Long-Term Exposure to Low-Dose Di-(2-ethylhexyl) Phthalate Impairs Cholesterol Metabolism in Hepatic Stellate Cells and Exacerbates Liver Fibrosis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Cell Proliferation Assay
2.3. Western Blot
2.4. RNA Extraction, Reverse Transcription, and Real-Time Polymerase Chain Reaction (PCR)
2.5. Flow Cytometry
2.6. Seahorse Assay
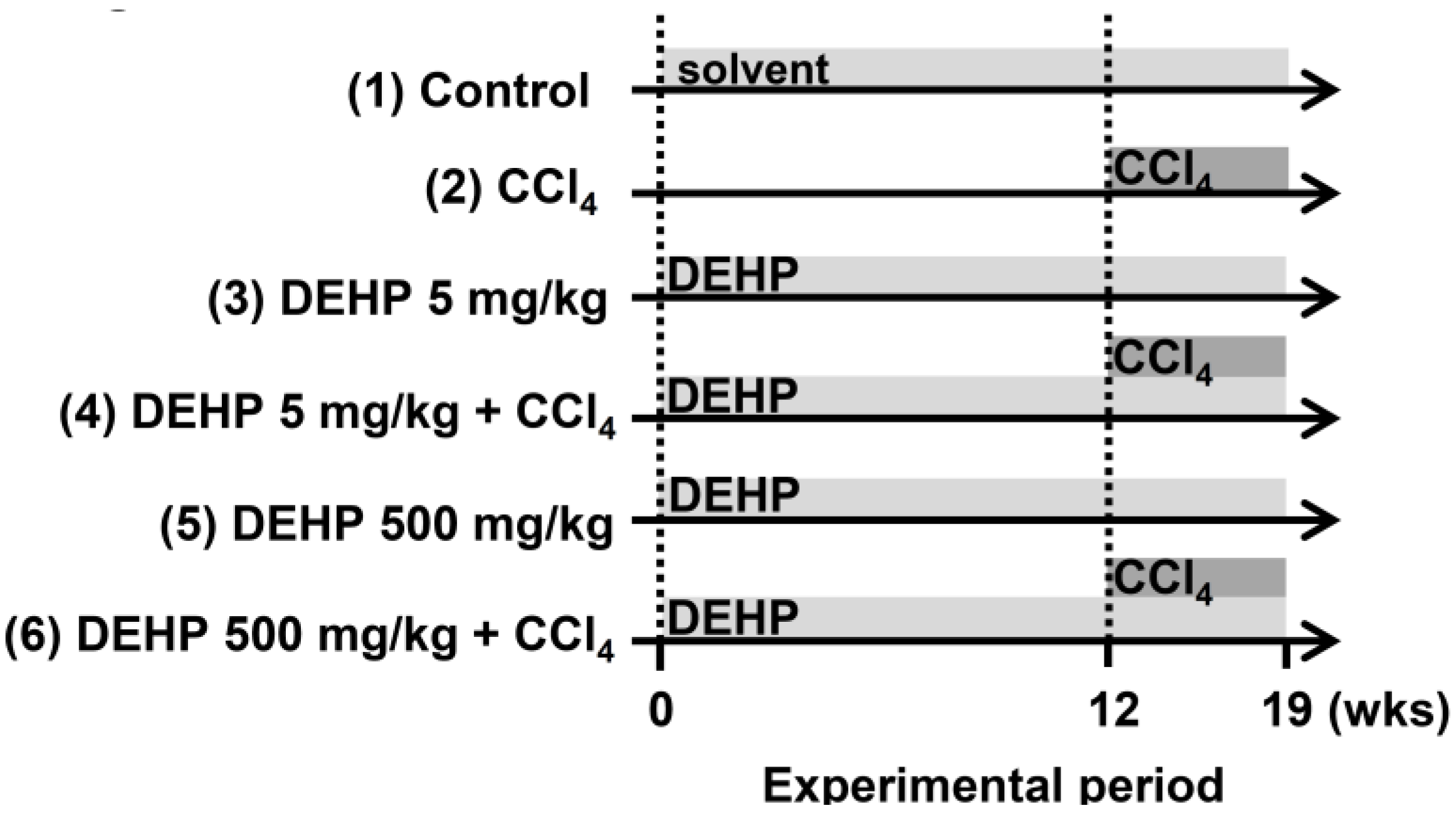
2.7. Established Long-Term, Low-dose, DEHP-exposed Mice for Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis
2.8. Immunohistochemical Staining and Cholesterol Quantification
2.9. Statistical Analyses
3. Results
3.1. Cytotoxicity Effects of DEHP in Hepatic Stellate Cells
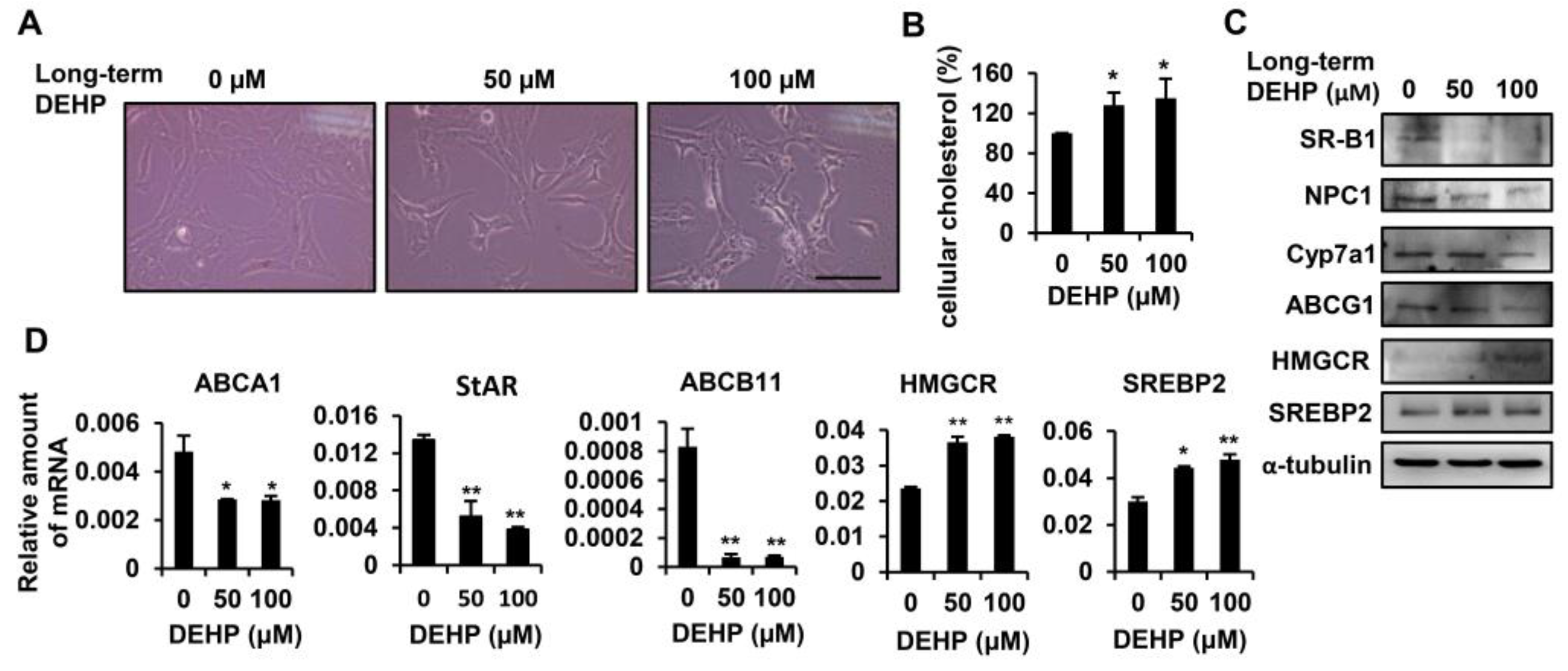
3.2. Long-Term Exposure to Low-Dose DEHP Disturbs Cholesterol Metabolism and Synthesis in Hepatic Stellate Cells
3.3. Long-Term Exposure to Low-Dose DEHP Attenuates PDGF-BB-Induced Cell Proliferation in Hepatic Stellate Cells
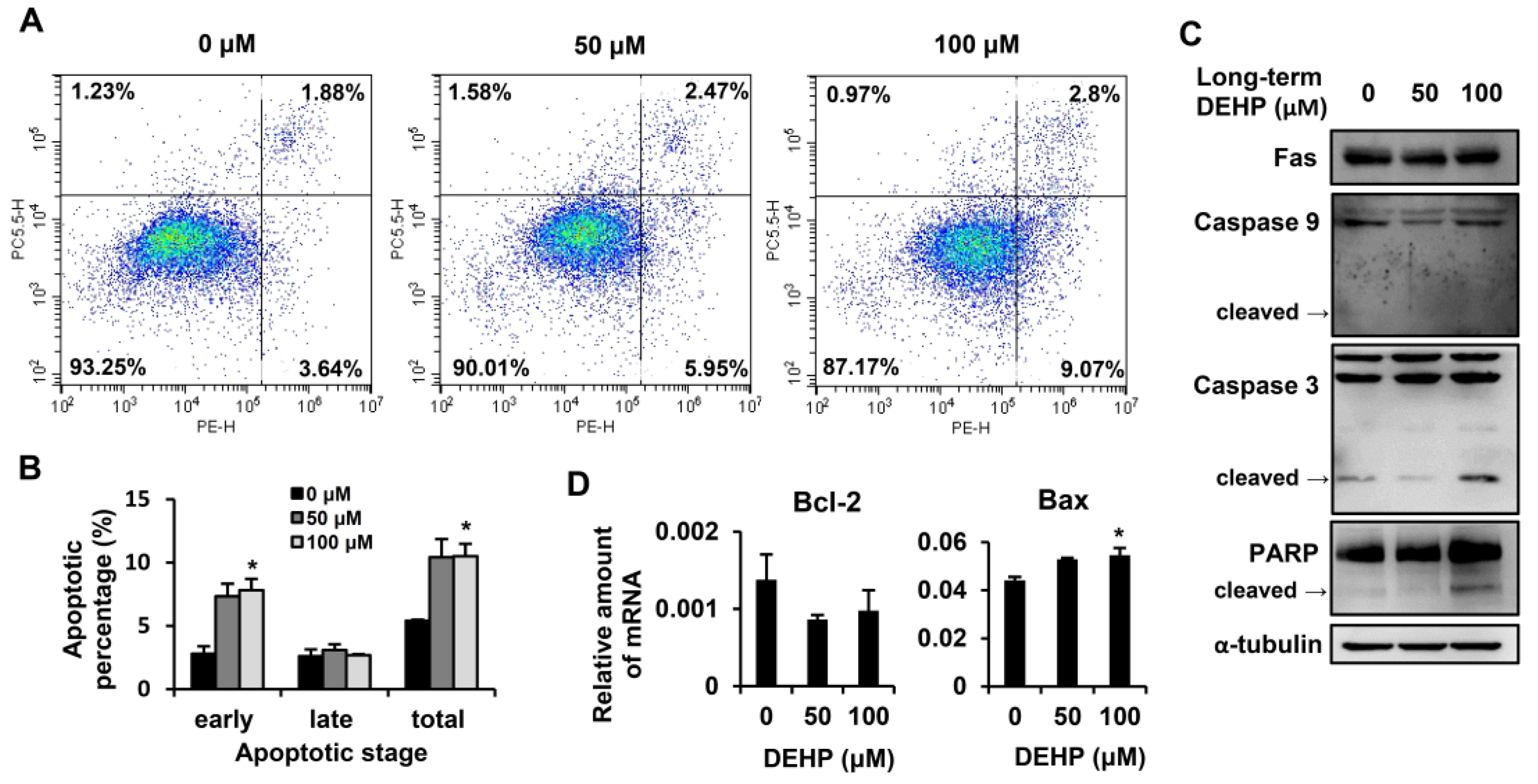
3.4. Long-Term Exposure to Low-Dose DEHP Triggers Apoptosis Signals in Hepatic Stellate Cells
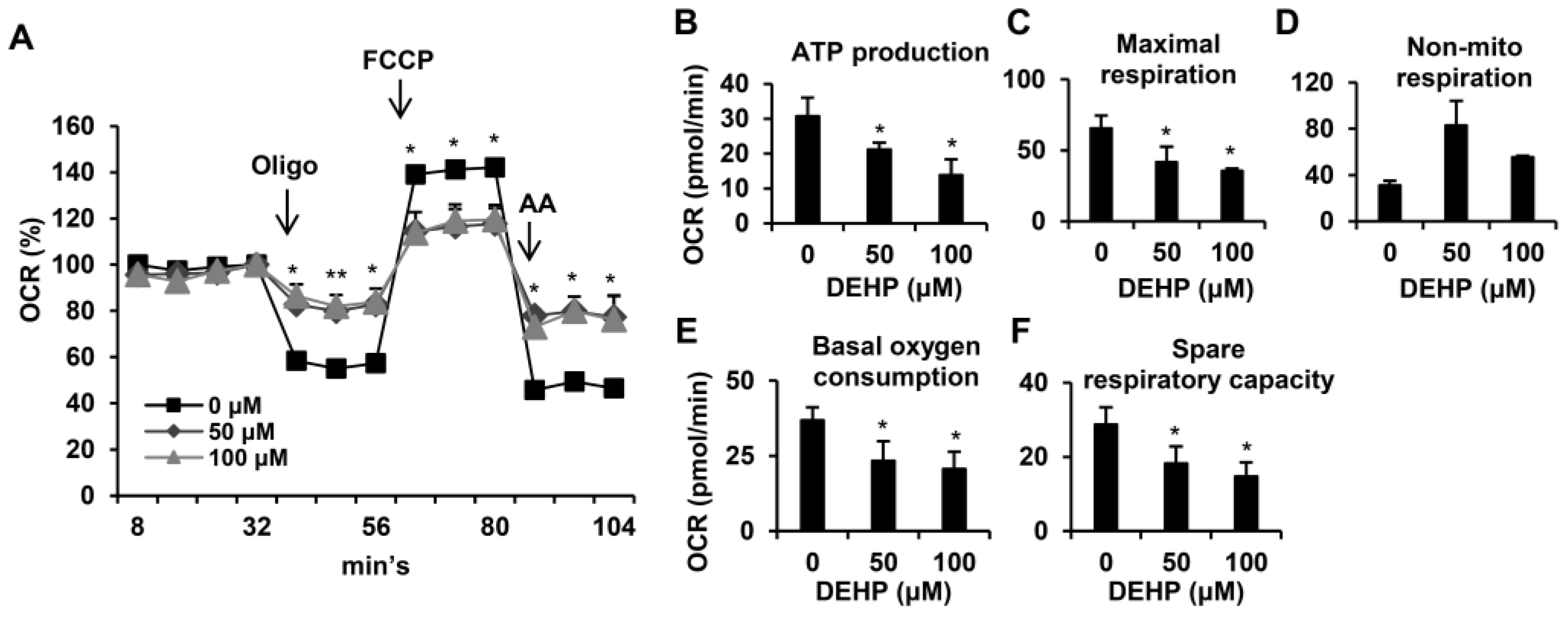
3.5. Long-Term Exposure to Low-Dose DEHP Impairs Mitochondrial Respiration Function in Hepatic Stellate Cells
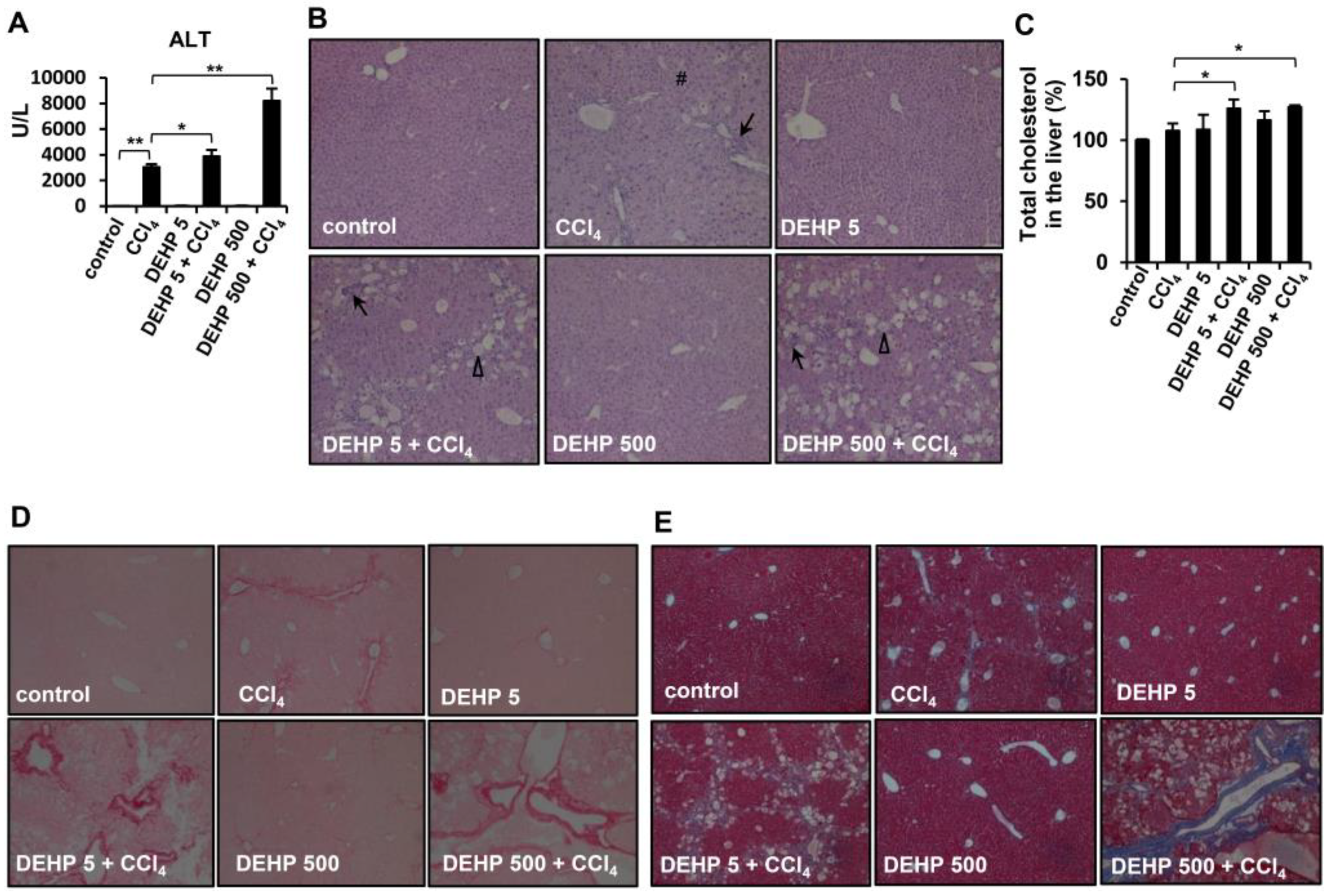
3.6. Chronic Long-Term Exposure to Low-Dose DEHP Accelerates CCl4-induced Liver Damage and Fibrosis in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Halden, R.U. Plastics and health risks. Annu. Rev. Public Health 2010, 31, 179–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-I.; Chiang, C.-W.; Lin, H.-C.; Zhao, J.-F.; Li, C.-T.; Shyue, S.-K.; Lee, T.-S. Maternal exposure to di-(2-ethylhexyl) phthalate exposure deregulates blood pressure, adiposity, cholesterol metabolism and social interaction in mouse offspring. Arch. Toxicol. 2015, 90, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.F.; Hsiao, S.H.; Hsu, M.H.; Pao, K.C.; Kou, Y.R.; Shyue, S.K.; Lee, T.S. Di-(2-ethylhexyl) phthalate accelerates atherosclerosis in apolipoprotein E-deficient mice. Arch. Toxicol. 2016, 90, 181–190. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Rui, B.B.; Yang, S.M.; Xu, W.P.; Wei, W. Di(2-ethylhexyl) phthalate exacerbates non-alcoholic fatty liver in rats and its potential mechanisms. Environ Toxicol. Pharmacol. 2016, 42, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Tickner, J.A.; Schettler, T.; Guidotti, T.; McCally, M.; Rossi, M. Health risks posed by use of Di-2-ethylhexyl phthalate (DEHP) in PVC medical devices: A critical review. Am. J. Ind. Med. 2001, 39, 100–111. [Google Scholar] [CrossRef]
- Chang, J.W.; Lee, C.C.; Pan, W.H.; Chou, W.C.; Huang, H.B.; Chiang, H.C.; Huang, P.C. Estimated Daily Intake and Cumulative Risk Assessment of Phthalates in the General Taiwanese after the 2011 DEHP Food Scandal. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Zuo, Y.Z.; Du, Z.H.; Xia, J.; Zhang, C.; Wang, H.; Li, X.N.; Li, J.L. Di (2-ethylhexyl) phthalate (DEHP)-induced hepatotoxicity in quails (Coturnix japonica) via triggering nuclear xenobiotic receptors and modulating cytochrome P450 systems. Food Chem. Toxicol. 2018, 120, 287–293. [Google Scholar] [CrossRef]
- Hanioka, N.; Isobe, T.; Kinashi, Y.; Tanaka-Kagawa, T.; Jinno, H. Hepatic and intestinal glucuronidation of mono(2-ethylhexyl) phthalate, an active metabolite of di(2-ethylhexyl) phthalate, in humans, dogs, rats, and mice: An in vitro analysis using microsomal fractions. Arch. Toxicol. 2016, 90, 1651–1657. [Google Scholar] [CrossRef]
- Koch, H.M.; Bolt, H.M.; Angerer, J. Di(2-ethylhexyl)phthalate (DEHP) metabolites in human urine and serum after a single oral dose of deuterium-labelled DEHP. Arch. Toxicol. 2004, 78, 123–130. [Google Scholar] [CrossRef]
- Yang, O.; Kim, H.L.; Weon, J.I.; Seo, Y.R. Endocrine-disrupting Chemicals: Review of Toxicological Mechanisms Using Molecular Pathway Analysis. J. Cancer Prev. 2015, 20, 12–24. [Google Scholar] [CrossRef]
- Wittassek, M.; Koch, H.M.; Angerer, J.; Bruning, T. Assessing exposure to phthalates–the human biomonitoring approach. Mol. Nutr. Food Res. 2011, 55, 7–31. [Google Scholar] [CrossRef]
- Doull, J.; Cattley, R.; Elcombe, C.; Lake, B.G.; Swenberg, J.; Wilkinson, C.; Williams, G.; van Gemert, M. A cancer risk assessment of di(2-ethylhexyl)phthalate: Application of the new U.S. EPA Risk Assessment Guidelines. Regul. Toxicol. Pharmacol. 1999, 29, 327–357. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Wei, L.; Guan, X.; Li, L.; Liu, C. p53-dependent apoptosis contributes to di-(2-ethylhexyl) phthalate-induced hepatotoxicity. Environ. Pollut. 2016, 208, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Praveena, S.M.; Teh, S.W.; Rajendran, R.K.; Kannan, N.; Lin, C.C.; Abdullah, R.; Kumar, S. Recent updates on phthalate exposure and human health: A special focus on liver toxicity and stem cell regeneration. Environ. Sci. Pollut. Res. Int. 2018, 25, 11333–11342. [Google Scholar] [CrossRef] [PubMed]
- Foulds, C.E.; Trevino, L.S.; York, B.; Walker, C.L. Endocrine-disrupting chemicals and fatty liver disease. Nat. Rev. Endocrinol. 2017, 13, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Gaitantzi, H.; Hakenberg, P.; Theobald, J.; Heinlein, H.; Cai, C.; Loff, S.; Wolfl, S.; Ebert, M.P.; Breitkopf-Heinlein, K.; Subotic, U. Di (2-Ethylhexyl) Phthalate and Its Role in Developing Cholestasis: An In Vitro Study on Different Liver Cell Types. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 28–35. [Google Scholar] [CrossRef]
- Kushman, M.E.; Kraft, A.D.; Guyton, K.Z.; Chiu, W.A.; Makris, S.L.; Rusyn, I. A systematic approach for identifying and presenting mechanistic evidence in human health assessments. Regul. Toxicol. Pharmacol. 2013, 67, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Rusyn, I.; Kadiiska, M.B.; Dikalova, A.; Kono, H.; Yin, M.; Tsuchiya, K.; Mason, R.P.; Peters, J.M.; Gonzalez, F.J.; Segal, B.H.; et al. Phthalates rapidly increase production of reactive oxygen species in vivo: Role of Kupffer cells. Mol. Pharmacol. 2001, 59, 744–750. [Google Scholar] [CrossRef]
- Ghosh, J.; Das, J.; Manna, P.; Sil, P.C. Hepatotoxicity of di-(2-ethylhexyl)phthalate is attributed to calcium aggravation, ROS-mediated mitochondrial depolarization, and ERK/NF-kappaB pathway activation. Free Radic. Biol. Med. 2010, 49, 1779–1791. [Google Scholar] [CrossRef]
- Wei, N.; Feng, X.; Xie, Z.; Zhang, Y.; Feng, Y. Long-term di (2-ethylhexyl)-phthalate exposure promotes proliferation and survival of HepG2 cells via activation of NFkappaB. Toxicol. In Vitro 2017, 42, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis–from bench to bedside. J. Hepatol. 2003, 38, 38–53. [Google Scholar] [CrossRef]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Invest. 2007, 117, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Twu, Y.C.; Lee, T.S.; Lin, Y.L.; Hsu, S.M.; Wang, Y.H.; Liao, C.Y.; Wang, C.K.; Liang, Y.C.; Liao, Y.J. Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation. Int. J. Mol. Sci. 2016, 17, 1122. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, E.F.; Rathmell, J.C. Cell metabolism: An essential link between cell growth and apoptosis. Biochim. Biophys. Acta 2011, 1813, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Lo, D.; Wang, Y.T.; Wu, M.C. Hepatoprotective effect of silymarin on di(2-ethylhexyl)phthalate (DEHP) induced injury in liver FL83B cells. Environ. Toxicol. Pharmacol. 2014, 38, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Schettler, T. Human exposure to phthalates via consumer products. Int. J. Androl. 2006, 29, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.H.; Ko, Y.C. Plasticizer incident and its health effects in Taiwan. Kaohsiung J. Med. Sci. 2012, 28, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.T.; Wu, C.F.; Wu, J.R.; Chen, B.H.; Chen, E.K.; Chao, M.C.; Liu, C.K.; Ho, C.K. The public health threat of phthalate-tainted foodstuffs in Taiwan: The policies the government implemented and the lessons we learned. Environ. Int. 2012, 44, 75–79. [Google Scholar] [CrossRef]
- Mettang, T.; Thomas, S.; Kiefer, T.; Fischer, F.P.; Kuhlmann, U.; Wodarz, R.; Rettenmeier, A.W. Uraemic pruritus and exposure to di(2-ethylhexyl) phthalate (DEHP) in haemodialysis patients. Nephrol. Dial. Transplant. 1996, 11, 2439–2443. [Google Scholar] [CrossRef]
- Zhao, Z.B.; Ji, K.; Shen, X.Y.; Zhang, W.W.; Wang, R.; Xu, W.P.; Wei, W. Di(2-ethylhexyl) phthalate promotes hepatic fibrosis by regulation of oxidative stress and inflammation responses in rats. Environ. Toxicol. Pharmacol. 2019, 68, 109–119. [Google Scholar] [CrossRef]
- Rigamonti, E.; Helin, L.; Lestavel, S.; Mutka, A.L.; Lepore, M.; Fontaine, C.; Bouhlel, M.A.; Bultel, S.; Fruchart, J.C.; Ikonen, E.; et al. Liver X receptor activation controls intracellular cholesterol trafficking and esterification in human macrophages. Circ. Res. 2005, 97, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Ikonen, E. Mechanisms for cellular cholesterol transport: Defects and human disease. Physiol. Rev. 2006, 86, 1237–1261. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 2002, 110, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiong, S.L.; Yi, G.H. ABCA1, ABCG1, and SR-BI: Transit of HDL-associated sphingosine-1-phosphate. Clin. Chim. Acta 2012, 413, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Patterson, A.D.; Wang, Y.; Krausz, K.W.; Neale, G.; Thomas, S.; Nachagari, D.; Vogel, P.; Vore, M.; et al. Abcb11 deficiency induces cholestasis coupled to impaired beta-fatty acid oxidation in mice. J. Biol. Chem. 2012, 287, 24784–24794. [Google Scholar] [CrossRef] [Green Version]
- Gelissen, I.C.; Harris, M.; Rye, K.A.; Quinn, C.; Brown, A.J.; Kockx, M.; Cartland, S.; Packianathan, M.; Kritharides, L.; Jessup, W. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 534–540. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequences (5’–3’) | Reverse Sequences (5’–3’) |
|---|---|---|
| Bcl-2 | AGGAAGTGAACATTTCGGTGAC | GCTCAGTTCCAGGACCAGGC |
| Bax | GATCCAGGATCGAGCAGA | AAGTAGAAGAGGGCAACCAC |
| ABCA1 | CCCCTGCTTCCGTTATCCA | GGACCTTGTGCATGTCCTTAATG |
| ABCB11 | CAGAACATGACAAACGGAACAAG | CCTGCGTATGCCAGAAAATT |
| HMGCR | TGTGGTTTGTGAAGCCGTCAT | TCAACCATAGCTTCCGTAGTTGTC |
| SREBP2 | CAGACAGCCGCCCTTCAAGT | GCTGTTCATTGACCTTCTCCCG |
| GAPDH | TCACCACCATGGAGAAGGC | GCTAAGCAGTTGGTGGTGCA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-Y.; Suk, F.-M.; Twu, Y.-C.; Liao, Y.-J. Long-Term Exposure to Low-Dose Di-(2-ethylhexyl) Phthalate Impairs Cholesterol Metabolism in Hepatic Stellate Cells and Exacerbates Liver Fibrosis. Int. J. Environ. Res. Public Health 2020, 17, 3802. https://doi.org/10.3390/ijerph17113802
Lee C-Y, Suk F-M, Twu Y-C, Liao Y-J. Long-Term Exposure to Low-Dose Di-(2-ethylhexyl) Phthalate Impairs Cholesterol Metabolism in Hepatic Stellate Cells and Exacerbates Liver Fibrosis. International Journal of Environmental Research and Public Health. 2020; 17(11):3802. https://doi.org/10.3390/ijerph17113802
Chicago/Turabian StyleLee, Chun-Ya, Fat-Moon Suk, Yuh-Ching Twu, and Yi-Jen Liao. 2020. "Long-Term Exposure to Low-Dose Di-(2-ethylhexyl) Phthalate Impairs Cholesterol Metabolism in Hepatic Stellate Cells and Exacerbates Liver Fibrosis" International Journal of Environmental Research and Public Health 17, no. 11: 3802. https://doi.org/10.3390/ijerph17113802