Histopathology of Barrett’s Esophagus and Early-Stage Esophageal Adenocarcinoma: An Updated Review

Department of Pathology, Immunology, and Laboratory Medicine, College of Medicine, University of Florida, Gainesville, FL 32610, USA
*
Author to whom correspondence should be addressed.
Gastrointest. Disord. 2019, 1(1), 147-163; https://doi.org/10.3390/gidisord1010011
Submission received: 24 October 2018
/
Revised: 20 November 2018
/
Accepted: 22 November 2018
/
Published: 27 November 2018
Abstract
:Esophageal adenocarcinoma carries a very poor prognosis. For this reason, it is critical to have cost-effective surveillance and prevention strategies and early and accurate diagnosis, as well as evidence-based treatment guidelines. Barrett’s esophagus is the most important precursor lesion for esophageal adenocarcinoma, which follows a defined metaplasia–dysplasia–carcinoma sequence. Accurate recognition of dysplasia in Barrett’s esophagus is crucial due to its pivotal prognostic value. For early-stage esophageal adenocarcinoma, depth of submucosal invasion is a key prognostic factor. Our systematic review of all published data demonstrates a “rule of doubling” for the frequency of lymph node metastases: tumor invasion into each progressively deeper third of submucosal layer corresponds with a twofold increase in the risk of nodal metastases (9.9% in the superficial third of submucosa (sm1) group, 22.0% in the middle third of submucosa (sm2) group, and 40.7% in deep third of submucosa (sm3) group). Other important risk factors include lymphovascular invasion, tumor differentiation, and the recently reported tumor budding. In this review, we provide a concise update on the histopathological features, ancillary studies, molecular signatures, and surveillance/management guidelines along the natural history from Barrett’s esophagus to early stage invasive adenocarcinoma for practicing pathologists.
1. Introduction
Esophageal carcinoma is the eighth most common cancer and the sixth most common cause of cancer-associated mortality worldwide [1]. In the United States, the majority of esophageal carcinomas are adenocarcinomas. While the prevalence of esophageal squamous cell carcinoma is dropping worldwide, the incidence rate of esophageal adenocarcinoma (EAC) has kept over the past three to four decades [2].
It is established that the most important precursor lesion for EAC is Barrett’s esophagus (BE) [3]. BE is the result of a metaplastic process where the squamous mucosa in the distal esophagus is replaced by intestinal type mucosa [4,5]. Gastroesophageal reflux is the pivotal risk factor for BE and EAC [6]. Other risk factors for EAC include obesity, older age, male gender, and Caucasian race. On the other hand, factors such as physical exercise, a diet rich in vegetables and medication such as protein pump inhibitor, nonsteroidal anti-inflammatory drugs, and statin might have protective effects [7]. Ironically Helicobacter pylori, a known oncogenic microorganism for gastric adenocarcinoma, has been shown to be protective against the development of BE and EAC [8]. The current recommendation for BE surveillance is to target high-risk population, i.e., males aged over 60 years with chronic uncontrolled reflux [9].
In this review, we discuss the histopathological features, ancillary studies, and molecular signatures of BE and early stage invasive adenocarcinoma; we also briefly introduce current guidelines regarding its surveillance and treatment.
2. Histopathology of BE
The diagnosis of BE requires correlation of findings from both upper endoscopy and histopathology [10]. The distal esophagus normally is covered with squamous mucosa that is white or pale pink in color. With repeated inflammatory stimuli such as reflux, the squamous mucosa can undergo a metaplastic process and become salmon colored mucosa on endoscopy (Figure 1). Endoscopic identification of the gastroesophageal junction (the proximal end of the gastric fold) and squamocolumnar junction (regular, slightly irregular, or undulated line, or so-called Z-line) is essential [11]. Normally there is an overlap between gastroesophageal junction and squamocolumnar junction. The progress of columnar metaplasia will move the squamocolumnar junction more proximally. There are three histologic types of columnar metaplasia: cardiac-type, fundic-type, and intestinal-type (Figure 2A–C). The clinical significance of cardiac- and fundic-type columnar metaplasia is unclear. However, intestinal-type columnar metaplasia is considered the hallmark of the BE.
By current definition, BE is the presence of longer than 1 cm salmon colored columnar mucosa above the gastroesophageal junction endoscopically which is proven to harbor intestinal metaplasia on histology [12]. The term “long segment BE” is defined as the length of columnar mucosa >3 cm, and “short segment BE” is used when the length is between 1 and 3 cm. Increased length of columnar mucosa is associated with increased incidence of intestinal metaplasia and about 46% increase in the risk of cancer progression for every 1 cm increase in the BE length [13]. For BE diagnosis, the American College of Gastroenterology (ACG) recommends four random biopsies every 2 cm, or 8 random biopsies to maximize the yield of intestinal metaplasia on histology. For cases with suspected “short segment BE”, taking a minimum of four biopsies for every 1 cm of circumferential columnar mucosa and one biopsy for every 1 cm in tongue of columnar mucosa is suggested. If subtle mucosal abnormalities such as ulceration and nodule are identified in BE, they should be sampled separately. If the mucosal abnormalities are present in BE with dysplasia, ideally they should be treated with endoscopic mucosal resection [12].
In the United States, identification of goblet cells is required for the diagnosis of BE [14]. Identification of goblet cells is usually straightforward. In the routine hematoxylin and eosin (H&E) stain, the goblet cells are simple columnar cells with a small narrow nucleus in the base portion of the cell, and a large mucin-filling globule in the cytoplasm. The mucin is acidic in nature, and appears bluish in routine H&E stain. Goblet cells are usually dispersed randomly within the columnar mucosa. From a practical point of view, it’s important to distinguish goblet cells from so-called “pseudogoblet cells”, which are barrel-shaped columnar cells with prominent cytoplasmic mucin [15]. Unlike the true goblet cells, pseudogoblet cells often line up in cluster, and the mucin is usually light pink and less bluish as compared to the true goblet cells. Another histological clue is that the nucleus of the pseudogoblet cells is not triangle shaped as the one in true goblet cells. Although it has been considered unnecessary for BE diagnosis, Alcian Blue (AB) (pH 2.5)/Periodic Acid Schiff (PAS) could be performed when in doubt, and goblet cells will be bright blue or dark purple.
Patients with initial diagnosis of BE usually enter surveillance program so that neoplasia can be detected and treated earlier to reduce the incidence and mortality of Barrett’s adenocarcinoma. BE surveillance biopsy interpretation remains a diagnostic challenge. Surveillance biopsy from BE patients should be evaluated using the previously published schema: negative for dysplasia, indefinite for dysplasia, low-grade dysplasia, and high-grade dysplasia [14].
2.1. Negative for Dysplasia (ND)
The epithelium in the category of BE with ND appears reactive in nature, demonstrates surface maturation and has low nucleus/cytoplasm (N/C) ratio. The nucleus has smooth contour, is uniform and evenly spaced, and parallels to each other with normal polarity (Figure 3A). If there is mild atypia, it is limited to the crypt and basal portion of the epithelium. Nuclear hyperchromasia and polymorphism are not features for reactive epithelium. The risk of cancer progression for ND is approximately 0.12–0.5%/year, about 11.3 times higher than the risk for general population [16,17,18], and regular endoscopic surveillance is recommended every three to five years. Endoscopic therapy is not routinely used to treat patients with ND due to its low risk of cancer progression.
2.2. Indefinite for Dysplasia (IND)
This is a “gray-zone” diagnosis. The columnar epithelium may have certain features concerning for dysplasia, such as cytological atypia and distorted architecture, but quantitatively or qualitatively it’s difficult to make a definite diagnosis of dysplasia. A common scenario is atypia, either mild or marked, in a background of extensive active inflammation making impossible the distinction between reactive versus dysplastic. The risk of cancer progression for IND is approximately 0.84–1.4%/year [19,20,21]. Unfortunately, the interobserver agreement on BE with IND is poor [22]. Once an IND diagnosis is rendered, the patient should receive appropriate proton pump inhibitor (PPI) treatment for 3 to 6 months, followed by repeat biopsy.
2.3. Low-Grade Dysplasia (LGD)
In LGD, the nucleus becomes hyperchromatic, enlarged, and may be stratified, and extends to the surface (Figure 3B). There might be a sharp transition between the normal and dysplastic epithelium. The dysplastic cells still maintain their nuclear polarity. The architecture is usually well preserved without cribriform glands. Extremely pleomorphic nucleus and complex architecture are not features for LGD. The interobserver agreement for LGD is poor-moderate based on several studies [23]. LGD is associated with higher risk of cancer progression (0.5–1.4%/year) when compared to ND [16,24]. For LGD, endoscopic therapy is preferred, alternatively patient could receive endoscopic surveillance every 12 months.
2.4. High-Grade Dysplasia (HGD)
HGD is characterized by more profound cytological and architectural abnormalities than LGD (Figure 3C). Dysplastic changes extend to the surface and the basement membranes of neoplastic glands are intact. Cytological abnormalities in HGD include enlarged hyperchromatic and pleomorphic nucleus, prominent nucleoli, frequent mitosis and atypical mitotic figure, increased N/C ratio, irregular nuclear contour, with loss of nuclear polarity and pleomorphism being the two most objective abnormalities. Architectural abnormalities in HGD include glandular cribriforming and papillary configuration. The risk of cancer progression in HGD is about 6%/year [25,26], and endoscopic therapy is currently recommended.
Loss of surface maturation is a key evidence to support the diagnosis of dysplasia in most BE surveillance biopsies. However certain practical dilemmas do exist. “Deep crypt dysplasia” is a situation that low-grade or even high-grade dysplasia is present but limited within the deep crypt, while the surface maturation is well maintained [27,28]. In the case of deep crypt dysplasia, conventional dysplasia actually is present in about 47% of cases. And deep crypt dysplasia may share similar molecular signature, such as aneuploidy and mutated p53 expression pattern, similar to conventional dysplasia [27]. Therefore it’s important to recognize this variant of dysplasia in the case with surface maturation. When in doubt, deep sections and close examination of the specimen should be applied, and it’s reasonable to at least label the case with “indefinite for dysplasia” (Figure 3D) so that close follow-up could be indicated. Comparison to previous biopsy might be helpful in some cases as well as p53 immunohistochemical stain.
Another commonly encountered dilemma in real-life practice is to distinguish HGD from intramucosal carcinoma. To address this controversial issue, “HGD with marked glandular architectural distortion, cannot exclude intramucosal carcinoma (HGD/MAD)” has been proposed to categorize lesions with: (1) glandular crowding with loss of lamina propria, or so-called “back-to-back” glands; (2) cribriform, or so-called “gland-in-gland” architecture; and (3) at least three dilated glands with intraluminal debris [29]. Another study proposed “HGD with features ‘suspicious’ for invasive carcinoma (HGD/S)” which include solid/cribriform architecture, ulcerated HGD, dilated dysplastic glands with intraluminal debris, prominent neutrophils within HGD, and dysplastic glands incorporating into overlying squamous epithelium [30]. HGD/MAD and HGD/S have excellent agreement with each other, and among HGD cases, the presence of more than three dilated glands with intraluminal debris is the only independent predictor of adenocarcinoma in multivariable analysis [31].
3. Histopathology of Intramucosal Carcinoma (IMC)
Generally speaking, IMC is a transitional process when tumor cells start to demonstrate its invasive potential, but it’s still within the boundary of muscularis mucosae. The hallmarks of IMC is invasion of the neoplastic cells through the basement membrane into the lamina propria but not beyond the muscularis mucosae. Histologically, IMC demonstrates the following patterns: solid growth pattern, individual cells infiltrating the lamina propria but not beyond the muscularis mucosae, or an anastomosing “never ending” glandular pattern (Figure 4A–C).
By definition, the recognition of the muscularis mucosae layer is of great value when diagnosing IMC. In the case of BE, duplicated muscularis mucosae is very common, and present in about 92% of BE resections [32]. The deep layer is the original muscularis mucosae, and the superficial layer is the newly formed muscle layer. In endoscopic mucosal resection (EMR) specimen, the deeper layer of the muscularis mucosae is not always seen or well preserved, which is a potential pitfall for overstaging. When tumor invades into the interspace between these two layers of muscularis mucosae, it behaves similar to IMC invading the lamina propria and/or inner layer of muscularis mucosae in terms of lymph node metastasis and recurrence-free survival [33]. In poorly oriented endoscopic resection specimens, identification of submucosal esophageal glands and ducts and large caliber arteries can be of value to decide the depth of invasion and pathological staging.
When subcategorizing IMC based on the depth of invasion (m1: lack of tumor invasion through basement membrane, i.e. HGD; m2: tumor invades through basement membrane and into lamina propria; m3: tumor invades into muscularis mucosae), studies demonstrated that m1–m3 behaves similar and were unlikely to involve lymph node, locoregional recurrence, or distant recurrence [33], with only 1 T1m3 case (out of 22) developing lymph node metastasis. Just like HGD, endoscopic therapy is the preferred treatment for IMC. However, IMC with lymphovascular invasion, a signet ring cell feature, and poor differentiation may be treated with esophagectomy due to high risk of nodal metastasis [34].
4. Histopathology of Submucosal Adenocarcinoma (SAC)
Grossly SAC can be polypoid, flat, or ulcerated. Similar to carcinoma in the gastric cardia, most of the esophageal SAC has a tubular or papillary growth pattern. Submucosal esophageal glands/ducts and thick-walled arteries, if present, could serve as a marker for submucosal determination. Stromal desmoplasia is another important clue. Microscopically the tumor has features of gland forming, and could be categorized as well differentiated (more than 95% gland formation), moderately differentiated (50–95% gland formation), or poorly differentiated carcinoma (less than 50% gland formation). Rarely poorly differentiated components such as signet ring cell carcinoma can be seen, which usually associates with adverse prognosis. Other uncommon microscopic pattern includes mucinous adenocarcinoma, mucoepidermoid carcinoma, adenoid cystic carcinoma, and adenosquamous carcinoma. Some patients may not have a prior BE history, however BE with HGD is often a common finding in the background esophageal tissue. As an early stage esophageal adenocarcinoma, SAC is becoming commonly recognized due to the widespread use of endoscopic surveillance in BE patients.
Unfortunately, even with a relatively superficial tumor location, SAC is associated with significant cancer mortality. The depth of submucosal invasion is the prognostic key. SAC could be subcategorized into three risk groups based on the depth of invasion: sm1, where the tumor invades into upper one-third of the submucosa; sm2, where the tumor invades into middle third of the submucosa; and sm3, where the tumor invades into lower one third of submucosa. Studies showed that there’s essentially no significant difference between T1a m3 and T1b sm1 in terms of lymph node metastases, recurrence-free period, and 5-year survival rate. Endoscopic therapy is the preferred management for T1b sm1 esophageal carcinoma, similar to T1a IMC. On the contrary, pT1b sm2–3 carcinoma carries a much worse prognosis, with lymph node metastases in 44% and a recurrence-free period only in 57% [35]. For pT1b sm2–3, surgical resection is considered as the treatment of choice [36,37]. From a practical point of view, subdivision of submucosa in endoscopically resection specimen is not always possible due to resection procedure and the sample orientation, thus the absolute invasion depths of 500 µm has been proposed to separate sm1 carcinoma from sm2–3. In a recent study, three risk factors (depth of invasion >500 µm into the submucosa, lymphovascular invasion, and poor differentiation) have been shown to be associated with lymph node metastasis. For the low risk SAC, which was defined as the one that invades <500 µm, has no lymphovascular invasion, and is well or moderately differentiated, there’s no reported lymph node involvement. The frequency of lymph node metastasis increased to 25% with one risk factor, 33% with two risk factors, and in high risk SAC with all three risk factors, the lymph node involvement was present in 50% of cases [37]. A summary of current studies evaluating the depth of invasion in esophageal SAC is presented in Table 1 [32,35,36,37,38,39,40,41,42,43,44,45,46,47]. Even though few studies argued against the “safe zone” of superficial submucosal involvement [44,45], most studies support the clinical significance of depth of submucosal invasion (Table 1). Our systemic review of all published data demonstrates a “rule of doubling” for the frequency of lymph node metastases: tumor invasion into each progressively deeper one third of submucosal layer corresponds with a twofold increase in the risk of nodal metastases (9.9% in the sm1 group, 22.0% in the sm2 group, and 40.7% in the sm3 group). It should be noted that superficial submucosal invasion does carry certain risk of nodal metastases (9.9% in sm1 group), therefore any SAC should be treated surgically if there are any coexisted major risk factors such as lymphovascular involvement or features of poor tumor differentiation (Figure 5A,B).
Tumor budding is a newly recognized prognostic factor for EAC. Following epithelial-mesenchymal transition (EMT), epithelial tumor cells may present as tumor buds that are single cells or small clusters with up to 5 tumor cells ahead of the invasive front (Figure 5C). Recent studies have shown that high-grade tumor budding is associated with higher overall TNM-stage, lymph node involvement, poor differentiation, incomplete excision, and poor overall survival [48,49,50,51,52,53,54,55]. Table 2 provides an overview of current understanding towards this important subject. Interestingly the depth of submucosal invasion may also play a role in tumor budding [56]. However, this feature has not been assessed and reported on routine pathology practice.
5. Ancillary Biomarkers for BE, Dysplasia, and Early Stage EAC
Biomarkers for BE and early stage EAC include mucin glycoprotein MUC2, CDX-2, Das-1, SOX-9, Villin, and Hep Par 1. MUC2 was proposed as a highly specific marker of goblet cell metaplasia in BE [57,58]. CDX-2, the key regulator of intestinal differentiation in the colon and small bowel, is also a potential regulator of BE. Its expression increases during the development of intestinal metaplasia, but subsequently decreases through the progression of dysplasia and carcinoma [59]. MUC2, CDX-2, Das-1, Villin, and Hep Par 1 are all considered as biomarkers for intestinal metaplasia. However currently histopathology is still the gold standard, and these markers have not been recommended to aid in the diagnosis of BE based on recent consensus from the Rodger C. Haggitt Gastrointestinal Pathology Society [60].
Extensive studies have been conducted to explore the potential ancillary biomarkers for the diagnosis of BE-related dysplasia. Among them, p53 expression is the most promising diagnostic and prognostic marker [61,62]. In one study, positive p53 expression was found in 0% of BE ND cases, 9% of LGD cases, 55% of HGD cases, and 87% of those with EAC [63]. In a recent follow-up study, p53 expression levels were surveyed on 275 BE patients who had no HGD or EAC, and result showed that no patient (0%) with scattered positive cells progressed to HGD/EAC, while 31.25% patients with aggregates of positive cells and 75% of those with multifocal aggregates of positive cells progressed to HGD/EAC [64]. Despite these promising results, p53 as a biomarker for diagnosing dysplasia is still controversial due to the high false positive and negative rate.
Alpha-methylacyl-CoA-racemase (AMACR) is also a potential practical biomarker for dysplasia. AMACR was not expressed in non-dysplastic esophagus, but was positive in 38% of LGD, 81% of HGD, and 72% of EAC [65]. One recent report demonstrated that AMACR expression in BE was associated with increased risk of cancer progression, but as a prognostic predictor it’s not much better than histopathological diagnosis of LGD [66]. In another report, AMACR expression level was not correlated with prevalent or incident neoplasia in BE patients with IND [67]. Insulin-like growth factor-II mRNA-binding protein 3 (IMP3) is another promising biomarker in detecting dysplasia in BE and early stage EAC with a better sensitivity as compared to AMACR [68,69,70]. However its validation still requires more stringent investigation.
Her2, which encodes human epithelial growth factor receptor 2, is one of the validated therapeutic biomarkers for EAC. The decision of whether to use or not the anti-Her2 monoclonal antibody Trastuzumab targeted therapy heavily depends on tumor’s expression level of Her2, whose overexpression has been described in about 15–30% of esophageal primary adenocarcinoma [71]. Currently combination of Trastuzumab with a chemotherapy regimen is the standard first-line treatment for Her2 positive metastatic esophageal adenocarcinoma. Her2 testing methods and algorithm for EAC is similar to those in gastric adenocarcinoma, first by immunohistochemistry (IHC) followed by in situ hybridization (ISH) when immunohistochemistry result is 2+ (equivocal). Positive (3+) or negative (0 or 1+) immunohistochemistry results do not require further ISH testing [72]. Her2 IHC is graded as negative 0 (on membranous staining in any of the tumor cells), negative 1+ (faint/barely perceptible membranous staining), equivocal 2+ (weak to moderate, complete, basolateral and lateral membranous staining), and positive 3+ (strong, complete, basolateral and lateral membranous staining) and the cutoff of at least 10% of tumor resection specimen or a small cluster of at least 5 tumor cells in biopsy specimen is used [72]. Cases with Her2 equivocal 2+ IHC result should be tested by ISH for Her2 amplification with positivity defined as Her2/CEP17 ratio ≥2. However, the use of Her2 in early stage EAC has not been explored.
6. Genetic and Molecular Signature of BE and Early Stage EAC
The BE-dysplasia-adenocarcinoma sequence is associated with well-defined genetic and molecular signatures. EAC have frequent chromosome loss of 17p (100%), 5q (80%), 9p (64%), 13q (43%), 18q (43%), and 1p (41%) [73]. In another study, 17p loss was found in 14% BE, 42% LGD, 79% HGD, and 75% EAC. 18q loss was found in 32% BE, 42% LGD, 73% HGD, and 69% EAC [74]. One recent study has demonstrated the potential clinical application of DNA flow cytometry in the diagnosis and risk stratification for BE patients [75].
Loss of both 17p and 18q are frequently identified in EAC patients with poor prognosis. The potential tumor suppressor genes corresponding to each chromosomes are p53 (17p), SMAD4 (18q), APC (5q), p16 (9q), RB1 (13q) and ARID1a (1p). Actually a recent study with whole-genome sequencing analysis in 129 EAC cases confirmed that the most frequent somatic mutation and indel (insertion or deletion) events were TP53 (81%), ARID1a (17%), SMAD4 (16%), p16 (15%), KCNQ3 (12%), CCDC102B (9%), and CYP7B1 (7%) [76]. Previously, 9q (p16) loss was thought to be the earliest event followed by 17p (p53) and 18q (SMAD4) loss [77], and subsequently followed by 5q (APC) loss. However a recent study suggest that TP53 mutations may be earlier events than previously recognized [78]. With TP53 mutation, BE will follow a distinct pathway to develop EAC via catastrophic aneuploidy and oncogene amplification. Through genome-wide mapping method, Orloff et al. identified germline mutations of 3 candidate genes (MSR1, ASCC1, and CTHRC1) in about 11% of patients with BE and EAC [79]. Other research areas of interest for BE include epigenetic regulation, miRNA profiling, and microbiome study.
7. Conclusions
Extensive research on BE, dysplasia, and early stage EAC have greatly shaped our understanding and clinical practice, and it will continue to do so. Despite that, many important questions remain unanswered. For example, what is the best and practical definition for BE? What is the key molecular event driving the initiation and progression of intestinal metaplasia and dysplasia? Are there any reliable prognostic markers to predict disease and neoplasia progression during surveillance? What is the best practice for surveillance as far as cost-effectiveness is concerned? Will there be personalized surveillance protocol for BE and personalized treatment strategy for early stage EAC? Answers to these questions will no doubt have dramatic implications for the development of early disease detection and clinical management.
Author Contributions
Writing-Original Draft Preparation: F.Y.; Writing-Review & Editing: D.H.G., J.L.; Supervision: X.L.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Y. Epidemiology of esophageal cancer. World J. Gastroenterol. 2013, 19, 5598–5606. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P.; Whiteman, D.C. The incidence of esophageal adenocarcinoma continues to rise: Analysis of period and birth cohort effects on recent trends. Ann Oncol. 2012, 23, 3155–3162. [Google Scholar] [CrossRef] [PubMed]
- Bytzer, P.; Christensen, P.B.; Damkier, P.; Vinding, K.; Seersholm, N. Adenocarcinoma of the esophagus and Barrett’s esophagus: A population-based study. Am. J. Gastroenterol. 1999, 94, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Spaander, M.C. Natural History of Barrett’s Esophagus. Dig. Dis. Sci. 2018, 63, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Graham, T.A.; Jansen, M. Insights into the Pathophysiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H. From reflux esophagitis to Barrett’s esophagus and esophageal adenocarcinoma. World J. Gastroenterol. 2015, 21, 5210–5219. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal cancer. Nat. Rev. Dis. Primers 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed]
- Erőss, B.; Farkas, N.; Vincze, Á.; Tinusz, B.; Szapáry, L.; Garami, A.; Balaskó, M.; Sarlós, P.; Czopf, L.; Alizadeh, H.; et al. Helicobacter pylori infection reduces the risk of Barrett’s esophagus: A meta-analysis and systematic review. Helicobacter 2018, 23, e12504. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.; Moayyedi, P.; Corley, D.A.; DeCaestecker, J.; Falck-Ytter, Y.; Falk, G.; Vakil, N.; Sanders, S.; Vieth, M.; Inadomi, J.; et al. BOB CAT: A Large-Scale Review and Delphi Consensus for Management of Barrett’s Esophagus with No Dysplasia, Indefinite for, or Low-Grade Dysplasia. Am. J. Gastroenterol. 2015, 110, 662–682. [Google Scholar] [CrossRef] [PubMed]
- Naini, B.V.; Chak, A.; Ali, M.A.; Odze, R.D. Barrett’s oesophagus diagnostic criteria: Endoscopy and histology. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Wallner, B.; Sylvan, A.; Stenling, R.; Janunger, K.G. The esophageal Z-line appearance correlates to the prevalence of intestinal metaplasia. Scand. J. Gastroenterol. 2000, 35, 17–22. [Google Scholar] [PubMed]
- Shaheen, N.J.; Falk, G.W.; Iyer, P.G.; Gerson, L.B.; American College of Gastroenterology. ACG Clinical Guideline: Diagnosis and Management of Barrett’s Esophagus. Am. J. Gastroenterol. 2016, 111, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.D.; Chaves, P. Low risk of adenocarcinoma and high-grade dysplasia in patients with non-dysplastic Barrett’s esophagus: Results from a cohort from a country with low esophageal adenocarcinoma incidence. United Eur. Gastroenterol. J. 2016, 4, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Riddell, R.H.; Odze, R.D. Definition of Barrett’s esophagus: Time for a rethink—Is intestinal metaplasia dead? Am. J. Gastroenterol. 2009, 104, 2588–2594. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Ertan, A.; Ergun, G.; Verm, R.; Bridges, M.; Woods, K.; Meriano, F.; Schmulen, A.C.; Colman, R.; Johnson, C.; et al. Goblet cell mimickers in esophageal biopsies are not associated with an increased risk for dysplasia. Arch. Pathol. Lab. Med. 2007, 131, 571–575. [Google Scholar] [PubMed]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sørensen, H.T.; Funch-Jensen, P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.B.; Falk, G.W.; Richter, J.E. The incidence of adenocarcinoma and dysplasia in Barrett’s esophagus: Report on the Cleveland Clinic Barrett’s Esophagus Registry. Am. J. Gastroenterol. 1999, 94, 2037–2042. [Google Scholar] [PubMed]
- Drewitz, D.J.; Sampliner, R.E.; Garewal, H.S. The incidence of adenocarcinoma in Barrett’s esophagus: A prospective study of 170 patients followed 4.8 years. Am. J. Gastroenterol. 1997, 92, 212–215. [Google Scholar] [PubMed]
- Sinh, P.; Anaparthy, R.; Young, P.E.; Gaddam, S.; Thota, P.; Balasubramanian, G.; Singh, M.; Higbee, A.D.; Wani, S.; Gupta, N.; et al. Clinical outcomes in patients with a diagnosis of “indefinite for dysplasia” in Barrett’s esophagus: A multicenter cohort study. Endoscopy 2015, 47, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Kestens, C.; Leenders, M.; Offerhaus, G.J.; van Baal, J.W.; Siersema, P.D. Risk of neoplastic progression in Barrett’s esophagus diagnosed as indefinite for dysplasia: A nationwide cohort study. Endoscopy 2015, 47, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Shroff, S.; Feldman, M.; DeMarshall, M.; Price, C.; Tierney, A.; Falk, G.W. Risk of malignant progression in Barrett’s esophagus indefinite for dysplasia. Dis. Esophagus 2017, 30, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Salomao, M.A.; Lam-Himlin, D.; Pai, R.K. Substantial Interobserver Agreement in the Diagnosis of Dysplasia in Barrett Esophagus Upon Review of a Patient’s Entire Set of Biopsies. Am. J. Surg. Pathol. 2018, 42, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Vennalaganti, P.; Kanakadandi, V.; Goldblum, J.R.; Mathur, S.C.; Patil, D.T.; Offerhaus, G.J.; Meijer, S.L.; Vieth, M.; Odze, R.D.; Shreyas, S.; Parasa, S.; Gupta, N.; Repici, A.; Bansal, A.; Mohammad, T.; Sharma, P. Discordance Among Pathologists in the United States and Europe in Diagnosis of Low-Grade Dysplasia for Patients with Barrett’s Esophagus. Gastroenterology 2017, 152, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Coleman, H.G.; Yousef, F.; Johnston, B.T.; McManus, D.T.; Gavin, A.T.; Murray, L.J. Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J. Natl. Cancer Inst. 2011, 103, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spechler, S.J. Barrett esophagus and risk of esophageal cancer: a clinical review. JAMA 2013, 310, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, A.; Puli, S.; El-Serag, H.B.; Bansal, A.; Wani, S.; Sharma, P. Incidence of esophageal adenocarcinoma in patients with Barrett’s esophagus and high-grade dysplasia: A meta-analysis. Gastrointest. Endosc. 2008, 67, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Lomo, L.C.; Blount, P.L.; Sanchez, C.A.; Li, X.; Galipeau, P.C.; Cowan, D.S.; Ayub, K.; Rabinovitch, P.S.; Reid, B.J.; Odze, R.D. Crypt dysplasia with surface maturation: A clinical, pathologic, and molecular study of a Barrett’s esophagus cohort. Am. J. Surg. Pathol. 2006, 30, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Coco, D.P.; Goldblum, J.R.; Hornick, J.L.; Lauwers, G.Y.; Montgomery, E.; Srivastava, A.; Wang, H.; Odze, R.D. Interobserver variability in the diagnosis of crypt dysplasia in Barrett esophagus. Am. J. Surg. Pathol. 2011, 35, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Appelman, H.D.; Greenson, J.K.; Ramsburgh, S.R.; Orringer, M.B.; Chang, A.C.; McKenna, B.J. A histologically defined subset of high-grade dysplasia in Barrett mucosa is predictive of associated carcinoma. Am. J. Clin. Pathol. 2009, 132, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Downs-Kelly, E.; Mendelin, J.E.; Bennett, A.E.; Castilla, E.; Henricks, W.H.; Schoenfield, L.; Skacel, M.; Yerian, L.; Rice, T.W.; Rybicki, L.A.; et al. Poor interobserver agreement in the distinction of high-grade dysplasia and adenocarcinoma in pretreatment Barrett’s esophagus biopsies. Am. J. Gastroenterol. 2008, 103, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.T.; Goldblum, J.R.; Rybicki, L.; Plesec, T.P.; Mendelin, J.E.; Bennett, A.E.; Castilla, E.A.; Henricks, W.H.; Schoenfield, L.; Skacel, M.; et al. Prediction of adenocarcinoma in esophagectomy specimens based upon analysis of preresection biopsies of Barrett esophagus with at least high-grade dysplasia: A comparison of 2 systems. Am. J. Surg. Pathol. 2012, 36, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.C.; Krasinskas, A.M.; Correa, A.M.; Hofstetter, W.L.; Ajani, J.A.; Swisher, S.G.; Wu, T.T. Duplication of the muscularis mucosae in Barrett esophagus: An underrecognized feature and its implication for staging of adenocarcinoma. Am. J. Surg. Pathol. 2007, 31, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Estrella, J.S.; Hofstetter, W.L.; Correa, A.M.; Swisher, S.G.; Ajani, J.A.; Lee, J.H.; Bhutani, M.S.; Abraham, S.C.; Rashid, A.; Maru, D.M. Duplicated muscularis mucosae invasion has similar risk of lymph node metastasis and recurrence-free survival as intramucosal esophageal adenocarcinoma. Am. J. Surg. Pathol. 2011, 35, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rice, T.W.; Liu, X.; Goldblum, J.R.; Williams, S.J.; Rybicki, L.A.; Murthy, S.C.; Mason, D.P.; Raymond, D.P.; Blackstone, E.H. Intramucosal esophageal adenocarcinoma: primum non nocere. J. Thorac. Cardiovasc. Surg. 2013, 145, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Koppert, L.B.; Buskens, C.J.; Tilanus, H.W.; ten Kate, F.J.; Bergman, J.J.; Siersema, P.D.; van Dekken, H.; van Lanschot, J.J. Outcome of surgical treatment for early adenocarcinoma of the esophagus or gastro-esophageal junction. Virchows Arch. 2005, 446, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Boys, J.A.; Worrell, S.G.; Chandrasoma, P.; Vallone, J.G.; Maru, D.M.; Zhang, L.; Blackmon, S.H.; Dickinson, K.J.; Dunst, C.M.; Hofstetter, W.L.; et al. Can the Risk of Lymph Node Metastases Be Gauged in Endoscopically Resected Submucosal Esophageal Adenocarcinomas? A Multi-Center Study. J. Gastrointest. Surg. 2016, 20, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Manner, H.; Wetzka, J.; May, A.; Pauthner, M.; Pech, O.; Fisseler-Eckhoff, A.; Stolte, M.; Vieth, M.; Lorenz, D.; Ell, C. Early-stage adenocarcinoma of the esophagus with mid to deep submucosal invasion (pT1b sm2–3): The frequency of lymph-node metastasis depends on macroscopic and histological risk patterns. Dis. Esophagus 2017, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Buskens, C.J.; Westerterp, M.; Lagarde, S.M.; Bergman, J.J.; ten Kate, F.J.; van Lanschot, J.J. Prediction of appropriateness of local endoscopic treatment for high-grade dysplasia and early adenocarcinoma by EUS and histopathologic features. Gastrointest. Endosc. 2004, 60, 703–710. [Google Scholar] [CrossRef]
- Bollschweiler, E.; Baldus, S.E.; Schröder, W.; Prenzel, K.; Gutschow, C.; Schneider, P.M.; Hölscher, A.H. High rate of lymph-node metastasis in submucosal esophageal squamous-cell carcinomas and adenocarcinomas. Endoscopy 2006, 38, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Sepesi, B.; Watson, T.J.; Zhou, D.; Polomsky, M.; Litle, V.R.; Jones, C.E.; Raymond, D.P.; Hu, R.; Qiu, X.; Peters, J.H. Are endoscopic therapies appropriate for superficial submucosal esophageal adenocarcinoma? An analysis of esophagectomy specimens. J. Am. Coll. Surg. 2010, 210, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, A.H.; Bollschweiler, E.; Schröder, W.; Metzger, R.; Gutschow, C.; Drebber, U. Prognostic impact of upper, middle, and lower third mucosal or submucosal infiltration in early esophageal cancer. Ann. Surg. 2011, 254, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Ancona, E.; Rampado, S.; Cassaro, M.; Battaglia, G.; Ruol, A.; Castoro, C.; Portale, G.; Cavallin, F.; Rugge, M. Prediction of lymph node status in superficial esophageal carcinoma. Ann. Surg. Oncol. 2008, 15, 3278–3288. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, D.; Origer, J.; Pauthner, M.; Graupe, F.; Fisseler-Eckhoff, A.; Stolte, M.; Pech, O.; Ell, C. Prognostic risk factors of early esophageal adenocarcinomas. Ann. Surg. 2014, 259, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Leers, J.M.; DeMeester, S.R.; Oezcelik, A.; Klipfel, N.; Ayazi, S.; Abate, E.; Zehetner, J.; Lipham, J.C.; Chan, L.; Hagen, J.A.; et al. The prevalence of lymph node metastases in patients with T1 esophageal adenocarcinoma a retrospective review of esophagectomy specimens. Ann. Surg. 2011, 253, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.P.; Jones, M.; Brown, I.; Gotley, D.C.; Martin, I.; Thomas, J.; Clouston, A.; Smithers, B.M. Risk stratification for early esophageal adenocarcinoma: analysis of lymphatic spread and prognostic factors. Ann. Surg. Oncol. 2010, 17, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Zemler, B.; May, A.; Ell, C.; Stolte, M. Early Barrett’s carcinoma: The depth of infiltration of the tumour correlates with the degree of differentiation, the incidence of lymphatic vessel and venous invasion. Virchows Arch. 2010, 456, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Badreddine, R.J.; Prasad, G.A.; Lewis, J.T.; Lutzke, L.S.; Borkenhagen, L.S.; Dunagan, K.T.; Wang, K.K. Depth of submucosal invasion does not predict lymph node metastasis and survival of patients with esophageal carcinoma. Clin. Gastroenterol. Hepatol. 2010, 8, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hofstetter, W.L.; Rashid, A.; Swisher, S.G.; Correa, A.M.; Ajani, J.A.; Hamilton, S.R.; Wu, T.T. Significance of the depth of tumor invasion and lymph node metastasis in superficially invasive (T1) esophageal adenocarcinoma. Am. J. Surg. Pathol. 2005, 29, 1079–1085. [Google Scholar] [PubMed]
- Alvarez Herrero, L.; Pouw, R.E.; van Vilsteren, F.G.; ten Kate, F.J.; Visser, M.; van Berge Henegouwen, M.I.; Weusten, B.L.; Bergman, J.J. Risk of lymph node metastasis associated with deeper invasion by early adenocarcinoma of the esophagus and cardia: Study based on endoscopic resection specimens. Endoscopy 2010, 42, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, R.; Oyama, T.; Abe, S.; Takahashi, H.; Ono, H.; Fujisaki, J.; Kaise, M.; Goda, K.; Kawada, K.; Koike, T.; et al. Risk of metastasis in adenocarcinoma of the esophagus: A multicenter retrospective study in a Japanese population. J. Gastroenterol. 2017, 52, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.S.; Hastings, S.M.; Foxwell, T.J.; Luketich, J.D.; Nason, K.S.; Davison, J.M. Tumor budding is associated with an increased risk of lymph node metastasis and poor prognosis in superficial esophagealadenocarcinoma. Mod. Pathol. 2014, 27, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Sillah, K.; Griffiths, E.A.; Swindell, R.; West, C.M.; Page, R.D.; Welch, I.M.; Pritchard, S.A. Tumour budding and a low host inflammatory response are associated with a poor prognosis in oesophageal and gastro-oesphageal junction cancers. Histopathology 2010, 56, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Thies, S.; Guldener, L.; Slotta-Huspenina, J.; Zlobec, I.; Koelzer, V.H.; Lugli, A.; Kröll, D.; Seiler, C.A.; Feith, M.; Langer, R. Impact of peritumoral and intratumoral budding in esophageal adenocarcinomas. Hum. Pathol. 2016, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.B.; Schaeffer, D.F. Tumor budding as a standardized parameter in gastrointestinal carcinomas: More than just the colon. Mod. Pathol. 2018, 31, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Langer, R.; Zlobec, I.; Lugli, A. Tumor Budding in Upper Gastrointestinal Carcinoma. Front. Oncol. 2014, 4, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolte, M.; Kirtil, T.; Oellig, F.; Vogel, C.; Mueller, H.; May, A.; Ell, C.; Wittenberg, R. The pattern of invasion of early carcinomas in Barrett’s esophagus is dependent on the depth of infiltration. Pathol. Res. Pract. 2010, 206, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Glickman, J.N.; Wang, H.; Das, K.M.; Goyal, R.K.; Spechler, S.J.; Antonioli, D.; Odze, R.D. Phenotype of Barrett’s esophagus and intestinal metaplasia of the distal esophagus and gastroesophageal junction: An immunohistochemical study of cytokeratins 7 and 20, Das-1 and 45 MI. Am. J. Surg. Pathol. 2001, 25, 87–94. [Google Scholar] [CrossRef] [PubMed]
- McIntire, M.G.; Souc, G.; Vaughan, T.L.; Shahsafaei, A.; Odze, R.D. MUC2 is a highly specific marker of goblet cell metaplasia in the distal esophagus and gastroesophageal junction. Am. J. Surg. Pathol. 2011, 35, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Ahmed, S.; Clark, P. Immunohistochemical assessment for Cdx2 expression in the Barrett metaplasia-dysplasia-adenocarcinoma sequence. J. Clin. Pathol. 2011, 64, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Appelman, H.; Goldsmith, J.D.; Davison, J.M.; Hart, J.; Krasinskas, A.M. The Use of Ancillary Stains in the Diagnosis of Barrett Esophagus and Barrett Esophagus-associated Dysplasia: Recommendations from the Rodger C. Haggitt Gastrointestinal Pathology Society. Am. J. Surg. Pathol. 2017, 41, e8–e21. [Google Scholar] [CrossRef] [PubMed]
- Janmaat, V.T.; van Olphen, S.H.; Biermann, K.E.; Looijenga, L.H.J.; Bruno, M.B.; Spaander, M.C.W. Use of immunohistochemical biomarkers as independent predictor of neoplastic progression in Barrett’s oesophagus surveillance: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0186305. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M.; Noffsinger, A.; et al. Detection of Mutations in Barrett’s Esophagus Before Progression to High-Grade Dysplasia or Adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Lebovitz, R.M.; Lechago, L.V.; Lechago, J. p53 protein accumulation in Barrett’s metaplasia, dysplasia, and carcinoma: a follow-up study. Gastroenterology 1993, 105, 1637–1642. [Google Scholar] [CrossRef]
- Younes, M.; Brown, K.; Lauwers, G.Y.; Ergun, G.; Meriano, F.; Schmulen, A.C.; Barroso, A.; Ertan, A. p53 protein accumulation predicts malignant progression in Barrett’s metaplasia: A prospective study of 275 patients. Histopathology 2017, 71, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Dorer, R.; Odze, R.D. AMACR immunostaining is useful in detecting dysplastic epithelium in Barrett’s esophagus, ulcerative colitis, and Crohn’s disease. Am. J. Surg. Pathol. 2006, 30, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, F.; Biermann, K.; Steyerberg, E.W.; Verheij, J.; Kalisvaart, M.; Looijenga, L.H.; Stoop, H.A.; Walter, L.; Kuipers, E.J.; Spaander, M.C.; et al. Value of α-methylacyl-CoA racemase immunochemistry for predicting neoplastic progression in Barrett’s oesophagus. Histopathology 2013, 63, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Horvath, B.; Singh, P.; Xie, H.; Thota, P.N.; Sun, X.; Liu, X. Expression of p53 predicts risk of prevalent and incident advanced neoplasia in patients with Barrett’s esophagus and epithelial changes indefinite for dysplasia. Gastroenterol. Rep. 2016, 4, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhou, Z.; Peters, J.H.; Khoury, T.; Zhai, Q.; Wei, Q.; Truong, C.D.; Song, S.W.; Tan, D. Expression of insulin-like growth factor II mRNA-binding protein 3 in human esophageal adenocarcinoma and its precursor lesions. Arch. Pathol Lab. Med. 2011, 135, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Vohra, P.; Chu, P.G.; Woda, B.; Rock, K.L.; Jiang, Z. An oncofetal protein IMP3: A new molecular marker for the detection of esophageal adenocarcinoma and high-grade dysplasia. Am. J. Surg. Pathol. 2009, 33, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Gadara, M.R.; Gonzalez, M.; Cartun, R.W.; Ligato, S. IMP3 Immunoreactivity is More Sensitive Than AMACR in Detecting Dysplastic Epithelium and Early Adenocarcinoma in Barrett Esophagus. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Koopman, T.; Smits, M.M.; Louwen, M.; Hage, M.; Boot, H.; Imholz, A.L. HER2 positivity in gastric and esophageal adenocarcinoma: Clinicopathological analysis and comparison. J. Cancer Res. Clin. Oncol. 2015, 141, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Bartley, A.N.; Washington, M.K.; Ventura, C.B.; Ismaila, N.; Colasacco, C.; Benson III, A.B.; Carrato, A.; Gulley, M.L.; Jain, D.; Kakar, S.; et al. HER2 testing and clinical decision making in gastroesophageal adenocarcinoma. Arch. Pathol. Lab. Med. 2016, 140, 1345–1363. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.T.; Galipeau, P.C.; Sanchez, C.A.; Emond, M.J.; Reid, B.J. Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene 1996, 12, 1873–1878. [Google Scholar] [PubMed]
- Wu, T.T.; Watanabe, T.; Heitmiller, R.; Zahurak, M.; Forastiere, A.A.; Hamilton, S.R. Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am. J. Pathol. 1998, 153, 287–294. [Google Scholar] [CrossRef]
- Choi, W.T.; Tsai, J.H.; Rabinovitch, P.S.; Small, T.; Huang, D.; Mattis, A.N.; Kakar, S. Diagnosis and risk stratification of Barrett’s dysplasia by flow cytometric DNA analysis of paraffin-embedded tissue. Gut 2018, 67, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galipeau, P.C.; Prevo, L.J.; Sanchez, C.A.; Longton, G.M.; Reid, B.J. Clonal expansion and loss of heterozygosity at chromosomes 9p and 17p in premalignant esophageal (Barrett’s) tissue. J. Natl. Cancer Inst. 1999, 91, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Orloff, M.; Peterson, C.; He, X.; Ganapathi, S.; Heald, B.; Yang, Y.R.; Bebek, G.; Romigh, T.; Song, J.H.; Wu, W.; et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA 2011, 306, 410–419. [Google Scholar] [CrossRef] [PubMed]
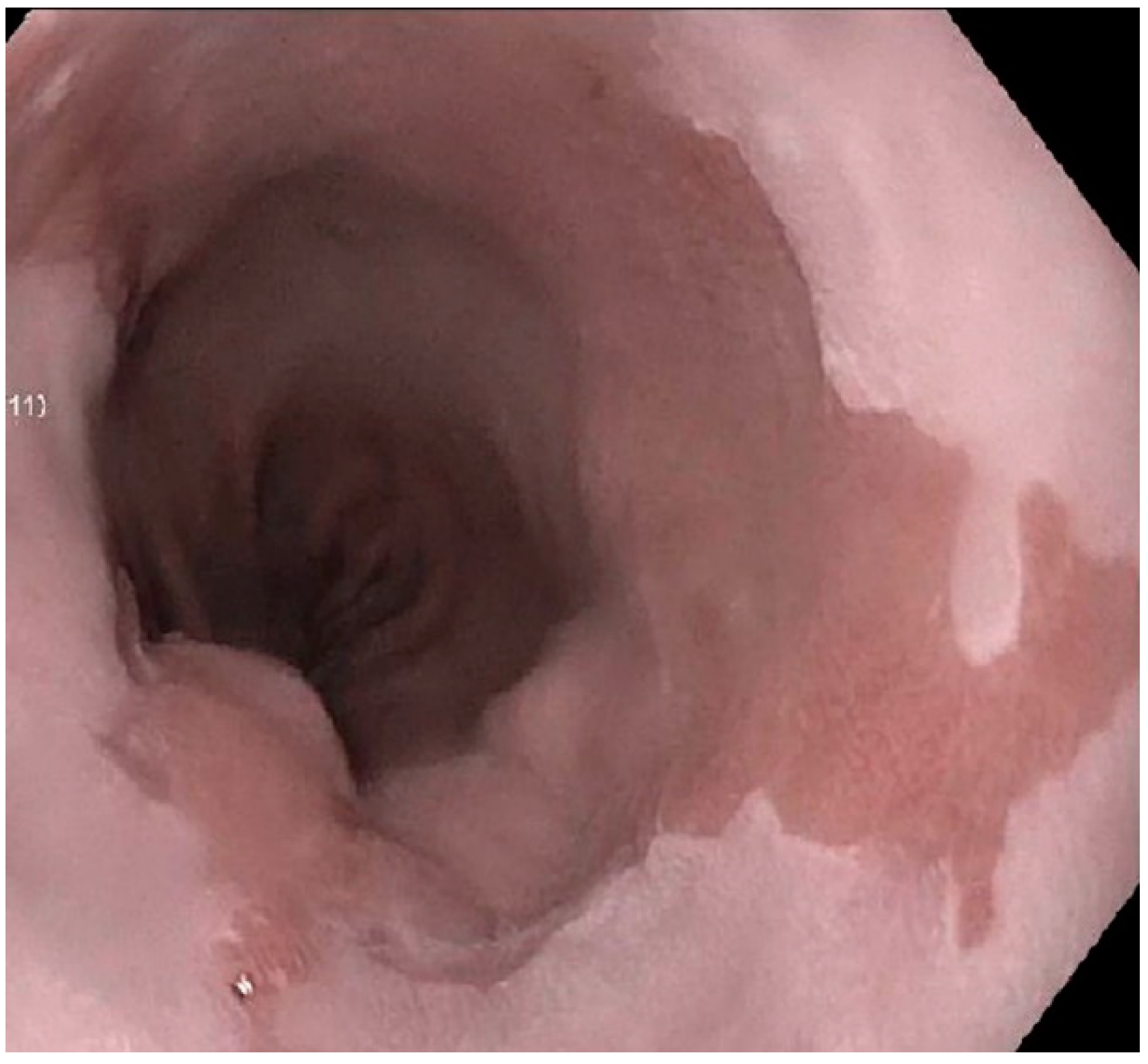
Figure 1.
Endoscopic appearance of salmon-color columnar mucosa in Barrett’s esophagus as a result of chronic reflux. Squamous mucosa of the esophagus appears white to pale pink on endoscopic examination.
Figure 1.
Endoscopic appearance of salmon-color columnar mucosa in Barrett’s esophagus as a result of chronic reflux. Squamous mucosa of the esophagus appears white to pale pink on endoscopic examination.

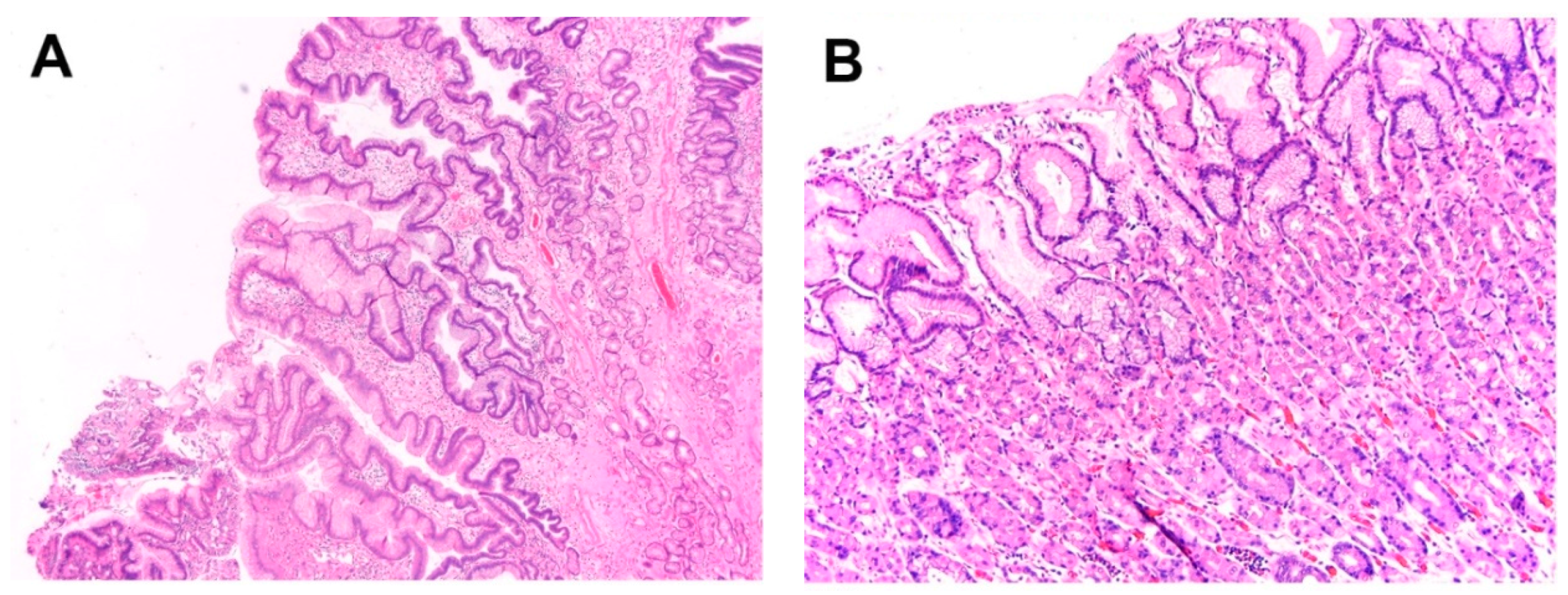
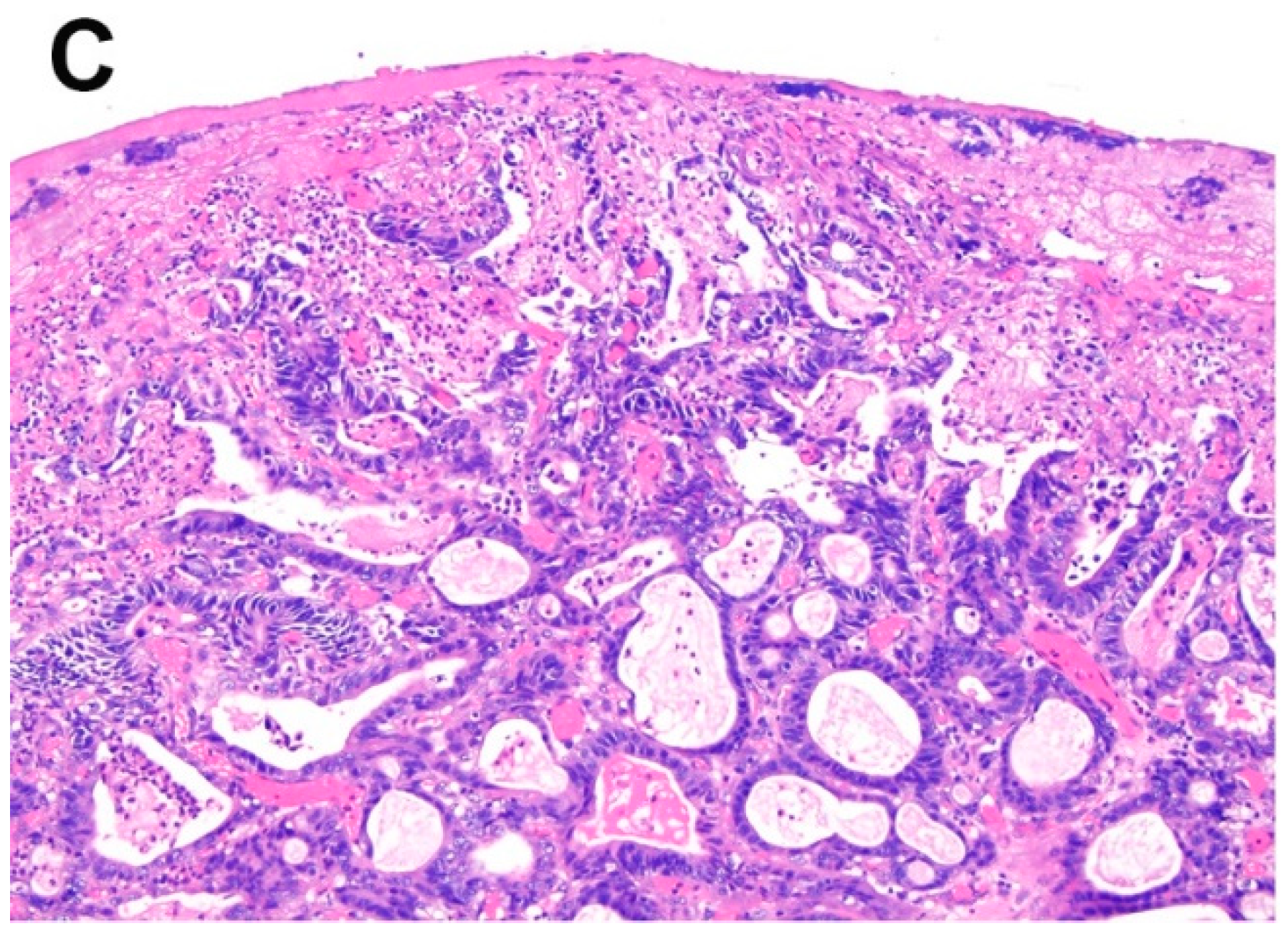
Figure 2.
Histologically, salmon-colored mucosa in the esophagus may correspond to (A) cardiac-type columnar mucosa, (B) fundic-type columnar mucosa, and (C) intestinal type columnar mucosa.
Figure 2.
Histologically, salmon-colored mucosa in the esophagus may correspond to (A) cardiac-type columnar mucosa, (B) fundic-type columnar mucosa, and (C) intestinal type columnar mucosa.


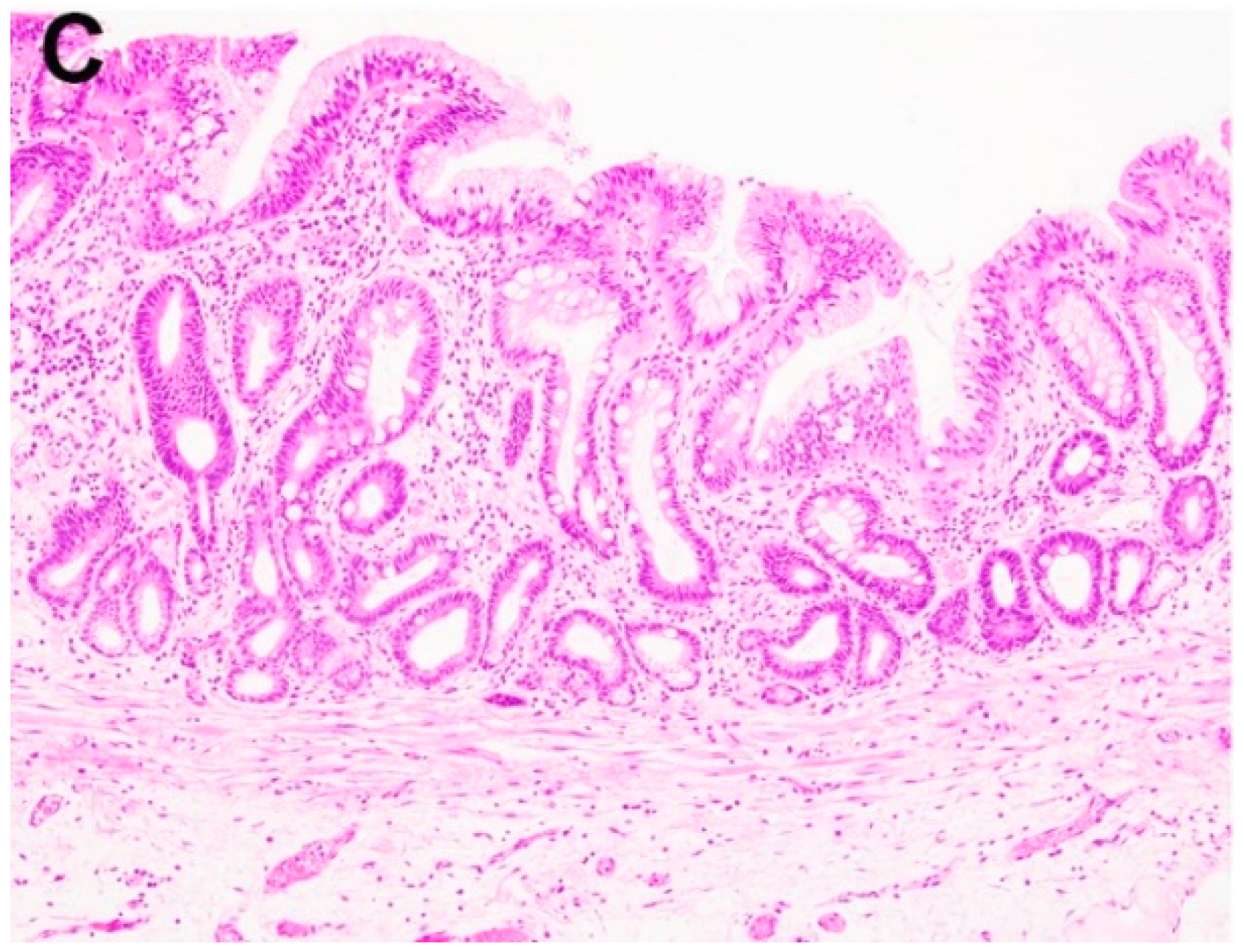
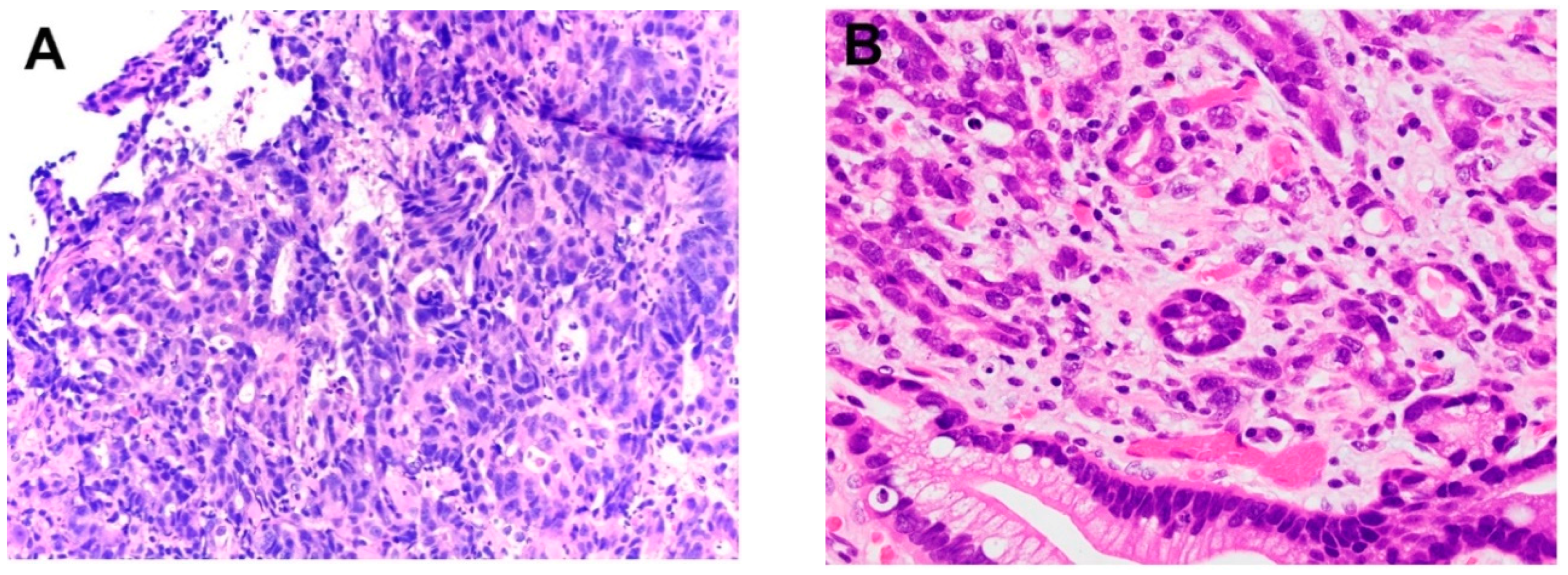
Figure 3.
Examples of Barrett’s esophagus surveillance biopsy without and with dysplasia. (A) Barrett’s esophagus, negative for dysplasia, (B) Barrett’s esophagus, positive for low-grade dysplasia, (C) Barrett’s esophagus, positive for high-grade dysplasia, (D) Barrett’s esophagus with epithelial change indefinite for dysplasia.
Figure 3.
Examples of Barrett’s esophagus surveillance biopsy without and with dysplasia. (A) Barrett’s esophagus, negative for dysplasia, (B) Barrett’s esophagus, positive for low-grade dysplasia, (C) Barrett’s esophagus, positive for high-grade dysplasia, (D) Barrett’s esophagus with epithelial change indefinite for dysplasia.

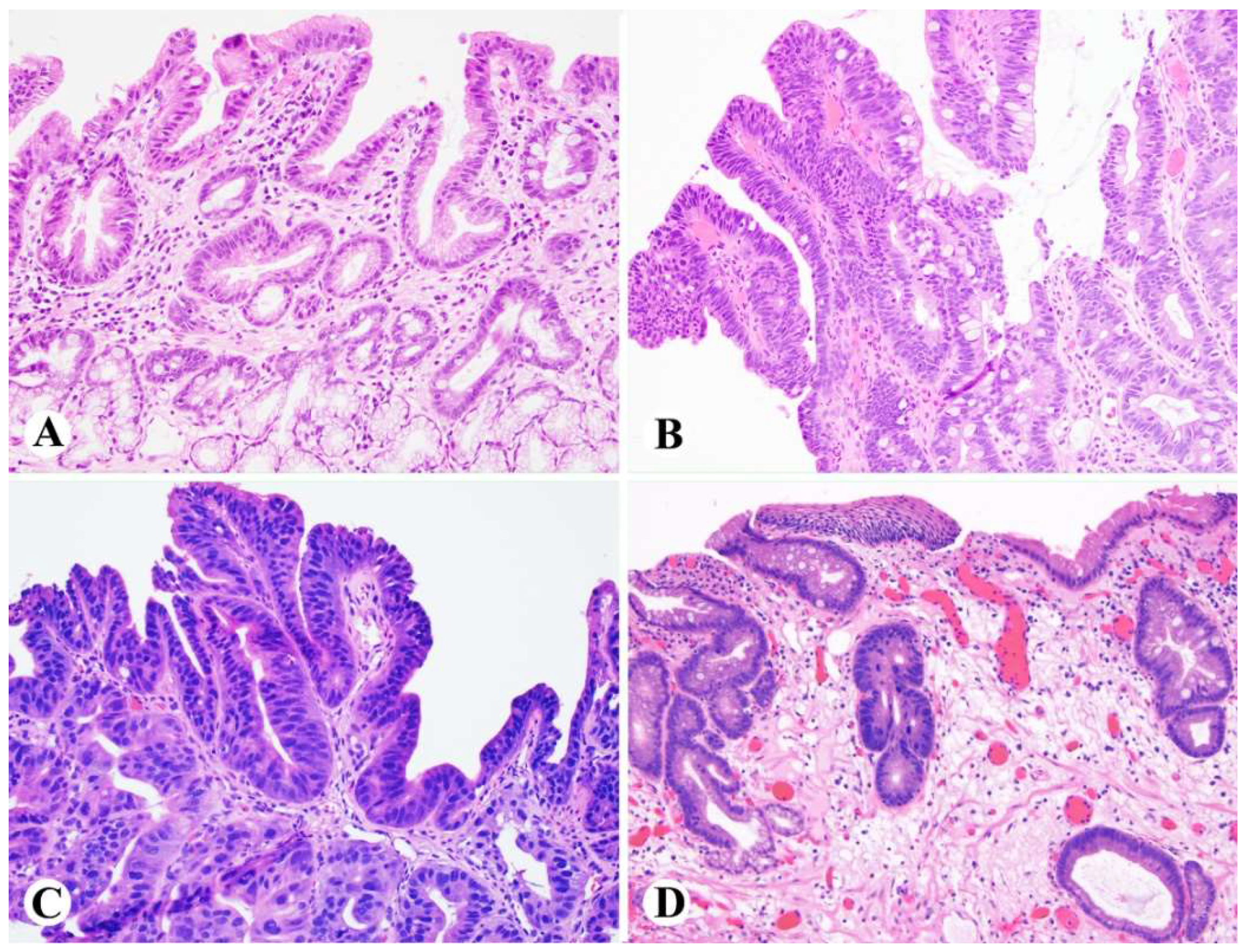
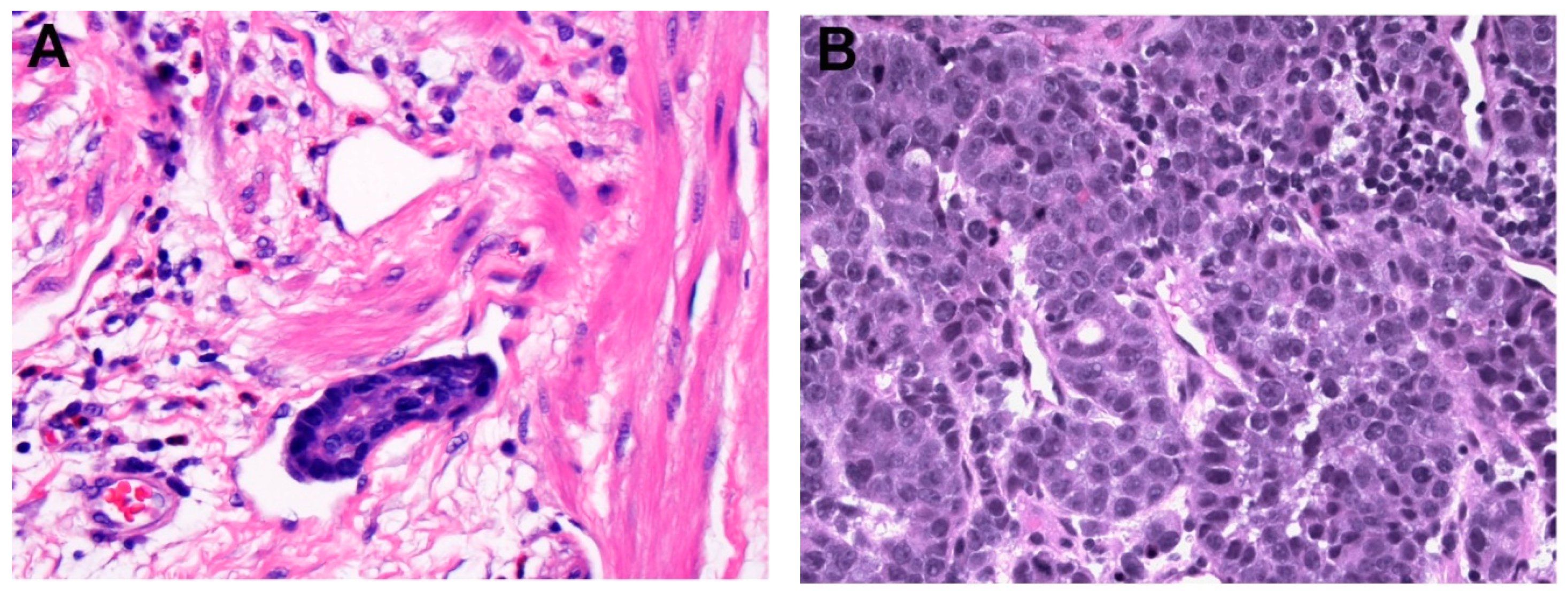
Figure 4.
Examples of intramucosal adenocarcinoma arising from Barrett’s esophagus. (A) Solid growth pattern, (B) Individual cells infiltrating beyond the lamina propria but not through the muscularis mucosae, (C) Anastomosing glandular pattern.
Figure 4.
Examples of intramucosal adenocarcinoma arising from Barrett’s esophagus. (A) Solid growth pattern, (B) Individual cells infiltrating beyond the lamina propria but not through the muscularis mucosae, (C) Anastomosing glandular pattern.


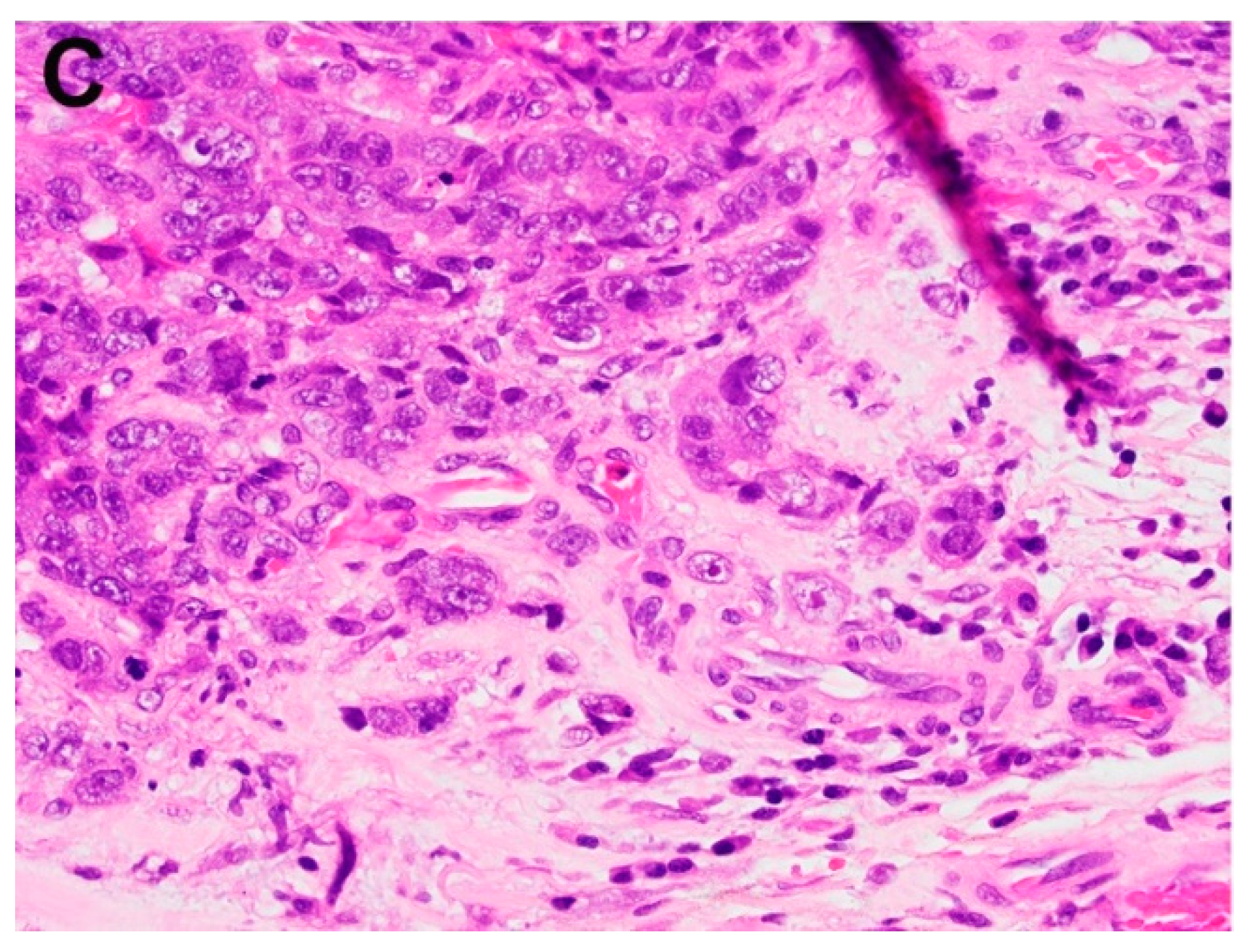
Figure 5.
Factors predictive of adverse prognosis in esophageal adenocarcinoma in endoscopic resection specimen in addition to depth of invasion include (A) lymphovascular invasion, (B) poor differentiation, and (C) tumor budding.
Figure 5.
Factors predictive of adverse prognosis in esophageal adenocarcinoma in endoscopic resection specimen in addition to depth of invasion include (A) lymphovascular invasion, (B) poor differentiation, and (C) tumor budding.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Studies evaluating depth of invasion in esophageal submucosal carcinoma.
| Reference | Procedure | Subgroup | n | LN + | L + | V + | Recurrence | Metastasis | Mortality | HR (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|
| Buskens et al., 2004 [35] | esophagectomy | sm1 | 16 | 0 (0%) | 0 (0%) | |||||
| sm2 | 13 | 3 (23.1%) | 4 (30.8%) | |||||||
| sm3 | 13 | 9 (69.2%) | 12 (92.3%) | |||||||
| Bollschweiler et al., 2006 [36] | esophagectomy | sm1 | 9 | 2 (22.2%) | ||||||
| sm2 | 4 | 0 (0%) | ||||||||
| sm3 | 9 | 7 (77.8%) | ||||||||
| Sepesi et al., 2010 [37] | esophagectomy | sm1 | 14 | 3 (21.4%) | 25% | |||||
| sm2 | 11 | 4 (36.4%) | 60% | |||||||
| sm3 | 4 | 2 (50.0%) | 100% | |||||||
| Westerterp et al., 2005 [32] | esophagectomy | sm1 | 25 | 0 (0%) | 0 (0%) | 4 (16%) | ||||
| sm2 | 23 | 6 (26.1%) | 4 (17.4%) | 11 (47.8%) | ||||||
| sm3 | 18 | 12 (66.7%) | 4 (22.2%) | 11 (61.1%) | ||||||
| Hölscher et al., 2011 [38] | esophagectomy | sm1 | 23 | 2 (8.6%) | ||||||
| sm2 | 15 | 2 (13.3%) | ||||||||
| sm3 | 28 | 12 (42.9%) | ||||||||
| Ancona et al., 2008 [39] | esophagectomy | sm1 | 36 | 3 (8.3%) | ||||||
| sm2 | 7 | 2 (28.6%) | ||||||||
| sm3 | 28 | 15 (53.6%) | ||||||||
| Lorenz et al., 2014 [40] | endoscopic resection and esophagectomy | sm1 | 37 | 3 (8.1%) | 12 (32.4%) | 3 (8.1%) | 3 (8.1%) | |||
| sm2 | 33 | 9 (27.3%) | 12 (36.4%) | 1 (3.0%) | 4 (12.1%) | |||||
| sm3 | 56 | 14 (25.0%) | 26 (46.4%) | 5 (8.9%) | 12 (21.4%) | |||||
| Leers et al., 2011 [41] | esophagectomy | sm1 | 19 | 4 (21.0%) | 1 | |||||
| sm2 | 9 | 1 (11.1%) | 1.07 (0.08–9.78) | |||||||
| sm3 | 23 | 6 (26.1%) | 0.75 (0.12–4.77) | |||||||
| Barbour et al., 2010 [42] | esophagectomy | sm1 | 12 | 2 (16.7%) | ||||||
| sm2 | 17 | 2 (11.8%) | ||||||||
| sm3 | 20 | 4 (20.0%) | ||||||||
| Summary of LN + Rate, [32,35,36,37,38,39,40,41,42] | sm1 | 191 | 19 (9.9%) | |||||||
| sm2 | 132 | 29 (22.0%) | ||||||||
| sm3 | 199 | 81 (40.7%) | ||||||||
| Zemler et al., 2010 [43] | endoscopic resection | sm1 | 61 | 15 (24.6%) | 2 (3.3%) | |||||
| sm2 | 19 | 8 (42.1%) | 0 (0%) | |||||||
| sm3 | 29 | 12 (41.4%) | 4 (13.8%) | |||||||
| Badreddine et al., 2010 [44] | esophagectomy | sm1 | 31 | 4 (12.9%) | 10 (32%) | 3 (9.6%) | 10 (32%) | |||
| sm2-3 | 49 | 10 (20%) | 17 (35%) | 4 (8.2%) | 21 (43%) | |||||
| Manner et al., 2017 [34] | endoscopic resection and esophagectomy | sm2 low risk | 12 | 0 (0%) | 0 (0%) | |||||
| sm2 high risk | 11 | 6 (54.5%) | 0 (0%) | |||||||
| sm3 low risk | 7 | 0 (0%) | 0 (0%) | |||||||
| sm3 high risk | 32 | 17 (53.1%) | 3 (9.4%) | |||||||
| Liu et al., 2005 [45] | esophagectomy | Superficial (<50%) | 12 | 1 (8.3%) | 3 (25.0%) | 3 (0.8–11.5) | ||||
| Deep (>50%) | 25 | 9 (36.0%) | 12 (48.0%) | 2.2 (0.7–7.1) | ||||||
| Alvarez Herrero et al., 2010 [46] | endoscopic resection | <500 μm | 12 | 0 (0%) | 0 (0%) | |||||
| >500 μm | 13 | 0 (0%) | 3 (23.1%) | |||||||
| Ishihara et al., 2017 [47] | endoscopic resection and esophagectomy | <500 μm | 59 | 6.80% | ||||||
| >500 μm | 195 | 22.9–50% |
LN +: lymph node involvement; L+: lymphatic vessel invasion; V+: blood vessel invasion; HR: hazard ratio; CI: confidence interval; sm1: tumor invades into upper one-third of the submucosa; sm2: tumor invades into middle third of the submucosa; sm3: tumor invades into lower one third of submucosa.
Table 2.
Studies of tumor budding in esophageal adenocarcinoma.
| Reference | Stage | Staining | Methods | Cut-off | n | G3 | LN + | L + | V + | PNI | Tumor ≥ 2 cm | Survival (Month) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Landau et al., 2014 [48] | pT1 | H&E | TBF: a 20X microscopic field | TBF (0–2) | 151 | 6% | 11% | 40% | ||||
| (0.785 mm2) with 5 or more TB | TBF (≥ 3) | 59 | 39% | 41% | 75% | |||||||
| Brown et al., 2010 [49] | pT1-pT4 | H&E | Count the total number of TB at | TB < 5 | 31 ± 5.55 | |||||||
| a 20X microscopic field | TB ≥ 5 | 15 ± 1.14 | ||||||||||
| Thies et al., 2016 [50] | pT1-pT4 | Pan-CK | PTB: TB count in 10 HPFs along | LG PTB < 130 TB | 100 | 32% | 42% | 37% | 8% | 13% | ||
| the invasion front | HG PTB ≥ 130 TB | 99 | 63% | 66% | 71% | 23% | 50% | |||||
| ITB: TB count in 10 HPF within | LG ITB < 80 TB | 100 | 29% | 44% | 38% | 8% | 11% | |||||
| the main tumor body | HG ITB ≥ 80 TB | 100 | 66% | 63% | 69% | 23% | 52% |
G3: poorly-differentiated carcinoma; LN +: lymph node involvement; L+: lymphatic vessel invasion; V+: blood vessel invasion; PNI: perineural invasion; TB: tumor budding; PTB: peritumoral tumor budding; ITB: intratumoral tumor budding, H&E: hematoxylin and eosin.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yin, F.; Hernandez Gonzalo, D.; Lai, J.; Liu, X. Histopathology of Barrett’s Esophagus and Early-Stage Esophageal Adenocarcinoma: An Updated Review. Gastrointest. Disord. 2019, 1, 147-163. https://doi.org/10.3390/gidisord1010011
AMA Style
Yin F, Hernandez Gonzalo D, Lai J, Liu X. Histopathology of Barrett’s Esophagus and Early-Stage Esophageal Adenocarcinoma: An Updated Review. Gastrointestinal Disorders. 2019; 1(1):147-163. https://doi.org/10.3390/gidisord1010011
Chicago/Turabian StyleYin, Feng, David Hernandez Gonzalo, Jinping Lai, and Xiuli Liu. 2019. "Histopathology of Barrett’s Esophagus and Early-Stage Esophageal Adenocarcinoma: An Updated Review" Gastrointestinal Disorders 1, no. 1: 147-163. https://doi.org/10.3390/gidisord1010011