
Long Noncoding RNA and mRNA Expression Profiles in the Thyroid Gland of Two Phenotypically Extreme Pig Breeds Using Ribo-Zero RNA Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Material
2.2. Serum THs Assays
2.3. RNA Isolation, Library Preparation, and Sequencing
2.4. Mapping to the Reference Genome
2.5. Transcriptome Assembly
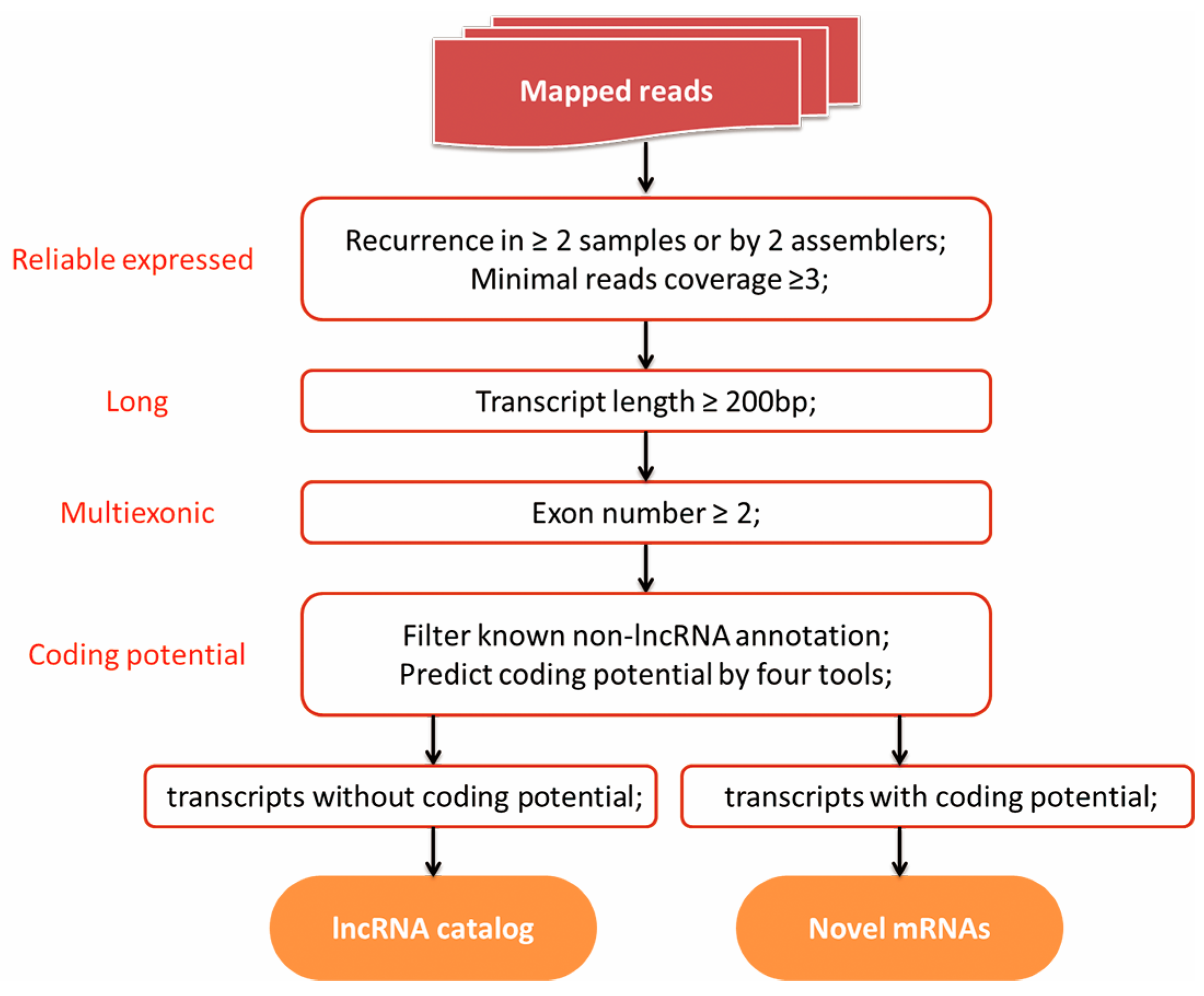
2.6. Identification of lncRNA and Novel mRNA
- The reads coverage of every transcript was calculated by Cufflinks, and those with reads coverage less than three were eliminated;
- Single exon transcripts and transcripts <200 bp were excluded;
- Transcripts blasted to known mRNA and belonged to pseudogenes, pre-microRNA, tRNA, rRNA, and snoRNA were removed;
2.7. Homology Analysis with lncRNAs in Human and Mouse
2.8. Differential Expression Analysis
2.9. Functional Enrichment Analysis
2.10. Target Gene Prediction
2.11. Validation of the Sequencing Data by qRT-PCR
3. Results
3.1. Body Index and THs Concentrations in the Serum of the Two Porcine Breeds
3.2. Characterization of the Thyroid Gland Transcriptome
3.3. Known mRNA Profiling in Pig Thyroid
3.4. Identification of lncRNA
3.5. Identification of Novel mRNAs
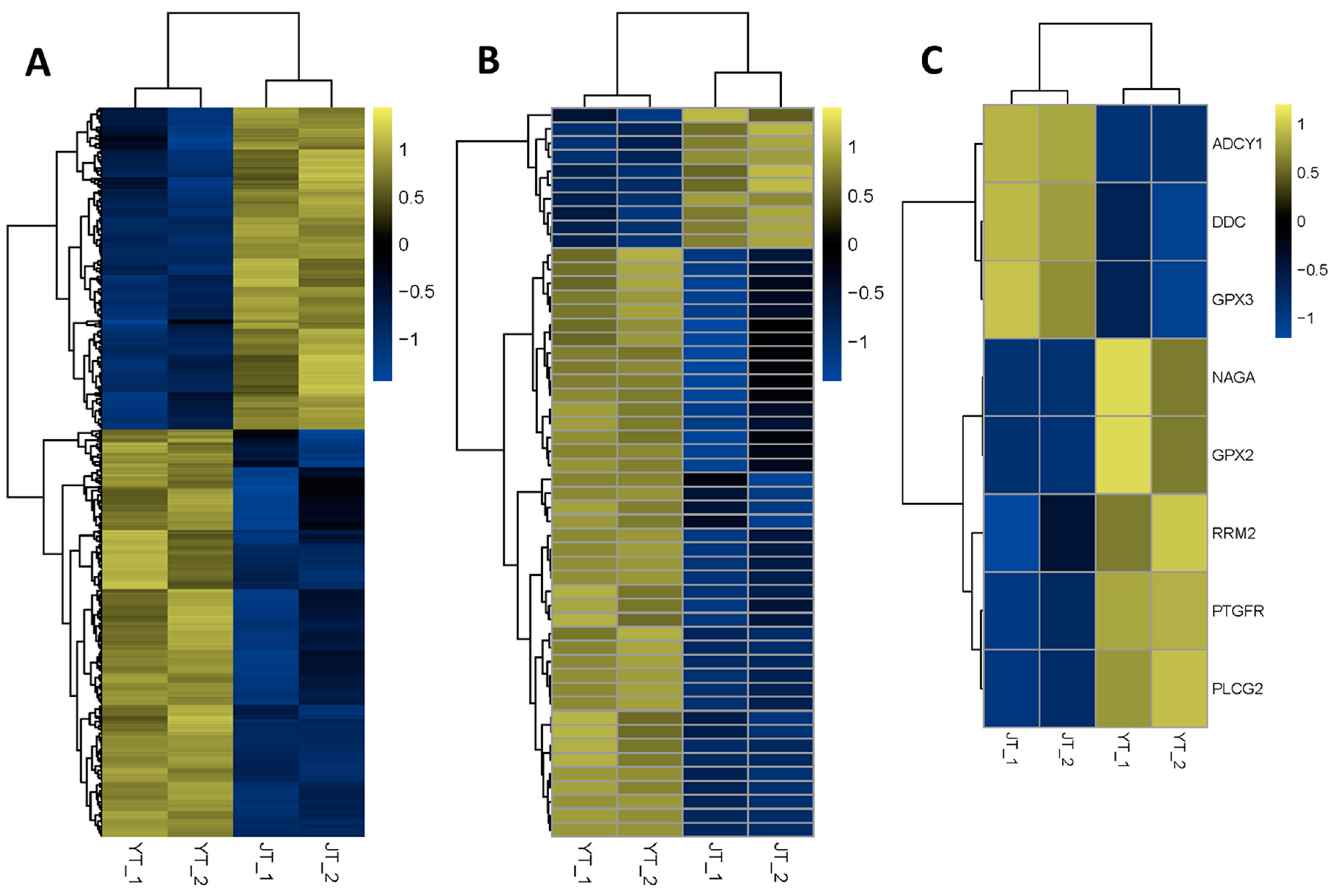
3.6. Differential Expression Analysis
3.7. Functional Enrichment of Differentially-Expressed mRNAs
3.8. Functional Prediction of lncRNAs
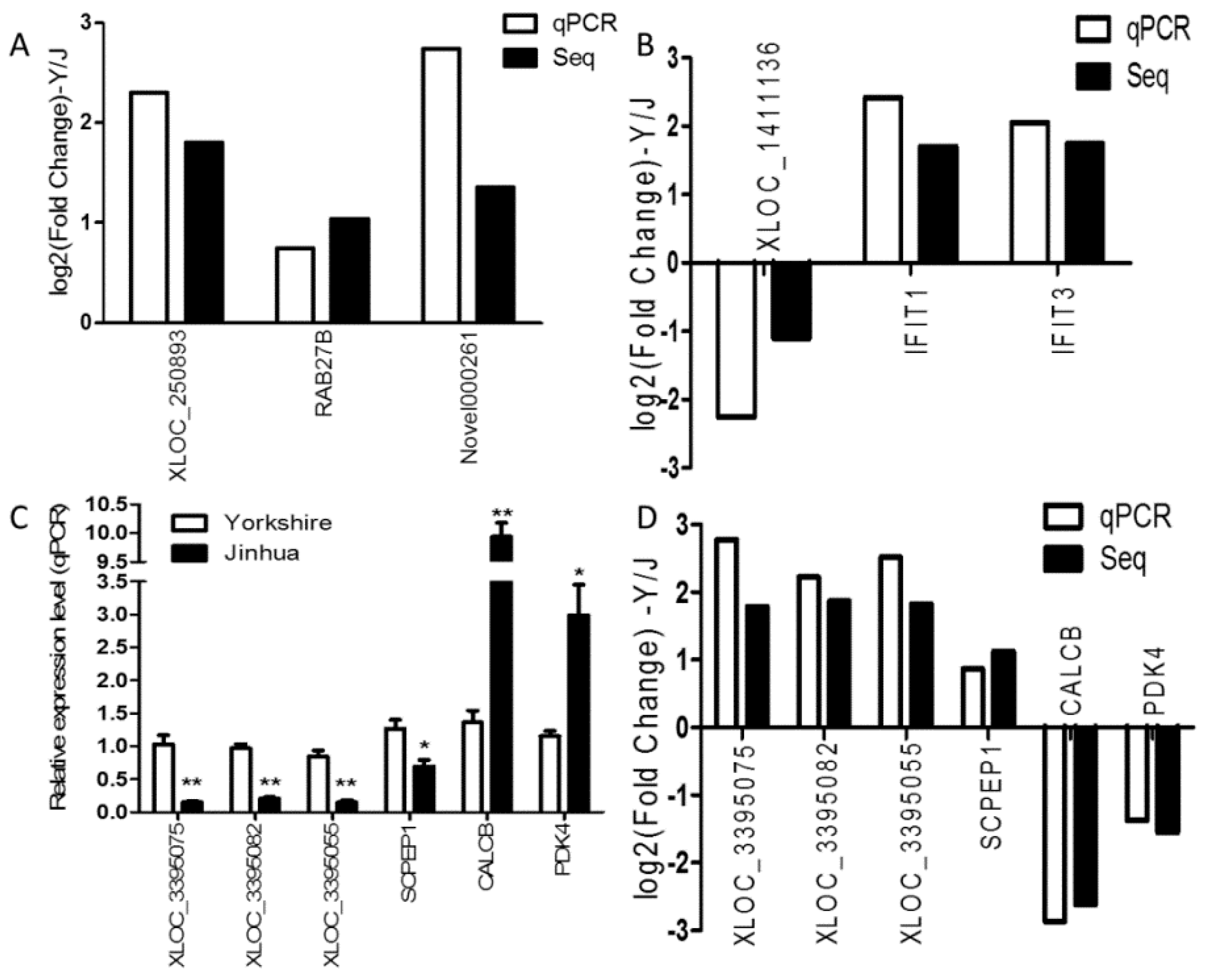
3.9. Quantitative Real-Time PCR Validation
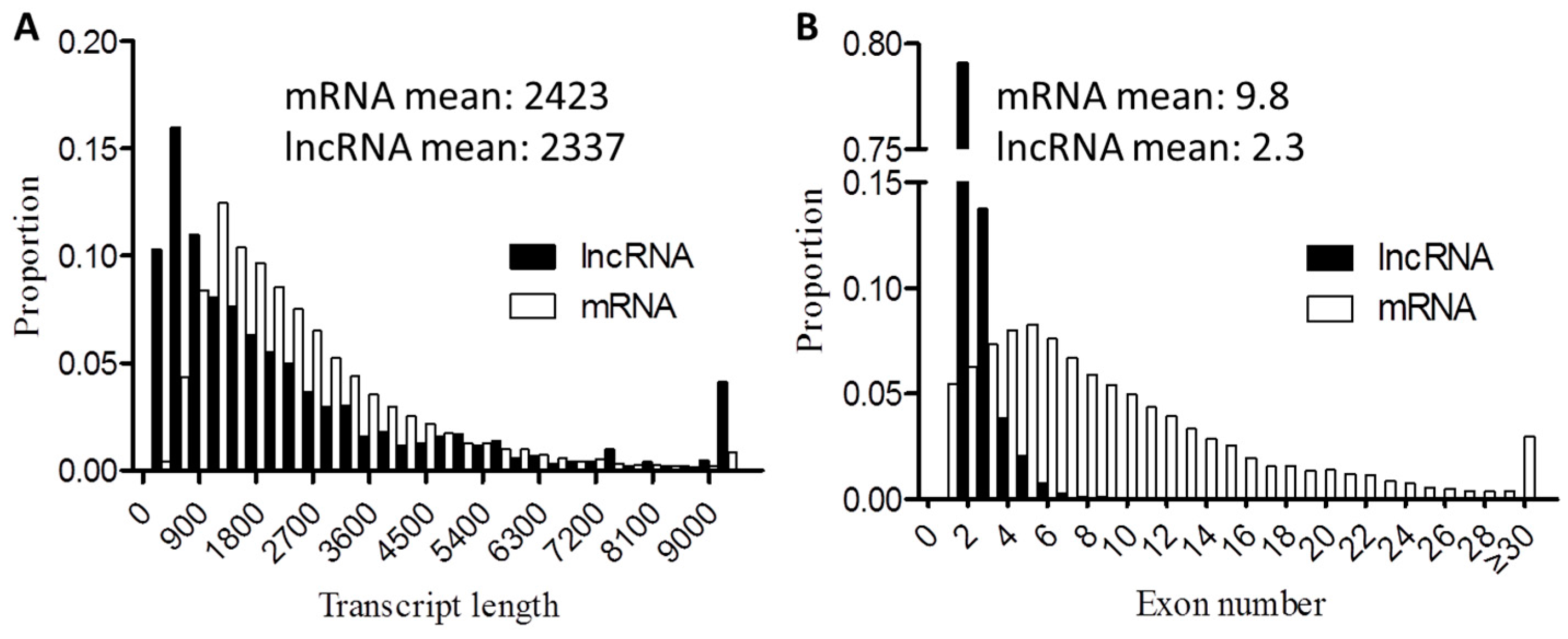
3.10. Genomic Features of lncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| T3 | Triiodothyronine |
| T4 | Tetraiodothyronine or Thyroxine |
| THs | Thyroid Hormones |
| PCR | Polymerase Chain Reaction |
| FPKM | Fragments per kilo-base of exon per million mapped fragments |
References
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [PubMed]
- Zhu, X.G.; Cheng, S.Y. New insights into regulation of lipid metabolism by thyroid hormone. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.-Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Chiamolera, M.I.; Wondisford, F.E. Thyrotropin-releasing hormone and the thyroid hormone feedback mechanism. Endocrinology 2009, 150, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Köhrle, J. On the importance of selenium and iodine metabolism for thyroid hormone biosynthesis and human health. Mol. Nutr. Food Res. 2008, 52, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Ortiga-Carvalho, T.; Oliveira, K.; Soares, B.; Pazos-Moura, C. The role of leptin in the regulation of TSH secretion in the fed state: In vivo and in vitro studies. J. Endocrinol. 2002, 174, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Mansourian, A. Metabolic pathways of tetraidothyronine and triidothyronine production by thyroid gland: A review of articles. Pak. J. Biol. Sci. 2011. [Google Scholar] [CrossRef]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Shkumatava, A. LncRNAs in vertebrates: Advances and challenges. Biochimie 2015, 117, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Knoll, M.; Lodish, H.F.; Sun, L. Long non-coding RNAs as regulators of the endocrine system. Nat. Rev. Endocrinol. 2015, 11, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Fei, T.; Verhaak, R.G.; Su, Z.; Zhang, Y.; Brown, M.; Chen, Y.; Liu, X.S. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat. Struct. Mol. Biol. 2013, 20, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Adiconis, X.; Borges-Rivera, D.; Satija, R.; DeLuca, D.S.; Busby, M.A.; Berlin, A.M.; Sivachenko, A.; Thompson, D.A.; Wysoker, A.; Fennell, T. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 2013, 10, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Fastqc. A quality control tool for high throughput sequence data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 8 July 2016).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A python framework to work with high-throughput sequencing data. Bioinformatics 2014. [Google Scholar] [CrossRef]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J. The Pfam protein families database. Nucleic Acids Res. 2011, 28, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.F.; Jungreis, I.; Kellis, M. Phylocsf: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; Chen, R. Noncode 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2015, 44, D203–D208. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S. Ensembl 2015. Nucleic Acids Res. 2015, 43, D662–D669. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Brown, G.R.; Hiatt, S.M.; Thibaud-Nissen, F.; Astashyn, A.; Ermolaeva, O.; Farrell, C.M.; Hart, J.; Landrum, M.J.; McGarvey, K.M. Refseq: An update on mammalian reference sequences. Nucleic Acids Res. 2014, 42, D756–D763. [Google Scholar] [CrossRef] [PubMed]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. LncRNAdb v2. 0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2014, 43, D168–D173. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G. The gencode v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Method gene ontology analysis for RNA-Seq: Accounting for selection bias. Genome Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Esteller, M. Cis-acting noncoding RNAs: Friends and foes. Nat. Struct. Mol. Biol. 2012, 19, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-δδct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Vilain, C.; Libert, F.; Venet, D.; Costagliola, S.; Vassart, G. Small amplified RNA-sage: An alternative approach to study transcriptome from limiting amount of mRNA. Nucleic Acids Res. 2003, 31, e24–e24. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, J.R.; Bai, Z.; Xu, D.; Yuan, B.; Lo, K.A.; Yoon, M.J.; Lim, Y.C.; Knoll, M.; Slavov, N.; Chen, S. De novo reconstruction of adipose tissue transcriptomes reveals long non-coding RNA regulators of brown adipocyte development. Cell Metab. 2015, 21, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.-G.; Wang, L.-J.; Xu, Z.-R.; Huang, J.-F.; Wang, Y.-R. Developmental patterns in hormone and lipid metabolism of growing jinhua and landrace gilts. Can. J. Anim. Sci. 2008, 88, 601–607. [Google Scholar] [CrossRef]
- Zhao, W.; He, X.; Hoadley, K.A.; Parker, J.S.; Hayes, D.N.; Perou, C.M. Comparison of RNA-Seq by poly (a) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics 2014. [Google Scholar] [CrossRef] [PubMed]
- Ran, M.; Chen, B.; Wu, M.; Liu, X.; He, C.; Yang, A.; Li, Z.; Xiang, Y.; Li, Z.; Zhang, S. Integrated analysis of miRNA and mRNA expression profiles in development of porcine testes. RSC Adv. 2015, 5, 63439–63449. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Q.; Chamba, Y.; Zhang, B.; Shang, P.; Zhang, H.; Wu, C. Identification of genes related to growth and lipid deposition from transcriptome profiles of pig muscle tissue. PLoS ONE 2015, 10, e0141138. [Google Scholar] [CrossRef] [PubMed]
- Marians, R.; Ng, L.; Blair, H.; Unger, P.; Graves, P.; Davies, T. Defining thyrotropin-dependent and-independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc. Natl. Acad. Sci. USA 2002, 99, 15776–15781. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Chiu, J.; Köhrle, J. Peroxides and peroxide-degrading enzymes in the thyroid. Antioxid. Redox Signal. 2008, 10, 1577–1592. [Google Scholar] [CrossRef] [PubMed]
- Drutel, A.; Archambeaud, F.; Caron, P. Selenium and the thyroid gland: More good news for clinicians. Clin. Endocrinol. 2013, 78, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Howie, A.F.; Walker, S.W.; Akesson, B.; Arthur, J.R.; Beckett, G.J. Thyroidal extracellular glutathione peroxidase: A potential regulator of thyroid-hormone synthesis. Biochem. J. 1995, 308, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Iosco, C.; Cosentino, C.; Sirna, L.; Romano, R.; Cursano, S.; Mongia, A.; Pompeo, G.; di Bernardo, J.; Ceccarelli, C.; Tallini, G. Anoctamin 1 is apically expressed on thyroid follicular cells and contributes to ATP-and calcium-activated iodide efflux. Cell. Physiol. Biochem. 2014, 34, 966–980. [Google Scholar] [CrossRef] [PubMed]
- Viitanen, T.M.; Sukumaran, P.; Löf, C.; Törnquist, K. Functional coupling of TRPC2 cation channels and the calcium-activated anion channels in rat thyroid cells: Implications for iodide homeostasis. J. Cell Physiol. 2013, 228, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Matsubara, T.; Seo, D.; Okamoto, M.; Nagashima, K.; Sasaki, Y.; Hayase, S.; Murata, T.; Liao, X.-H.; Hanson, J. Thyroid regeneration: Characterization of clear cells after partial thyroidectomy. Endocrinology 2012, 153, 2514–2525. [Google Scholar] [CrossRef] [PubMed]
- Stephen, J.K.; Chitale, D.; Narra, V.; Chen, K.M.; Sawhney, R.; Worsham, M.J. DNA methylation in thyroid tumorigenesis. Cancers 2011, 3, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Chung, K.-W.; Yang, S.K.; Park, M.J.; Min, H.S.; Kim, S.W.; Kang, H.S. DNA methylation of MAPK signal-inhibiting genes in papillary thyroid carcinoma. Anticancer Res. 2013, 33, 4833–4839. [Google Scholar] [PubMed]
- Planck, T.; Shahida, B.; Sjögren, M.; Groop, L.; Hallengren, B.; Lantz, M. Association of BTG2, CYR61, ZFP36, and SCD gene polymorphisms with Graves’ Disease and ophthalmopathy. Thyroid 2014, 24, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Yamamoto, T.; Sato, N.; Tanaka, K. Ventricular fibrillation associated with early repolarization in a patient with thyroid storm. J. Interv. Card. Electrophysiol. 2010, 29, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Kuang, M.; Wang, S.; Wu, M.; Ning, G.; Yao, Z.; Li, L. Expression of IFNα-inducible genes and modulation of HLA-DR and thyroid stimulating hormone receptors in Graves’ disease. Mol. Cell. Endocrinol. 2010, 319, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Van den Hove, M.-F.; Croizet-Berger, K.; Jouret, F.; Guggino, S.E.; Guggino, W.B.; Devuyst, O.; Courtoy, P.J. The loss of the chloride channel, CLC-5, delays apical iodide efflux and induces a euthyroid goiter in the mouse thyroid gland. Endocrinology 2006, 147, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Zhang, Q.; Mao, Z.; Chen, J.; Wang, X.; Qu, J.; Zhang, J.; Jin, D. CTGF is overexpressed in papillary thyroid carcinoma and promotes the growth of papillary thyroid cancer cells. Tumor Biol. 2011, 32, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Neve, P.; Willems, C.; Dumont, J.E. Involvement of the microtubule-microfilament system in thyroid secretion. Exp. Cell Res. 1970, 63, 457–460. [Google Scholar] [CrossRef]
- Apodaca, G. Endocytic traffic in polarized epithelial cells: Role of the actin and microtubule cytoskeleton. Traffic 2001, 2, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Weikard, R.; Hadlich, F.; Kuehn, C. Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genomics 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, S.; Liu, X.; Liu, H.; Hu, T.; Qiu, X.; Zhang, J.; Lei, M. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci. Rep. UK 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z.; Wang, L.; Liu, Y.; Liu, Y. ALDB: A domestic-animal long noncoding RNA database. PLoS ONE 2015, 10, e0124003. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Bartel, D.P. LincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-Y.; Li, A.-M.; Adeola, A.C.; Liu, Y.-H.; Irwin, D.M.; Xie, H.-B.; Zhang, Y.-P. Genome-wide identification of long intergenic noncoding RNA genes and their potential association with domestication in pigs. Genome Biol. Evol. 2014, 6, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Mou, Y.; Huang, Z.; Li, Y.; Han, L.; Zhang, Y.; Feng, Y.; Chen, Y.; Jiang, X.; Zhao, W. The sequence and analysis of a Chinese pig genome. GigaScience 2012. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Long noncoding RNAs may alter chromosome’s 3D structure. Science 2013, 340, 910–910. [Google Scholar] [CrossRef] [PubMed]
- Ran, M.; Chen, B.; Li, Z.; Wu, M.; Liu, X.; He, C.; Zhang, S.; Li, Z. Systematic identification of long non-coding RNAs in immature and mature porcine testes. Biol. Reprod. Biolreprod. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wu, J.; Schuster, A.S.; Hennig, G.W.; Yan, W. Expression profiling reveals developmentally regulated lncRNA repertoire in the mouse male germline. Biol. Reprod. 2013. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, K.; Shi, Z.; Zhang, J.; Zhu, J.; Zhu, S.; Zhang, A.; Jia, Z.; Wang, G.; Yu, S. LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int. J. Oncol. 2012, 40, 2004–2012. [Google Scholar] [PubMed]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A. Gene silencing in x-chromosome inactivation: Advances in understanding facultative heterochromatin formation. Nat. Rev. Genet. 2011, 12, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab gtpases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Gomi, H.; Mori, K.; Itohara, S.; Izumi, T. Rab27b is expressed in a wide range of exocytic cells and involved in the delivery of secretory granules near the plasma membrane. Mol. Biol. Cell. 2007, 18, 4377–4386. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, D.N.; Zembutsu, H.; Sechanov, T.; Vidinov, K.; Kee, L.S.; Ivanova, R.; Becheva, E.; Kocova, M.; Toncheva, D.; Nakamura, Y. Genome-wide gene expression profiles of thyroid carcinoma: Identification of molecular targets for treatment of thyroid carcinoma. Oncol. Rep. 2008, 20, 105–121. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Jinhua | Yorkshire | p-Value |
|---|---|---|---|
| Body weight (kg) | 29.33 ± 0.88 | 47.07 ± 1.93 | 0.001 |
| Thyroid gland weight (g) | 3.20 ± 0.06 | 4.93 ± 0.38 | 0.011 |
| Thyroid index (g/kg) | 0.109 ± 0.004 | 0.105 ± 0.009 | 0.702 |
| Serum total thyroxine level (nmol/L) | 30.36 ± 0.76 | 37.85 ± 1.73 | 0.017 |
| Serum total triiodothyronine level (nmol/L) | 0.53 ± 0.02 | 1.00 ± 0.12 | 0.020 |
| Serum free thyroxine level (pmol/L) | 9.34 ± 0.20 | 11.31 ± 0.73 | 0.058 |
| Serum free triiodothyronine level (pmol/L) | 1.84 ± 0.09 | 2.58 ± 0.33 | 0.096 |
| Sample | Total Reads | Total Mapped % | Uniquely Mapped % |
|---|---|---|---|
| Jinhua-1 | 89460072 | 81.83 | 70.63 |
| Jinhua-2 | 90102440 | 81.50 | 72.83 |
| Yorkshire-1 | 84160098 | 83.63 | 72.62 |
| Yorkshire-2 | 87285462 | 84.37 | 73.80 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Mao, H.; Huang, M.; Chen, L.; Chen, J.; Cai, Z.; Wang, Y.; Xu, N. Long Noncoding RNA and mRNA Expression Profiles in the Thyroid Gland of Two Phenotypically Extreme Pig Breeds Using Ribo-Zero RNA Sequencing. Genes 2016, 7, 34. https://doi.org/10.3390/genes7070034
Shen Y, Mao H, Huang M, Chen L, Chen J, Cai Z, Wang Y, Xu N. Long Noncoding RNA and mRNA Expression Profiles in the Thyroid Gland of Two Phenotypically Extreme Pig Breeds Using Ribo-Zero RNA Sequencing. Genes. 2016; 7(7):34. https://doi.org/10.3390/genes7070034
Chicago/Turabian StyleShen, Yifei, Haiguang Mao, Minjie Huang, Lixing Chen, Jiucheng Chen, Zhaowei Cai, Ying Wang, and Ningying Xu. 2016. "Long Noncoding RNA and mRNA Expression Profiles in the Thyroid Gland of Two Phenotypically Extreme Pig Breeds Using Ribo-Zero RNA Sequencing" Genes 7, no. 7: 34. https://doi.org/10.3390/genes7070034