubinemia in Patients of Chinese Descent. PLoS ONE 2015, 10, e0132034. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.H.; Chiu, Y.W.; Cheng, S.W.; Yang, C.Y. Risk assessment of gene variants for neonatal hyperbilirubinemia in Taiwan. BMC Pediatr. 2016, 16, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutzman, D.L.; Gatien, E.; Ajayi, S.; Wong, R.J. Heme oxygenase-1 genetic variants and the conundrum of hyperbilirubinemia in African-American newborns. J. Perinatol. 2018, 38, 345–350. [Google Scholar] [CrossRef]
- Poggi, C.; Giusti, B.; Gozzini, E.; Sereni, A.; Romagnuolo, I.; Kura, A.; Pasquini, E.; Abbate, R.; Dani, C. Genetic Contributions to the Development of Complications in Preterm Newborns. PLoS ONE 2015, 10, e0131741. [Google Scholar] [CrossRef] [PubMed]
- Askenazi, D.J.; Halloran, B.; Patil, N.; Keeling, S.; Saeidi, B.; Koralkar, R.; Ambalavanan, N. Genetic polymorphisms of heme-oxygenase 1 (HO-1) may impact on acute kidney injury, bronchopulmonary dysplasia, and mortality in premature infants. Pediatr. Res. 2015, 77, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Kaartokallio, T.; Klemetti, M.M.; Timonen, A.; Uotila, J.; Heinonen, S.; Kajantie, E.; Kere, J.; Kivinen, K.; Pouta, A.; Lakkisto, P.; et al. Microsatellite polymorphism in the heme oxygenase-1 promoter is associated with nonsevere and late-onset preeclampsia. Hypertension 2014, 64, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Sandrim, V.; Coeli-Lacchini, F.B.; Tanus-Santos, J.E.; Lacchini, R.; Cavalli, R.C. Circulating HO-1 levels are not associated with plasma sFLT-1 and GTn HMOX1 polymorphism in preeclampsia. Hypertens. Preg. 2019, 38, 73–77. [Google Scholar] [CrossRef]
- Sandrim, V.C.; Luizon, M.R.; Pilan, E.; Caldeira-Dias, M.; Coeli-Lacchini, F.B.; Kors, G.; Berndt, I.; Lacchini, R.; Cavalli, R.C. Interaction Between NOS3 and HMOX1 on Antihypertensive Drug Responsiveness in Preeclampsia. Rev. Bras. Ginecol. Obstet. 2020, 42, 460–467. [Google Scholar] [CrossRef]
- Lv, X.; Li, X.; Dai, X.; Liu, M.; Wu, C.; Song, W.; Wang, J.; Ren, X.; Cai, Y. Investigation heme oxygenase-1 polymorphism with the pathogenesis of preeclampsia. Clin. Exp. Hypertens. 2020, 42, 167–170. [Google Scholar] [CrossRef]
- Denschlag, D.; Marculescu, R.; Unfried, G.; Hefler, L.A.; Exner, M.; Hashemi, A.; Riener, E.K.; Keck, C.; Tempfer, C.B.; Wagner, O. The size of a microsatellite polymorphism of the haem oxygenase 1 gene is associated with idiopathic recurrent miscarriage. Mol. Hum. Reprod. 2004, 10, 211–214. [Google Scholar] [CrossRef]
- American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 2004, 114, 297–316. [Google Scholar]
- Stevenson, D.K.; Rodgers, P.A.; Vreman, H.J. The use of metalloporphyrins for the chemoprevention of neonatal jaundice. Am. J. Dis. Child. 1989, 143, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F.; Lin, Z. Exploring the genetic architecture of neonatal hyperbilirubinemia. Semin. Fetal. Neon. Med. 2010, 15, 169–175. [Google Scholar] [CrossRef]
- Johnson, A.D.; Kavousi, M.; Smith, A.V.; Chen, M.H.; Dehghan, A.; Aspelund, T.; Lin, J.P.; van Duijn, C.M.; Harris, T.B.; Cupples, L.A.; et al. Genome-wide association meta-analysis for total serum bilirubin levels. Hum. Mol. Genet. 2009, 18, 2700–2710. [Google Scholar] [CrossRef] [Green Version]
- Sanna, S.; Busonero, F.; Maschio, A.; McArdle, P.F.; Usala, G.; Dei, M.; Lai, S.; Mulas, A.; Piras, M.G.; Perseu, L.; et al. Common variants in the SLCO1B3 locus are associated with bilirubin levels and unconjugated hyperbilirubinemia. Hum. Mol. Genet. 2009, 18, 2711–2718. [Google Scholar] [CrossRef] [Green Version]
- D’Silva, S.; Borse, V.; Colah, R.B.; Ghosh, K.; Mukherjee, M.B. Association of (GT)n repeats promoter polymorphism of heme oxygenase-1 gene with serum bilirubin levels in healthy Indian adults. Genet. Test. Mol. Biom. 2011, 15, 215–218. [Google Scholar] [CrossRef]
- Lin, R.; Wang, X.; Wang, Y.; Zhang, F.; Wang, Y.; Fu, W.; Yu, T.; Li, S.; Xiong, M.; Huang, W.; et al. Association of polymorphisms in four bilirubin metabolism genes with serum bilirubin in three Asian populations. Hum. Mutat. 2009, 30, 609–615. [Google Scholar] [CrossRef]
- Kaplan, M.; Wong, R.J.; Stevenson, D.K. Heme oxygenase-1 promoter polymorphisms: Do they modulate neonatal hyperbilirubinemia? J. Perinatol. 2017, 37, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.F.; Luo, J.Y.; Zhu, W.B.; Yang, C.Y.; Zeng, Y.L.; Qiu, X.L. Association between genetic polymorphism of heme oxygenase 1 promoter and neonatal hyperbilirubinemia: A meta-analysis. J. Matern Fetal. Neon. Med. 2021, 34, 12–23. [Google Scholar] [CrossRef]
- Immenschuh, S.; Shan, Y.; Kroll, H.; Santoso, S.; Wossmann, W.; Bein, G.; Bonkovsky, H.L. Marked hyperbilirubinemia associated with the heme oxygenase-1 gene promoter microsatellite polymorphism in a boy with autoimmune hemolytic anemia. Pediatrics 2007, 119, e764–e767. [Google Scholar] [CrossRef]
- Tang, J.R.; Markham, N.E.; Lin, Y.J.; McMurtry, I.F.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L344–L351. [Google Scholar] [CrossRef]
- Bhandari, V.; Gruen, J.R. The genetics of bronchopulmonary dysplasia. Semin. Perinatol. 2006, 30, 185–191. [Google Scholar] [CrossRef]
- Wang, H.; St Julien, K.R.; Stevenson, D.K.; Hoffmann, T.J.; Witte, J.S.; Lazzeroni, L.C.; Krasnow, M.A.; Quaintance, C.C.; Oehlert, J.W.; Jelliffe-Pawlowski, L.L.; et al. A genome-wide association study (GWAS) for bronchopulmonary dysplasia. Pediatrics 2013, 132, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Egawa, T.; Morioka, I.; Morisawa, T.; Yokoyama, N.; Nakao, H.; Ohashi, M.; Matsuo, M. Ureaplasma urealyticum and Mycoplasma hominis presence in umbilical cord is associated with pathogenesis of funisitis. Kobe J. Med. Sci. 2007, 53, 241–249. [Google Scholar] [PubMed]
- Kinsella, J.P.; Greenough, A.; Abman, S.H. Bronchopulmonary dysplasia. Lancet 2006, 367, 1421–1431. [Google Scholar] [CrossRef]
- Wong, R.J.; Zhao, H.; Stevenson, D.K. A deficiency in haem oxygenase-1 induces foetal growth restriction by placental vasculature defects. Acta Paediatr. 2012, 101, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Biswasa, C.; Lin, Q.S.; La, P.; Namba, F.; Zhuang, T.; Muthu, M.; Dennery, P.A. Heme oxygenase-1 regulates postnatal lung repair after hyperoxia: Role of beta-catenin/hnRNPK signaling. Redox. Biol. 2013, 1, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Didon, L.; Roos, A.B.; Elmberger, G.P.; Gonzalez, F.J.; Nord, M. Lung-specific inactivation of CCAAT/enhancer binding protein alpha causes a pathological pattern characteristic of COPD. Eur. Respir. J. 2010, 35, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Guenegou, A.; Boczkowski, J.; Aubier, M.; Neukirch, F.; Leynaert, B. Interaction between a heme oxygenase-1 gene promoter polymorphism and serum beta-carotene levels on 8-year lung function decline in a general population: The European Community Respiratory Health Survey (France). Am. J. Epidemiol. 2008, 167, 139–144. [Google Scholar] [CrossRef]
- Zhao, H.; Wong, R.J.; Doyle, T.C.; Nayak, N.; Vreman, H.J.; Contag, C.H.; Stevenson, D.K. Regulation of maternal and fetal hemodynamics by heme oxygenase in mice. Biol. Reprod. 2008, 78, 744–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Wong, R.J.; Kalish, F.S.; Nayak, N.R.; Stevenson, D.K. Effect of heme oxygenase-1 deficiency on placental development. Placenta 2009, 30, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Azuma, J.; Kalish, F.; Wong, R.J.; Stevenson, D.K. Maternal heme oxygenase 1 regulates placental vasculature development via angiogenic factors in mice. Biol. Reprod. 2011, 85, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, A.; Strauss-Albee, D.M.; Leipold, M.; Kubo, J.; Nemat-Gorgani, N.; Dogan, O.C.; Dekker, C.L.; Mackey, S.; Maecker, H.; Swan, G.E.; et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl. Med. 2013, 5, 208ra145. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Disease | Author | Polymorphism | Sample Size | Ethnicity | (GT)n Repeat Length Categorization | Polymorphism Associated with Disease |
|---|---|---|---|---|---|---|
| Neonatal Jaundice | ||||||
| Hyperbilirubinemia needing phototherapy | Kanai 2003 [37] | (GT)n repeat | 211 | Japanese German | S ≤ 26 M 27–32 L ≥ 33 | No |
| Prolonged unconjugated hyperbilirubinemia | Bozkaya 2010 [38] | (GT)n repeat | 152 | Turkish | S < 24 M 24–29 L ≥ 30 | Yes |
| Hyperbilirubinemia during the first two weeks of life | Tiwari 2013 [39] | (GT)n repeat -413 A/T | 200 | Indian | S < 21 L ≥ 21 | Yes |
| TB values on the 3rd day of life | Kaplan 2014 [19] | (GT)n repeat | 168 | Jewish | S ≤ 24 M 25–33 L ≥ 34 | No |
| Hyperbilirubinemia on day 3 or later before discharge | Zhou 2014 [40] | (GT)n repeat rs9607267 rs2071749 | 949 | Chinese | S < 27 M 27–32 L ≥ 33 | No |
| Bilirubin levels and changes during the 1st week of life | Zhou 2014 [41] | (GT)n repeat | 988 | Chinese | S < 27 M 27–32 L ≥ 33 | No |
| Hyperbilirubinemia requiring phototherapy | Yang 2015 [42] | (GT)n repeat | 237 | Chinese | S ≤ 23 M 24–29 L ≥ 30 | No |
| Hyperbilirubinemia in the 1st week of life | Katayama 2015 [18] | (GT)n repeat | 108 | Japanese | S < 22 L ≥ 22 | Yes |
| Hyperbilirubinemia during hospital course | Weng 2016 [43] | (GT)n repeat | 444 | Taiwan | S < 24 L ≥ 24 | Yes |
| Bilirubin risk percentile prior to discharge | Schutzman 2018 [44] | (GT)n repeat | 180 | African-American | S < 25 M 25–33 L > 33 | No |
| Bronchopulmonary Dysplasia | ||||||
| BPD | Poggi 2015 [45] | (GT)n repeat | 342 | Italian | S < 25 L ≥ 25 | No |
| BPD | Askenazi 2015 [46] | (GT)n repeat -413 A/T | 117 | American | S ≤ 27 L > 27 | No |
| Others | ||||||
| Spina Bifida | Fujioka 2015 [17] | (GT)n repeat -413 A/T | 300 | American | S < 26 L ≥ 26 | No |
| Non-severe and Late-onset Preeclampsia | Kaartokallio 2014 [47] | (GT)n repeat | 1538 | Finnish | S ≤ 25 L > 25 | Yes |
| Preeclampsia | Sandrim 2019 [48] | (GT)n repeat | 181 | Brazilians | S ≤ 25 L > 25 | No |
| Responsiveness to antihypertensive drugs in Preeclampsia | Sandrim 2020 [49] | -413 A/T | 398 | Brazilians | - | Yes |
| Preeclampsia | Xianping 2020 [50] | -413 A/T | 2955 | Chinese | - | No |
| Idiopathic recurrent miscarriage | Denschlag 2004 [51] | (GT)n repeat | 291 | Caucasian | S ≤ 27 L > 28 | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakasone, R.; Ashina, M.; Abe, S.; Tanimura, K.; Van Rostenberghe, H.; Fujioka, K. The Role of Heme Oxygenase-1 Promoter Polymorphisms in Perinatal Disease. Int. J. Environ. Res. Public Health 2021, 18, 3520. https://doi.org/10.3390/ijerph18073520
Nakasone R, Ashina M, Abe S, Tanimura K, Van Rostenberghe H, Fujioka K. The Role of Heme Oxygenase-1 Promoter Polymorphisms in Perinatal Disease. International Journal of Environmental Research and Public Health. 2021; 18(7):3520. https://doi.org/10.3390/ijerph18073520
Chicago/Turabian StyleNakasone, Ruka, Mariko Ashina, Shinya Abe, Kenji Tanimura, Hans Van Rostenberghe, and Kazumichi Fujioka. 2021. "The Role of Heme Oxygenase-1 Promoter Polymorphisms in Perinatal Disease" International Journal of Environmental Research and Public Health 18, no. 7: 3520. https://doi.org/10.3390/ijerph18073520