Epigallocatechin-3 Gallate Inhibits STAT-1/JAK2/IRF-1/HLA-DR/HLA-B and Reduces CD8 MKG2D Lymphocytes of Alopecia Areata Patients



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Blood Samples
2.2. Cell Lines
2.3. Treatment with EGCG
2.4. Peripheral Blood Mononuclear Cell Separation and FACS Analysis
2.5. Western Blotting
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. RNA Extraction and cDNA Synthesis
2.8. Q-PCR Analysis of Gene Expression
2.9. Statistical Analysis
3. Results
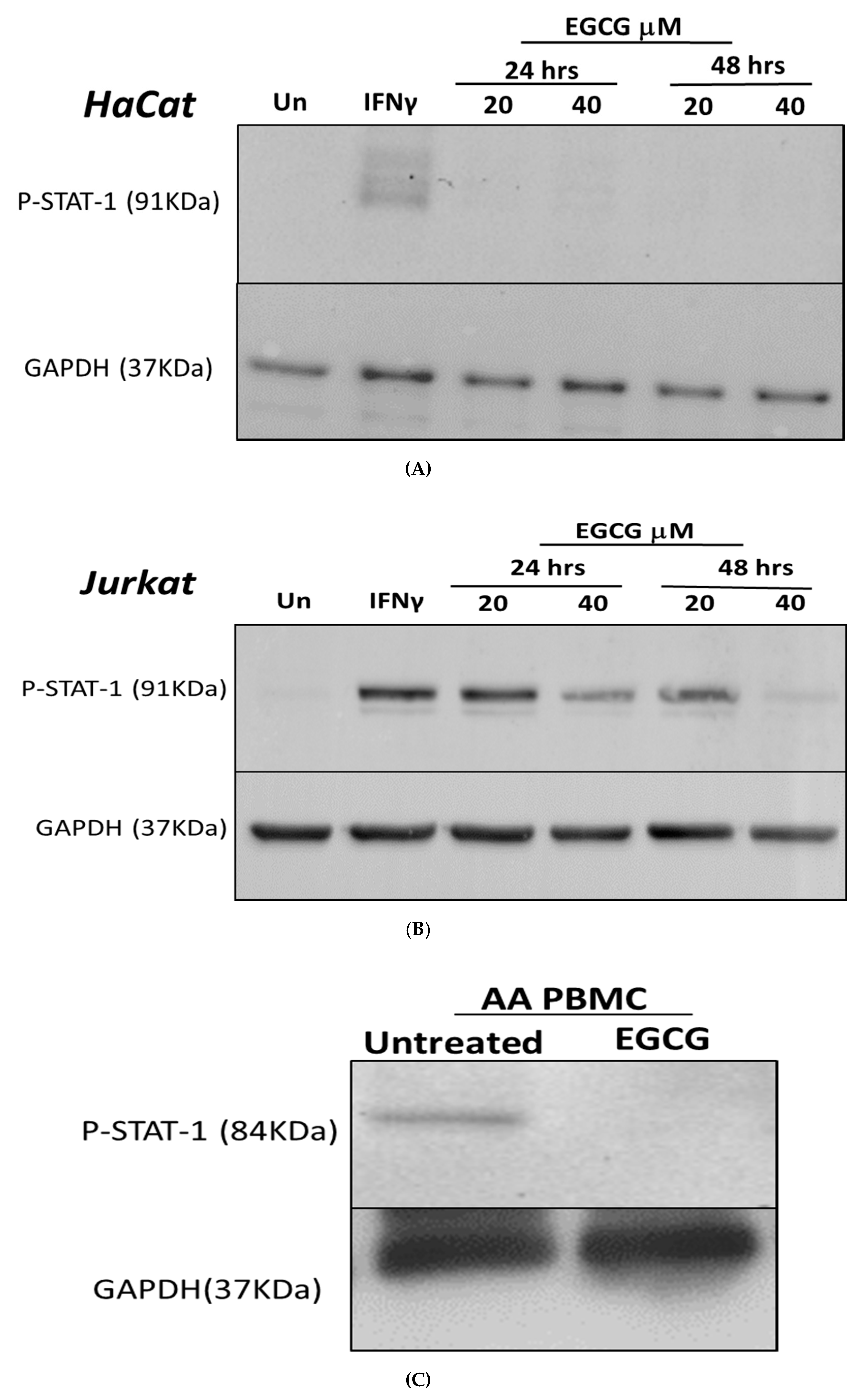
3.1. Inhibition of IFN-γ Signalling Pathway by EGCG
3.2. Effect of EGCG on IFN-γ Downstream Genes
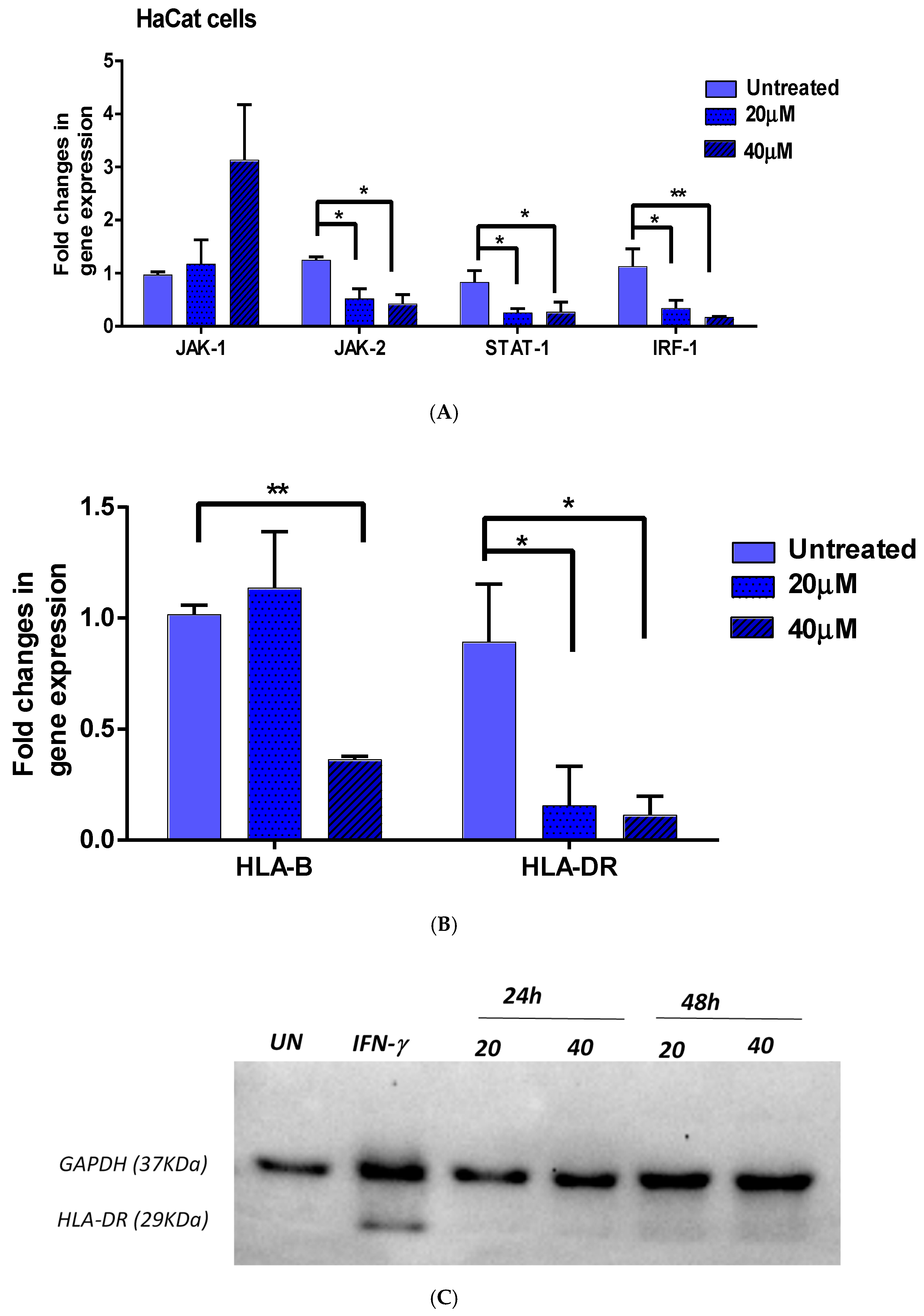
3.2.1. JAK1/JAK2/STAT1 and IRF-1
3.2.2. HLA-DR and HLA-B
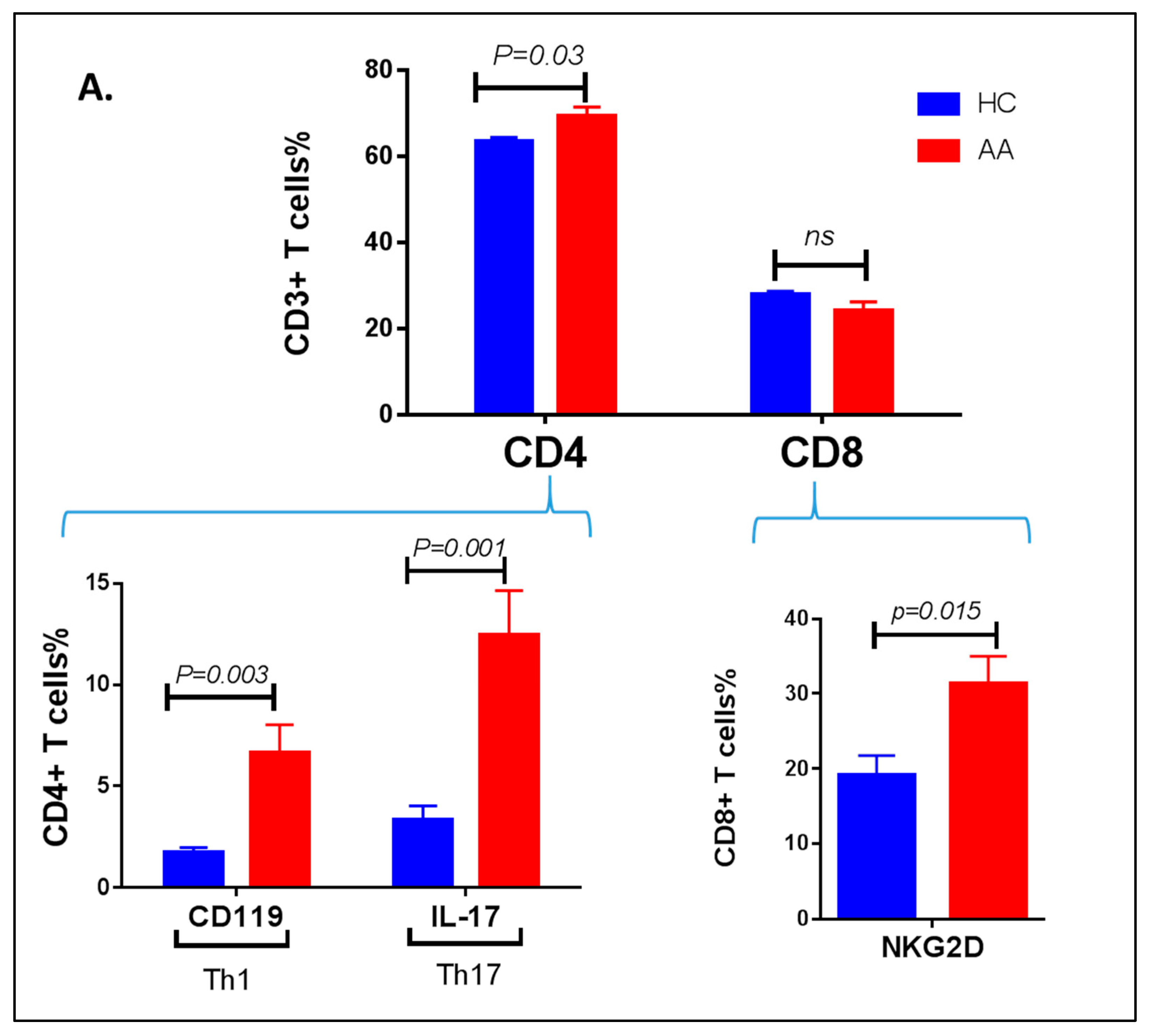
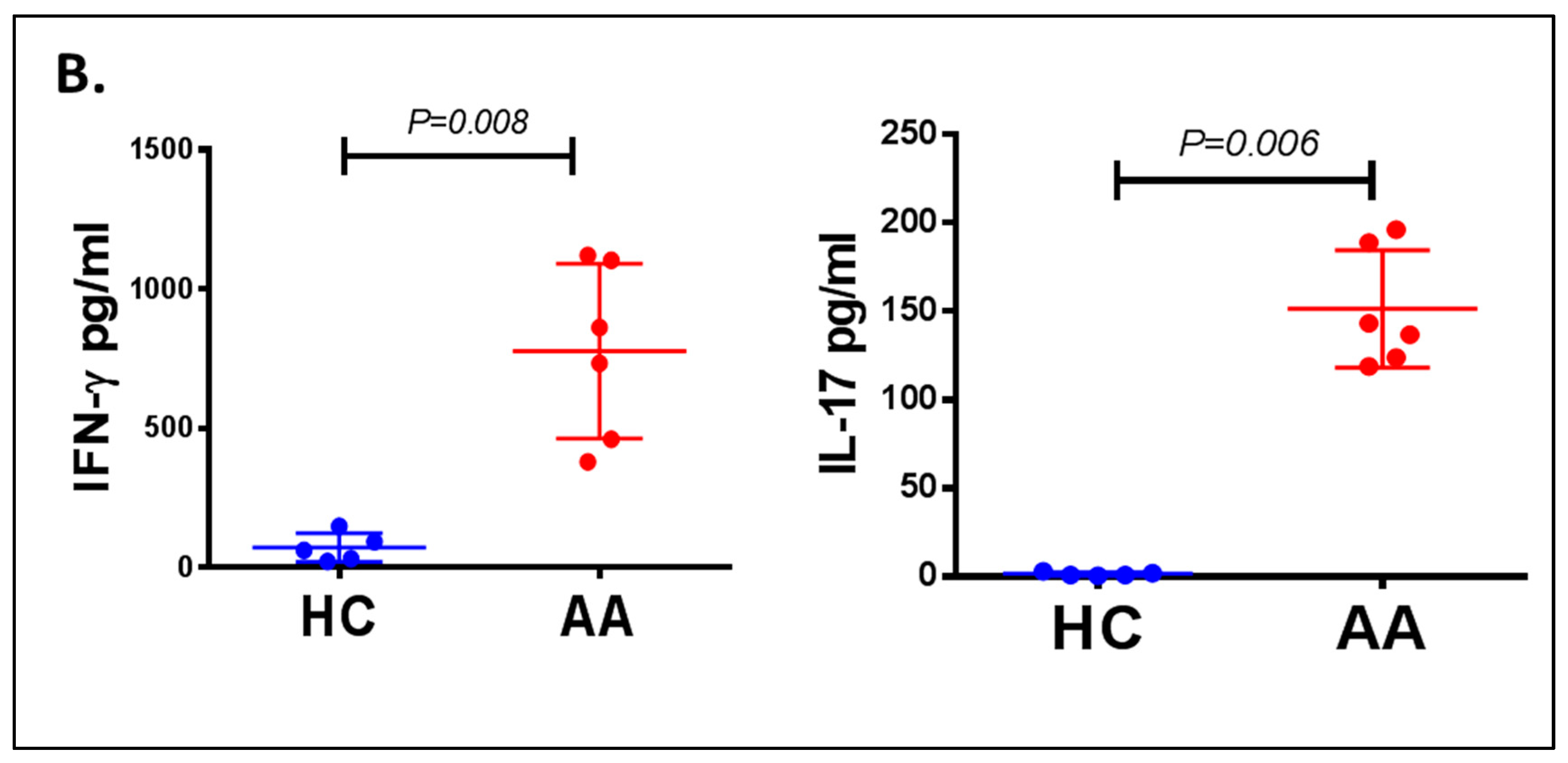
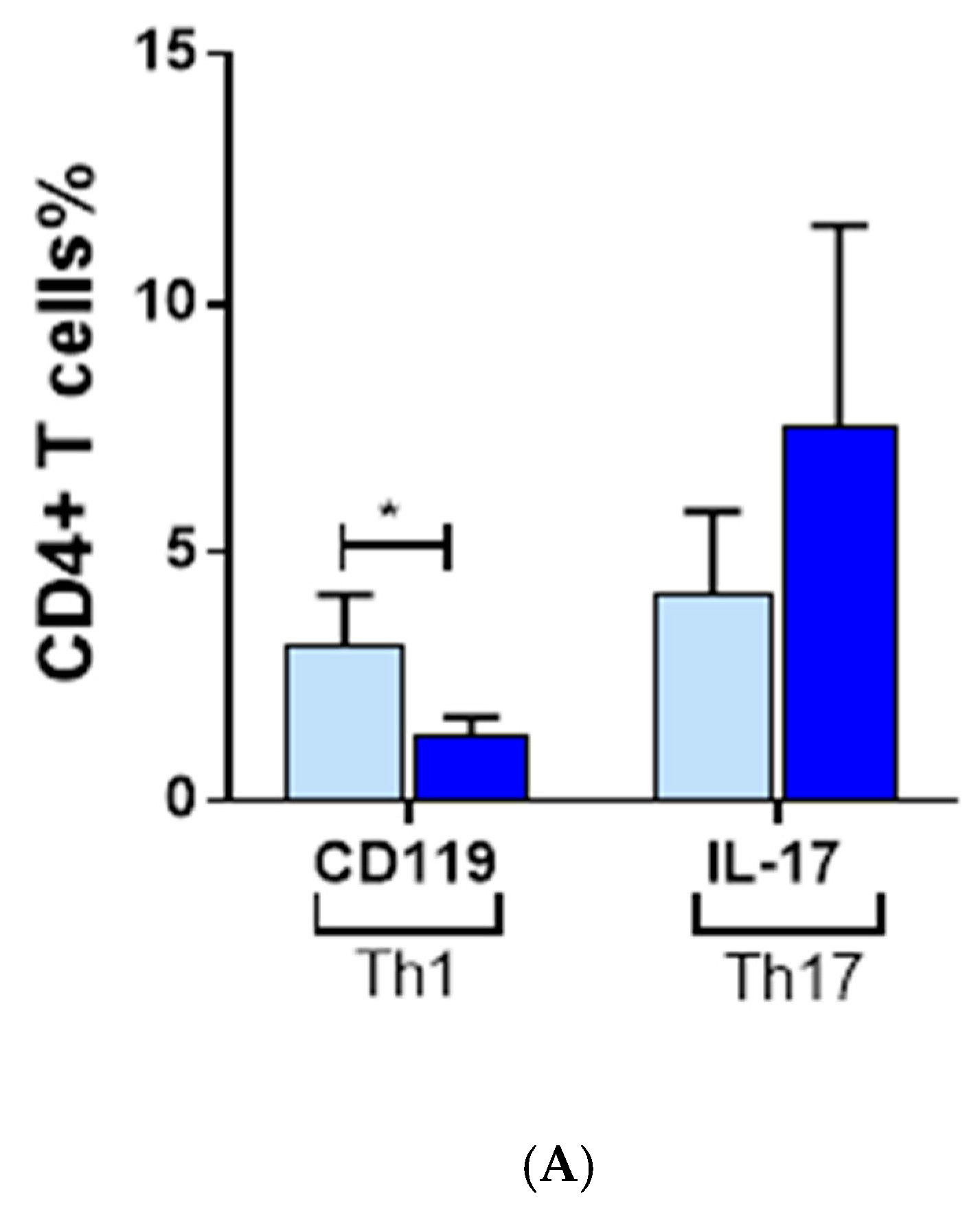
3.3. Inflammatory Cells Th1, NKG2D+ and Th17 and Their Cytokines in AA Patients
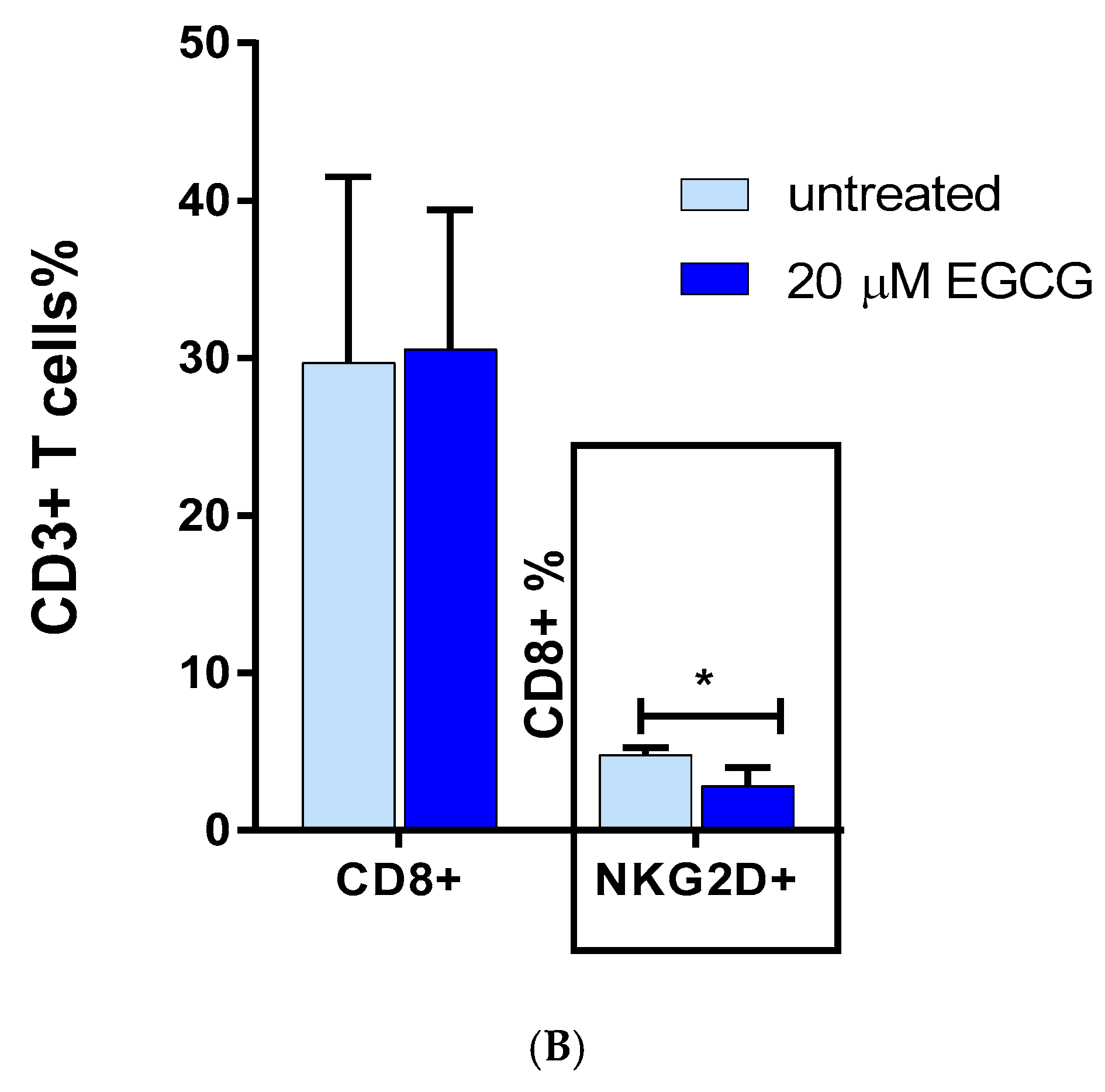
3.4. The Effect of EGCG on CD8 NKG2D Lymphocytes
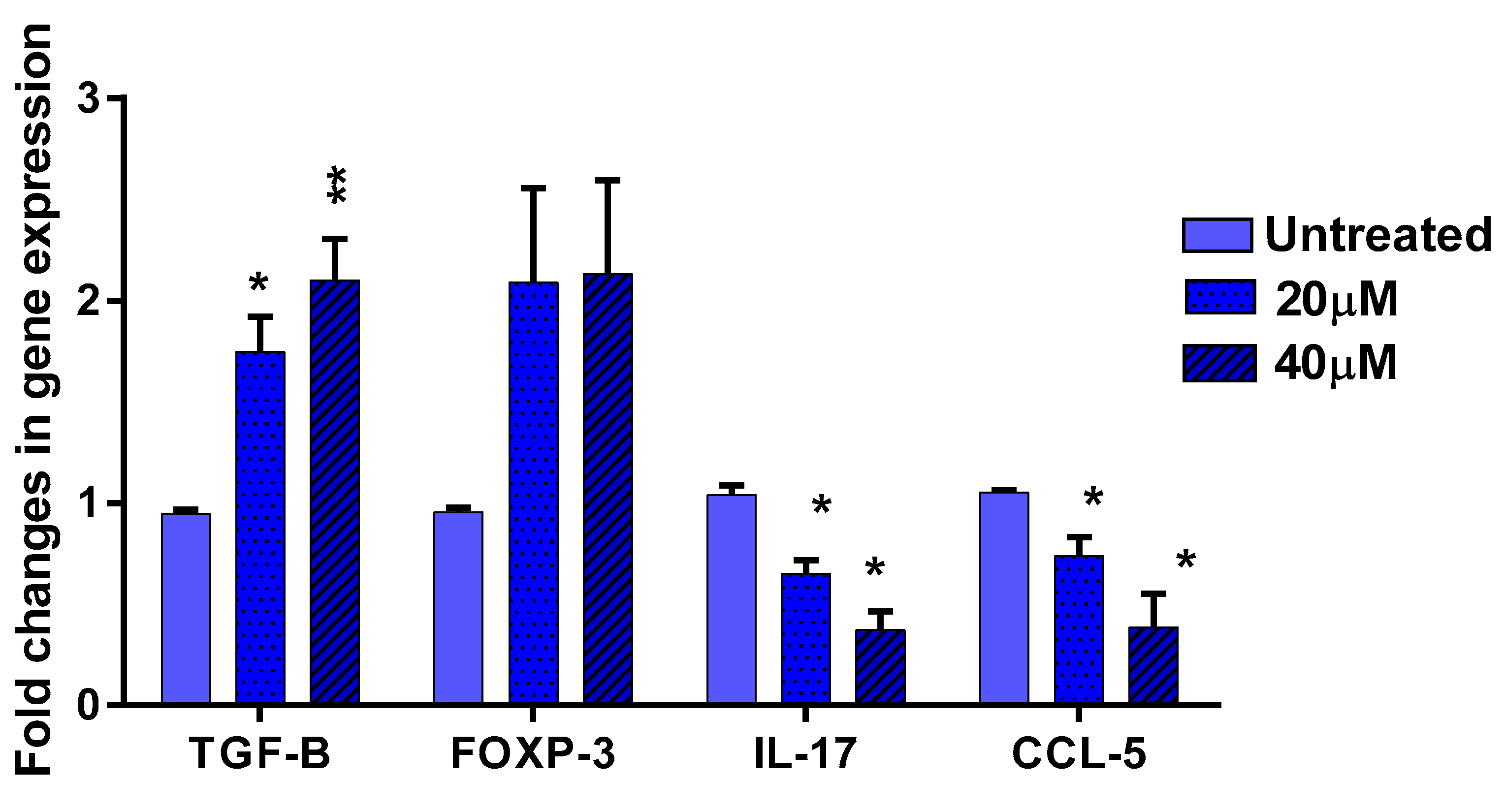
3.5. The Effect of EGCG on Pro- and Anti-Inflammatory Cytokines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- McDonagh, A.; Tazi-Ahnini, R. Epidemiology and genetics of alopecia areata. Clin. Exp. Dermatol. 2002, 27, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Petukhova, L.; Duvic, M.; Hordinsky, M.; Norris, D.; Price, V.; Shimomura, Y.; Kim, H.; Singh, P.; Lee, A.; Chen, W.V.; et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature 2010, 466, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Andrade, M.; Jackow, C.M.; Dahm, N.; Hordinsky, M.; Reveille, J.D.; Duvic, M. Alopecia areata in families: Association with the HLA locus. J. Investig. Dermatol. Symp. Proc. 1999, 4, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Barahmani, N.; De Andrade, M.; Slusser, J.P.; Zhang, Q.; Duvic, M. Major histocompatibility complex class I chain-related gene A polymorphisms and extended haplotypes are associated with familial alopecia areata. J. Investig. Dermatol. 2006, 126, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Tazi-Ahnini, R.; Cork, M.; Wengraf, D.; Wilson, A.G.; Gawkrodger, D.J.; Birch, M.P.; Messenger, A.G.; McDonagh, A.J. Notch4, a non-HLA gene in the MHC is strongly associated with the most severe form of alopecia areata. Hum. Genet. 2003, 112, 400–403. [Google Scholar]
- Collins, S.; Dominguez, M.; Ilmarinen, T.; Costigan, C.; Irvine, A.D. Dermatological manifestations of autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy syndrome. Br. J. Dermatol. 2006, 154, 1088–1093. [Google Scholar] [CrossRef]
- Tazi-Ahnini, R.; Cork, M.J.; Gawkrodger, D.J.; Birch, M.P.; Wengraf, D.; McDonagh, A.J.; Messenger, A.G. Role of the autoimmune regulator (AIRE) gene in alopecia areata: Strong association of a potentially functional AIRE polymorphism with alopecia universalis. Tissue Antigens 2002, 60, 489–495. [Google Scholar] [CrossRef]
- Wengraf, D.A.; McDonagh, A.J.; Lovewell, T.R.; Vasilopoulos, Y.; Macdonald-Hull, S.P.; Cork, M.J.; Messenger, A.G.; Tazi-Ahnini, R. Genetic analysis of autoimmune regulator haplotypes in alopecia areata. Tissue Antigens 2008, 71, 206–212. [Google Scholar] [CrossRef]
- Pforr, J.; Blaumeiser, B.; Becker, T.; Freudenberg-Hua, Y.; Hanneken, S.; Eigelshoven, S.; Cuyt, I.; De Weert, J.; Lambert, J.; Kruse, R.; et al. Investigation of the p.Ser278Arg polymorphism of the autoimmune regulator (AIRE) gene in alopecia areata. Tissue Antigens 2006, 68, 58–61. [Google Scholar] [CrossRef]
- Kemp, E.H.; McDonagh, A.J.; Wengraf, D.A.; Messenger, A.G.; Gawkrodger, D.J.; Cork, M.J.; Tazi-Ahnini, R. The non-synonymous C1858T substitution in the PTPN22 gene is associated with susceptibility to the severe forms of alopecia areata. Hum. Immunol. 2006, 67, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Betz, R.C.; König, K.; Flaquer, A.; Redler, S.; Eigelshoven, S.; Kortüm, A.K.; Hanneken, S.; Hillmer, A.; Tüting, T.; Lambert, J.; et al. The R620W polymorphism in PTPN22 confers general susceptibility for the development of alopecia areata. Br. J. Dermatol. 2008, 158, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Cork, M.J.; Tarlow, J.K.; Clay, F.E.; Crane, A.; Blakemore, A.I.; McDonagh, A.J.; Messenger, A.G.; Duff, G.W. An allele of the interleukin-1 receptor antagonist as a genetic severity factor in alopecia areata. J. Investig. Dermatol. 1995, 104, 15S–16S. [Google Scholar] [CrossRef] [PubMed]
- Tarlow, J.K.; Clay, F.E.; Cork, M.J.; Blakemore, A.I.; McDonagh, A.J.; Messenger, A.G.; Duff, G.W. Severity of alopecia areata is associated with a polymorphism in the interleukin-1 receptor antagonist gene. J. Investig. Dermatol. 1994, 103, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Barahamani, N.; De Andrade, M.; Slusser, J.; Zhang, Q.; Duvic, M. Interleukin-1 receptor antagonist allele 2 and familial alopecia areata. J. Investig. Dermatol. 2002, 118, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Tazi-Ahnini, R.; Cox, A.; McDonagh, A.J.; Nicklin, M.J.; di Giovine, F.S.; Timms, J.M.; Messenger, A.G.; Dimitropoulou, P.; Duff, G.W.; Cork, M.J. Genetic analysis of the interleukin-1 receptor antagonist and its homologue IL-1L1 in alopecia areata: Strong severity association and possible gene interaction. Eur. J. Immunogenet. 2002, 29, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Tazi-Ahnini, R.; di Giovine, F.S.; McDonagh, A.J.; Messenger, A.G.; Amadou, C.; Cox, A.; Duff, G.W.; Cork, M.J. Structure and polymorphism of the human gene for the interferon-induced p78 protein (MX1): Evidence of association with alopecia areata in the Down syndrome region. Hum. Genet. 2000, 106, 639–645. [Google Scholar]
- Pan, F.; Yu, H.; Dang, E.V. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science 2009, 325, 1142–1146. [Google Scholar] [CrossRef]
- Akar, A.; Arca, E.; Erbil, H.; Akay, C.; Sayal, A.; Gür, A.R. Antioxidant enzymes and lipid peroxidation in the scalp of patients with alopecia areata. J. Dermatol. Sci. 2002, 29, 85–90. [Google Scholar] [CrossRef]
- Karasawa, R.; Ozaki, S.; Nishioka, K.; Kato, T. Autoantibodies to peroxiredoxin I and IV in patients with systemic autoimmune diseases. Microbiol. Immunol. 2005, 49, 57–65. [Google Scholar] [CrossRef]
- Pielberg, G.R.; Golovko, A.; Sundström, E.; Curik, I.; Lennartsson, J.; Seltenhammer, M.H.; Druml, T.; Binns, M.; Fitzsimmons, C.; Lindgren, G.; et al. A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat. Genet. 2008, 40, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Singh, L.K.; Boucher, W.; Pang, X.; Letourneau, R.; Webster, E.; Chrousos, G. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology 1998, 139, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.M.; Liotiri, S.; Bodó, E.; Hagen, E.; Bíró, T.; Arck, P.C.; Paus, R. Probing the effects of stress mediators on the human hair follicle: Substance P holds central position. Am. J. Pathol. 2007, 171, 1872–1886. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.; Liberman, R. Local injection of nerve growth factor (NGF) triggers degranulation of mast cells in rat paw. Neurosci. Lett. 1997, 221, 129–132. [Google Scholar] [CrossRef]
- Ansel, J.; Brown, J.; Payan, D.; Brown, M.A. Substance P selectively activates TNF-α gene expression in murine mast cells. J. Immunol. 1993, 150, 4478–4485. [Google Scholar] [PubMed]
- Westgate, G.E.; Craggs, R.I.; Gibson, W.T. Immune privilege in hair-growth. J. Investig. Dermatol. 1991, 97, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Christoph, T.; Muller-Rover, S.; Audring, H.; Tobin, D.J.; Hermes, B.; Cotsarelis, G.; Rückert, R.; Paus, R. The human hair follicle immune system: Cellular composition and immune privilege. Br. J. Dermatol. 2000, 142, 862–873. [Google Scholar] [CrossRef]
- Moresi, J.M.; Horn, T.D. Distribution of langerhans cells in human hair follicle. J. Cutan. Pathol. 1997, 24, 636–640. [Google Scholar] [CrossRef]
- Paus, R.; Cotsarelis, G.; Epstein, F.H. The biology of hair follicles. New Eng. J. Med. 1999, 341, 491–497. [Google Scholar] [CrossRef]
- Gruschwitz, M.; Müller, P.U.; Sepp, N.; Hofer, E.; Fontana, A.; Wick, G. Transcription and expression of transforming growth factor type beta in the skin of progressive systemic sclerosis: A mediator of fibrosis? J. Investig. Dermatol. 1990, 94, 197–203. [Google Scholar] [CrossRef]
- Paus, R.; Slominski, A.; Czarnetzki, B.M. Is alopecia-areata an autoimmune-response against melanogenesis-related proteins, exposed by abnormal MHC class-I expression in the anagen hair bulb. Yale J. Biol. Med. 1993, 66, 541–554. [Google Scholar] [PubMed]
- Ito, T.; Ito, N.; Bettermann, A.; Tokura, Y.; Takigawa, M.; Paus, R. Collapse and restoration of MHC class-I-dependent immune privilege: Exploiting the human hair follicle as a model. Am. J. Pathol. 2004, 164, 623–634. [Google Scholar] [CrossRef]
- Messenger, A.G.; Bleehen, S.S. Expression of HLA-DR by anagen hair follicles in alopecia areata. J. Investig. Dermatol. 1985, 85, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Mc Donagh, A.J.; Snowden, J.A.; Stierle, C.; Elliott, K.; Messenger, A.G. HLA and ICAM-1 expression in alopecia areata in vivo and in vitro: The role of cytokines. Br. J. Dermatol. 1993, 129, 250–256. [Google Scholar] [CrossRef]
- Emilo, L.K.; Vera, H.P.; John, S.G. HLA-DR expression by hair follicle keratinocytes in alopecia areata: Evidence that it is secondary to the lymphoid infiltration. J. Investig. Dermatol. 1988, 90, 193. [Google Scholar]
- Taylor, A.W.; Streilein, J.W.; Cousins, S.W. Identification of alpha-melanocyte stimulating hormone as a potential immunosuppressive factor in aqueous-humor. Curr. Eye Res. 1992, 11, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Paus, R.; Ito, N.; Takigawa, M.; Ito, T. The hair follicle and immune privilege. J. Investig. Dermatol. Symp. Proc. 2003, 8, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Gilhar, A.; Paus, R.; Kalish, R.S. Lymphocytes, neuropeptides, and genes involved in alopecia areata. J. Clin. Investig. 2007, 117, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Fariñas, M.; Ungar, B.; Noda, S.; Shroff, A.; Mansouri, Y.; Fuentes-Duculan, J.; Czernik, A.; Zheng, X.; Estrada, Y.D.; Xu, H.; et al. Alopecia areata profiling shows Th1, Th2, and IL-23 cytokine activation without parallel Th17/Th22 skewing. J. Allergy. Clin. Immunol. 2015, 136, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Todes-Taylor, N.; Turner, R.; Wood, G.S. T cell subpopulations in alopecia areata. J. Am. Acad. Dermatol. 1984, 11, 216–223. [Google Scholar] [CrossRef]
- Perret, C.; Wiesner-Menzel, L.; Happle, R. Immunohistochemical analysis of T-cell subsets in the peribulbar and intrabulbar infiltrates of alopecia areata. Acta Derm. Venereol 1984, 64, 26–30. [Google Scholar] [PubMed]
- Zhu, J.F.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Romagnani, P.; Annunziato, F.; Piccinni, M.P.; Maggi, E.; Romagnani, S. Th1/Th2 cells, their associated molecules and role in pathophysiology. Eur. Cytokine Netw. 2000, 11, 510–511. [Google Scholar] [PubMed]
- Noble, A.; Staynov, D.Z.; Kemeny, D.M. Generation of rat Th2-like cells in-vitro is interleukin-4-dependent and inhibited by interferon-gamma. Immunology 1993, 79, 562–567. [Google Scholar] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Hirahara, K.; O’Shea, J.J. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011, 32, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levings, M.K.; Bacchetta, R.; Schulz, E.; Roncarolo, M.G. The role of IL-10 and TGF-beta in the differentiation and effector function of t regulatory cells. Int. Arch. Allergy Immunol. 2002, 129, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.R.M.; Legrand, N.; Papiernik, M.; Freitas, A.A. Homeostasis of peripheral CD4(+) T cells: IL-2R alpha and il-2 shape a population of regulatory cells that controls CD4(+) T cell numbers. J. Immunol. 2002, 169, 4850–4860. [Google Scholar] [CrossRef]
- Kuwano, Y.; Fujimoto, M.; Watanabe, R.; Ishiura, N.; Nakashima, H.; Ohno, Y.; Yano, S.; Yazawa, N.; Okochi, H.; Tamaki, K. Serum chemokine profiles in patients with alopecia areata. Br. J. Dermatol. 2007, 157, 466–473. [Google Scholar] [CrossRef]
- Teraki, Y.; Imanishi, K.; Shiohara, T. Cytokines in alopecia areata: Contrasting cytokine profiles in localized form and extensive form (alopecia universalis). Acta Dermatovenereol. Venereol. 1996, 76, 421–423. [Google Scholar]
- Tawfic, S.O.; Abdel-halim, M.R.E.; Shaker, O.G. Assessment of interleukin-17 in alopecia areata: A case–control pilot study. J. Egyp. Women’s Dermatol. Soc. 2014, 11, 20–23. [Google Scholar] [CrossRef]
- Tojo, G.; Fujimura, T.; Kawano, M.; Ogasawara, K.; Kambayashi, Y.; Furudate, S.; Mizuashi, M.; Aiba, S. Comparison of interleukin-17-producing cells in different clinical types of alopecia areata. Dermatology 2013, 227, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Tanemura, A.; Oiso, N.; Nakano, M.; Itoi, S.; Kawada, A.; Katayama, I. Alopecia areata: Infiltration of Th17 cells in the dermis, particularly around hair follicles. Dermatology 2013, 226, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Lew, B.I.; Cho, H.R.; Haw, S.; Kim, H.J.; Chung, J.H.; Sim, W.Y. Association between IL17a/IL17RA gene polymorphisms and susceptibility to alopecia areata in the korean population. Ann. Dermatol. 2012, 24, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Aytekin, N.; Akcali, C.; Pehlivan, S.; Kirtak, N.; Inaloz, S. Investigation of interleukin-12, interleukin-17 and interleukin-23 receptor gene polymorphisms in alopecia areata. J. Int. Med. Res. 2015, 43, 526–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Morsy, E.H.; Eid, A.A.; Ghoneim, H.; Al-Tameemi, K.A. Serum level of interleukin-17a in patients with alopecia areata and its relationship to age. Int. J. Dermatol. 2016, 55, 869–874. [Google Scholar] [CrossRef]
- Ito, T.; Ito, N.; Saatoff, M. Maintenance of hair follicle immune privilege is linked to prevention of nk cell attack. J. Investig. Dermatol. 2008, 128, 1196–1206. [Google Scholar] [CrossRef]
- Xing, I.Z.; Dai, Z.P.; Jabbari, A.; Cerise, J.E.; Higgins, C.A.; Gong, W.; de Jong, A.; Harel, S.; DeStefano, G.M.; Rothman, L.; et al. Alopecia areata is driven by cytotoxic t lymphocytes and is reversed by JAK inhibition. Nat. Med. 2014, 20, 1043–1049. [Google Scholar] [CrossRef]
- Horvath, C.M. The JAK-STAT pathway stimulated by interferon gamma. Sci. STKE 2004, 23, tr8. [Google Scholar]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. JAK-STAT pathways and transcriptional activation in response to ifns and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- White, I.C.; Wright, K.I.; Felix, N.J.; Ruffner, H.; Ruffner, H.; Reis, L.F.; Pine, R.; Ting, J.P. Regulation of LMP2 and TAP1 genes by IRF-1 explains the paucity of CD8 + t cells in Irf-1(-/-) mice. Immunity 1996, 5, 365–376. [Google Scholar] [CrossRef]
- Lechleitner, S.; Gille, J.; Johnson, D.R.; Petzelbauer, P. Interferon enhances tumor necrosis factor-induced vascular cell adhesion molecule 1 (CD106) expression in human endothelial cells by an interferon-related factor 1-dependent pathway. J. Exp. Med. 1998, 187, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kimura, T.; Kitagawa, M.; Pfeffer, K.; Kawakami, T.; Watanabe, N.; Kündig, T.M.; Amakawa, R.; Kishihara, K.; Wakeham, A.; et al. Targeted disruption of irf-1 or irf-2 results in abnormal type i ifn gene induction and aberrant lymphocyte development. Cell 1993, 75, 83–97. [Google Scholar] [CrossRef]
- Fragale, A.; Gabriele, I.; Stellacci, E.; Borghi, P.; Perrotti, E.; Ilari, R.; Lanciotti, A.; Remoli, A.L.; Venditti, M.; Belardelli, F.; et al. IFN regulatory factor-1 negatively regulates CD4 + CD25 + regulatory t cell differentiation by repressing foxp3 expression. J. Immunol. 2008, 181, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.Y.; Craiglow, B.G.; Dai, F.; King, B.A. Tofacitinib for the treatment of severe alopecia areata and variants: A study of 90 patients. J. Am. Acad. Dermatol. 2017, 76, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, A.; Dai, Z.; Xing, I.; Cerise, J.E.; Ramot, Y.; Berkun, Y.; Sanchez, G.A.; Goldbach-Mansky, R.; Christiano, A.M.; Clynes, R.; et al. Reversal of alopecia areata following treatment with the JAK1/2 inhibitor baricitinib. Ebiomedicine 2015, 2, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Shreberk-Hassidim, R.; Ramot, Y.; Zlotogorski, A. Janus kinase inhibitors in dermatology: A systematic review. J. Am. Acad. Dermatol. 2017, 76, 745–753.e19. [Google Scholar] [CrossRef]
- Yang, C.S.; Maliakal, M.; Meng, X. Inhibition of carcinogenesis by tea. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 25–54. [Google Scholar] [CrossRef]
- Cheng, C.W.; Shieh, P.C.; Lin, Y.C.; Chen, Y.J.; Lin, Y.H.; Kuo, D.P.; Liu, J.Y.; Kao, J.; Kao, M.C.; Way, T.D. Indoleamine 2,3-dioxygenase, an immunomodulatory protein, is suppressed by (-)-epigallocatechin-3-gallate via blocking of gamma-interferon-induced JAK-PKC-delta-STAT1 signaling in human oral cancer cells. Biochem. Biophys. Res. Commun. 2007, 354, 1004–1009. [Google Scholar]
- Ogawa, K.; Hara, T.; Shimizu, M.; Nagano, J.; Ohno, T.; Hoshi, M.; Ito, H.; Tsurumi, H.; Saito, K.; Seishima, M.; et al. (-)-Epigallocatechin gallate inhibits the expression of indoleamine 2,3-dioxygenase in human colorectal cancer cells. Oncol. Lett. 2012, 4, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Pae, M.; Ren, Z.H.; Meydani, M.; Shang, F.; Meydani, S.N.; Wu, D. Epigallocatechin-3-gallate directly suppresses t cell proliferation through impaired il-2 utilization and cell cycle progression. J. Nutr. 2010, 140, 1509–1515. [Google Scholar] [CrossRef]
- Aktas, O.; Prozorovski, T.; Smorodchenko, A.; Savaskan, N.E.; Lauster, R.; Kloetzel, P.M.; Infante-Duarte, C.; Brocke, S.; Zipp, F. Green tea epigallocatechin-3-gallate mediates t cellular nf-kappa b inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J. Immunol. 2004, 173, 5794–5800. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Guo, Z.; Ren, Z.; Guo, W.; Meydani, S.N. Green tea EGCG suppresses t cell proliferation through impairment of IL-2/IL-2 receptor signaling. Free Rad. Biol. Med. 2009, 47, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Matsui, M.S.; Elmets, C.A.; Mukhtar, H. Polyphenolic antioxidant (-)-epigallocatechin-3-gallate from green tea reduces uvb-induced inflammatory responses and infiltration of leukocytes in human skin. Photochem. Photobiol. 1999, 69, 148–153. [Google Scholar] [CrossRef]
- Chow, H.H.S.; Cai, Y.; Hakim, I.A.; Crowell, J.A.; Shahi, F.; Brooks, C.A.; Dorr, R.T.; Hara, Y.; Alberts, D.S. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon e in healthy individuals. Clin. Cancer Res. 2003, 9, 3312–3319. [Google Scholar] [PubMed]
- Scalia, S.; Trotta, V.; Bianchi, A. In vivo human skin penetration of (-)-epigallocatechin-3-gallate from topical formulations. Acta Pharm. 2004, 64, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.; Armstrong, A.W. JAK inhibitors: Treatment efficacy and safety profile in patients with psoriasis. J. Immunol. Res 2014, 2014, 283617. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin. Proc. 2011, 86, 1188–1191. [Google Scholar] [CrossRef]
- Zhao, H.; Zhu, W.; Jia, L.; Sun, X.; Chen, G.; Zhao, X.; Li, X.; Meng, X.; Kong, L.; Xing, L.; et al. Phase I study of topical epigallocatechin-3-gallate (EGCG) in patients with breast cancer receiving adjuvant radiotherapy. Br. J. Radiol. 2016, 89, 20150665. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, E.; Suzuki, H.; Menegazzi, M. Antiinflammatory action of egcg, the main component of green tea, through stat-1 inhibition. Ann. N. Y. Acad. Sci. 2002, 973, 435–437. [Google Scholar] [CrossRef]
- Watson, J.I.; Ansari, S.; Cameron, H.; Wang, A.; Akhtar, M.; McKay, D.M. Green tea polyphenol (-)-epigallocatechin gallate blocks epithelial barrier dysfunction provoked by IFN-gamma but not by IL-4. Am. J. Physiol. Gast Liver Physiol. 2004, 287, g954. [Google Scholar] [CrossRef] [PubMed]
- Zoller, M.; Mcelwee, K.J.; Vitacolonna, M.; Hoffmann, R. The progressive state, in contrast to the stable or regressive state of alopecia areata, is reflected in peripheral blood mononuclear cells. Exp. Dermatol. 2004, 13, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Tembhre, M.K.; Sharma, V.K. T-helper and regulatory T-cell cytokines in the peripheral blood of patients with active alopecia areata. Br. J. Dermatol. 2013, 169, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Ercan, A.R.C.A.; Musabak, U.; Ahmet, A.K.A.R.; Erbil, A.H.; Tatan, H.B. Interferon-gamma in alopecia areata. Eur. J. Dermatol. 2004, 14, 33–36. [Google Scholar]
- Shuai, K.; Stark, G.R.; Kerr, I.M.; Darnell, J.E. A single phosphotyrosine residue of stat91 required for gene activation by interferon-γ. Science 1993, 261, 1744–1746. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signalling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Chang, C.H.; Hammer, J.; Loh, J.E.; Fodor, W.L.; Flavell, R.A. The activation of major histocompatibility complex class I genes by Interferon Regulatory Factor-1 (IRF-1). Immunogenetics 1992, 35, 378–384. [Google Scholar] [CrossRef]
- Jarosinski, K.W.; Massa, P.T. Interferon regulatory factor-1 is required for interferon-γ-induced MHC class I genes in astrocytes. J. Neuroimmunol. 2002, 122, 74–84. [Google Scholar] [CrossRef]
- Girdlestone, J.; Isamat, M.; Gewert, D.; Milstein, C. Transcriptional regulation of HLA-A and -B: Differential binding of members of the REL and IRF families of transcription factors. Proc. Natl. Acad. Sci. USA 1993, 90, 11568. [Google Scholar] [CrossRef]
- Lin, A.; Guo, X.; Inman, R.D.; Sivak, J.M. Ocular inflammation in HLA-B27 transgenic mice reveals a potential role for MHC class I in corneal immune privilege. Mol. Vis. 2015, 21, 131. [Google Scholar]
- Harrist, T.J.; Ruiter, D.J.; Mihm, M.C., Jr.; Bhan, A.K. Distribution of major histocompatibility antigens in normal skin. Br. J. Dermatol. 1983, 109, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Wasilewska, A.; Winiarska, M.; Olszewska, M.; Rudnicka, L. Interleukin-17 inhibitors. A new era in treatment of psoriasis and other skin disease. Postep. Dermatol. Alergol. 2016, 33, 247–252. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamed, F.N.; McDonagh, A.J.G.; Almaghrabi, S.; Bakri, Y.; Messenger, A.G.; Tazi-Ahnini, R. Epigallocatechin-3 Gallate Inhibits STAT-1/JAK2/IRF-1/HLA-DR/HLA-B and Reduces CD8 MKG2D Lymphocytes of Alopecia Areata Patients. Int. J. Environ. Res. Public Health 2018, 15, 2882. https://doi.org/10.3390/ijerph15122882
Hamed FN, McDonagh AJG, Almaghrabi S, Bakri Y, Messenger AG, Tazi-Ahnini R. Epigallocatechin-3 Gallate Inhibits STAT-1/JAK2/IRF-1/HLA-DR/HLA-B and Reduces CD8 MKG2D Lymphocytes of Alopecia Areata Patients. International Journal of Environmental Research and Public Health. 2018; 15(12):2882. https://doi.org/10.3390/ijerph15122882
Chicago/Turabian StyleHamed, Fatma N., Andrew J. G. McDonagh, Sarah Almaghrabi, Youssef Bakri, Andrew G. Messenger, and Rachid Tazi-Ahnini. 2018. "Epigallocatechin-3 Gallate Inhibits STAT-1/JAK2/IRF-1/HLA-DR/HLA-B and Reduces CD8 MKG2D Lymphocytes of Alopecia Areata Patients" International Journal of Environmental Research and Public Health 15, no. 12: 2882. https://doi.org/10.3390/ijerph15122882