
High-Resolution Genotyping of Expressed Equine MHC Reveals a Highly Complex MHC Structure
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. PCR Amplification and Library Preparation
2.3. Sequencing and Bioinformatics Analysis
2.4. Nomenclature of Novel MHC Sequences and Haplotypes
3. Results
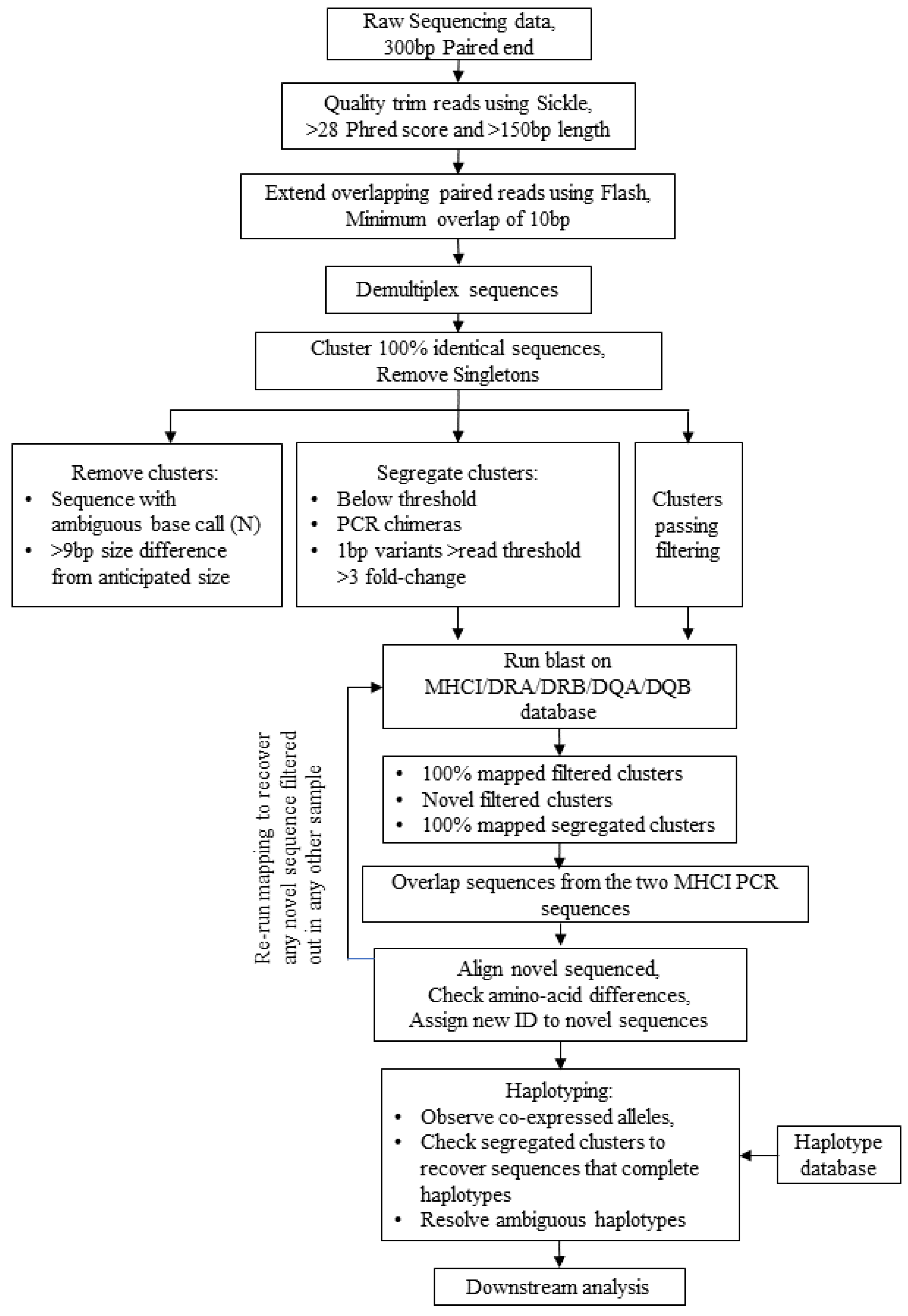
3.1. Design of an Equine MHC Sequencing Platform—Laboratory and Bioinformatics Workflow
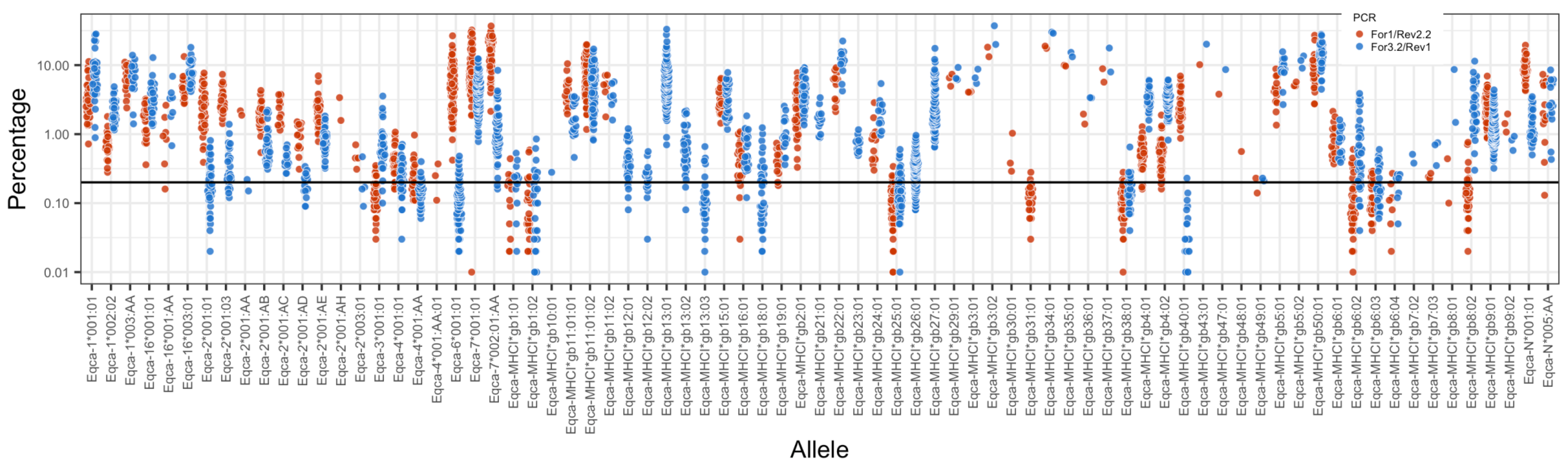
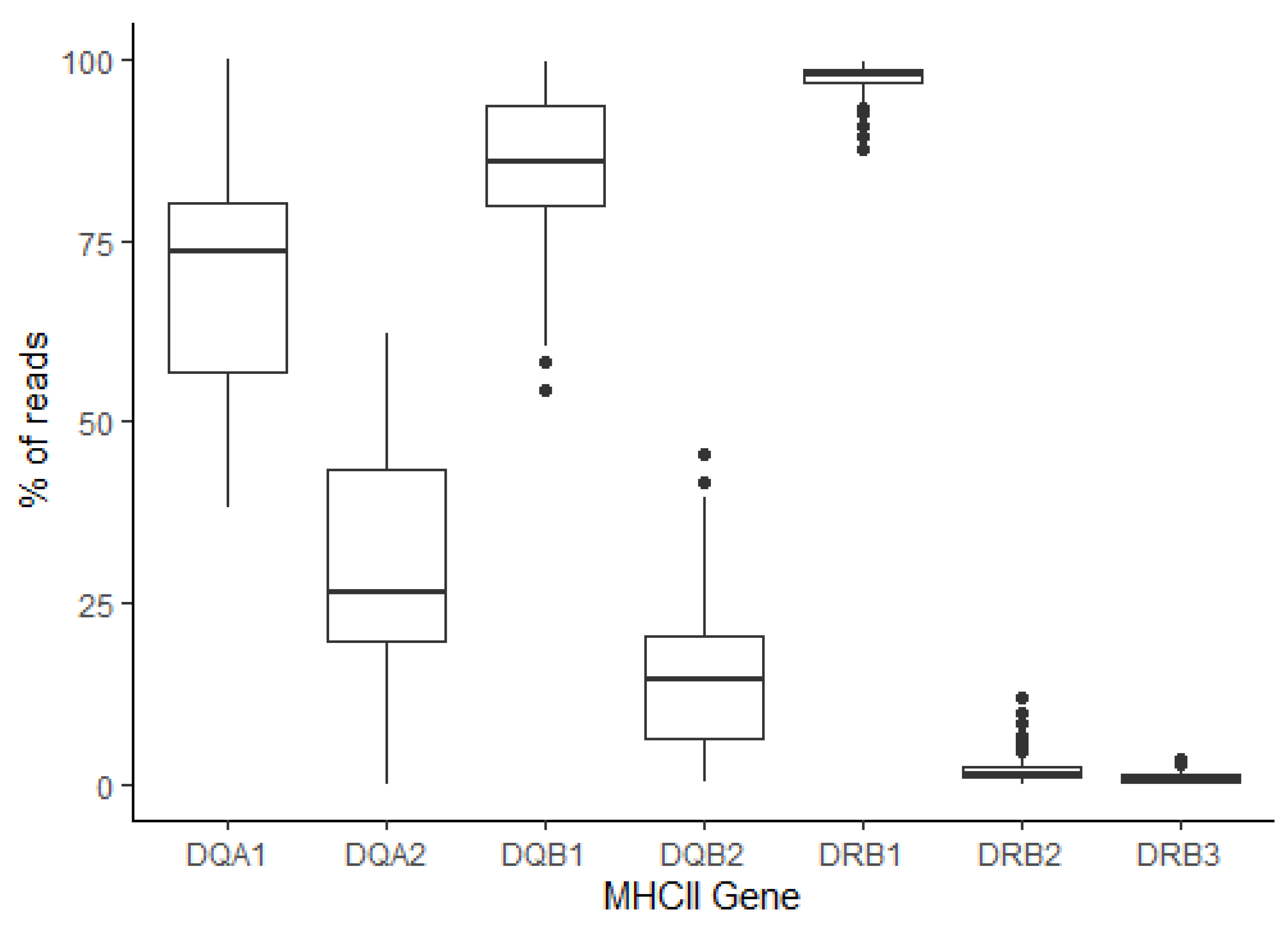
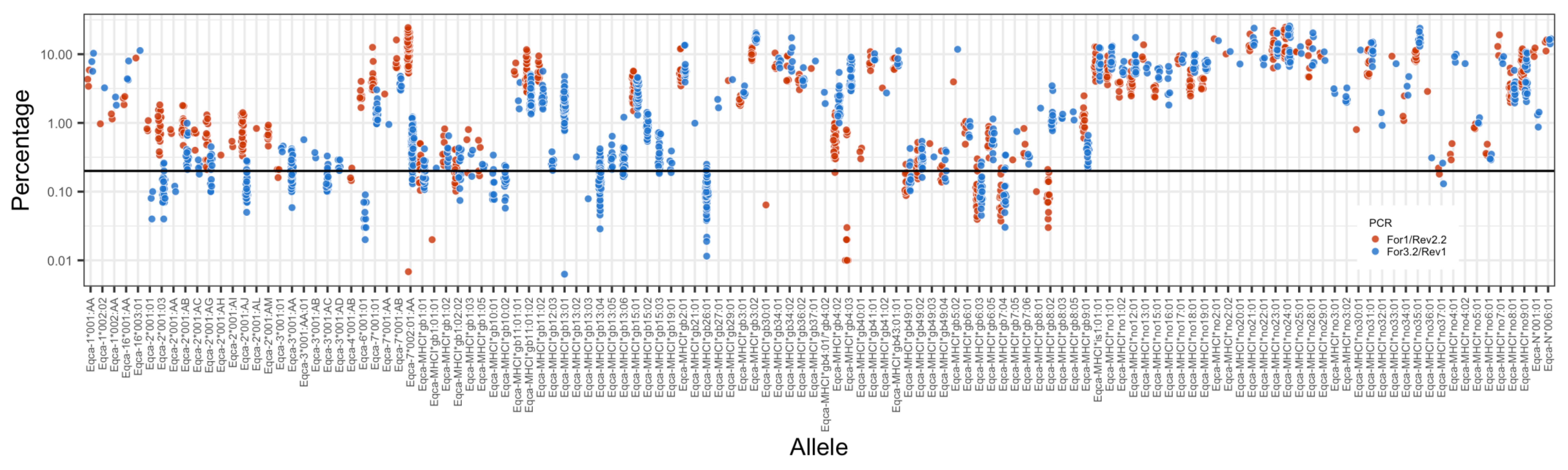
3.2. MHC Diversity in a Population of Thoroughbred Horses
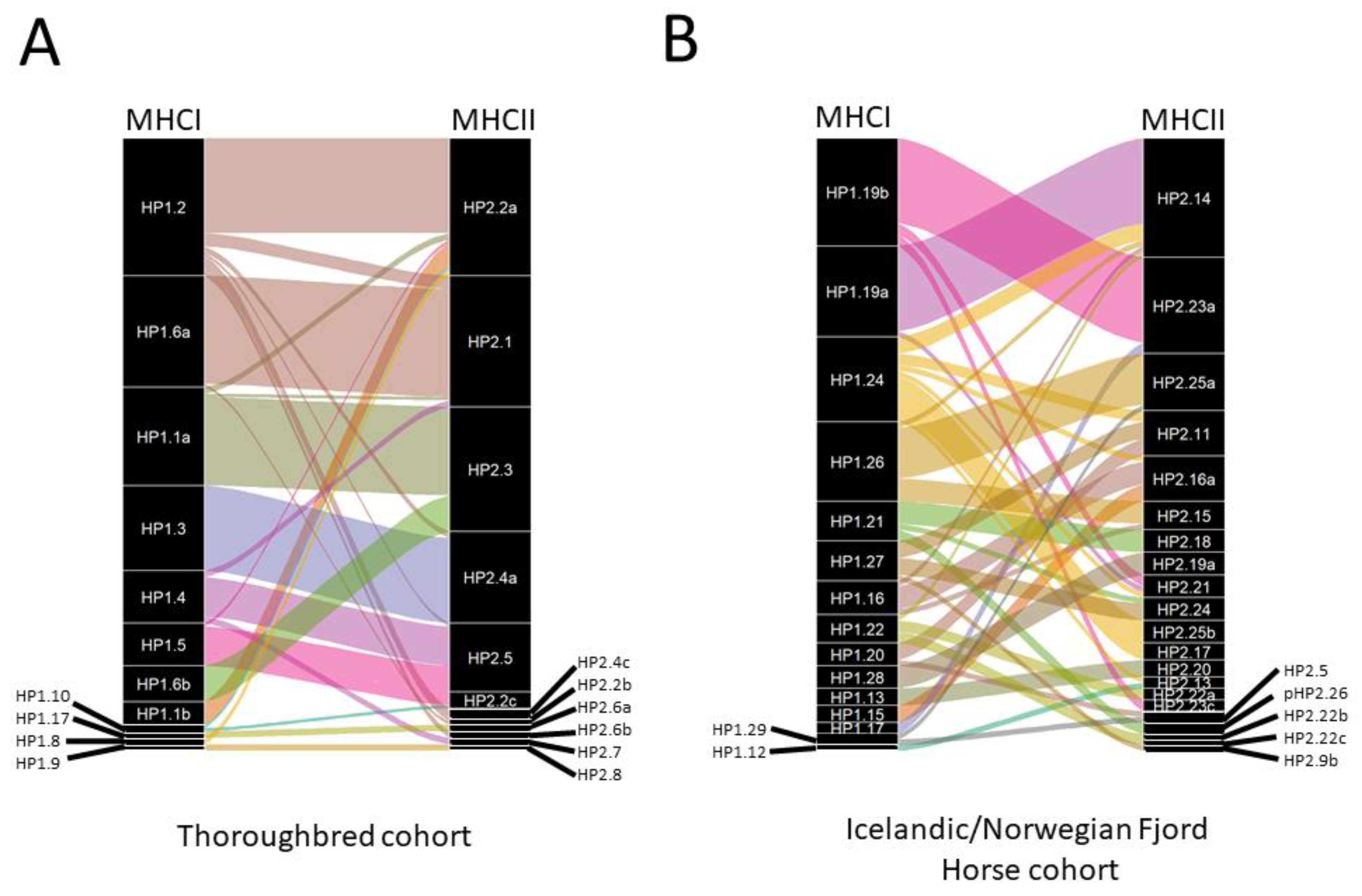
3.3. Analysis of the MHCI and MHCII Repertoires in Cohorts of Icelandic and Norwegian Fjord Horses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hofstetter, A.R.; Sullivan, L.C.; Lukacher, A.E.; Brooks, A.G. Diverse roles of non-diverse molecules: Mhc class ib molecules in host defense and control of autoimmunity. Curr. Opin. Immunol. 2011, 23, 104–110. [Google Scholar] [CrossRef]
- Radwan, J.; Babik, W.; Kaufman, J.; Lenz, T.L.; Winternitz, J. Advances in the evolutionary understanding of mhc polymorphism. Trends Genet. 2020, 36, 298–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, A.L.; Tallmadge, R.L.; Ramlachan, N.; Miller, D.; Bird, H.; Antczak, D.F.; Raudsepp, T.; Chowdhary, B.P.; Skow, L.C. An ordered bac contig map of the equine major histocompatibility complex. Cytogenet. Genome Res. 2003, 102, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Tallmadge, R.L.; Lear, T.L.; Antczak, D.F. Genomic characterization of mhc class i genes of the horse. Immunogenetics 2005, 57, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Viluma, A.; Mikko, S.; Hahn, D.; Skow, L.; Andersson, G.; Bergstrom, T.F. Genomic structure of the horse major histocompatibility complex class ii region resolved using pacbio long-read sequencing technology. Sci. Rep. 2017, 7, 45518. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.; Tallmadge, R.L.; Binns, M.; Zhu, B.; Mohamoud, Y.A.; Ahmed, A.; Brooks, S.A.; Antczak, D.F. Polymorphism at expressed dq and dr loci in five common equine mhc haplotypes. Immunogenetics 2017, 69, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbis, D.P.; Maher, J.K.; Stanek, J.; Klaunberg, B.A.; Antczak, D.F. Horse cdna clones encoding two mhc class i genes. Immunogenetics 1994, 40, 163. [Google Scholar] [CrossRef]
- Chung, C.; Leib, S.R.; Fraser, D.G.; Ellis, S.A.; McGuire, T.C. Novel classical mhc class i alleles identified in horses by sequencing clones of reverse transcription-pcr products. Eur. J. Immunogenet. 2003, 30, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Diaz, S.; Giovambattista, G.; Dulout, F.N.; Peral-Garcia, P. Genetic variation of the second exon of ela-drb genes in argentine creole horses. Anim. Genet. 2001, 32, 257–263. [Google Scholar] [CrossRef]
- Ellis, S.A.; Martin, A.J.; Holmes, E.C.; Morrison, W.I. At least four mhc class i genes are transcribed in the horse: Phylogenetic analysis suggests an unusual evolutionary history for the mhc in this species. Eur. J. Immunogenet. 1995, 22, 249–260. [Google Scholar] [CrossRef]
- Fraser, D.G.; Bailey, E. Demonstration of three drb loci in a domestic horse family. Immunogenetics 1996, 44, 441–445. [Google Scholar] [CrossRef]
- Fraser, D.G.; Bailey, E. Polymorphism and multiple loci for the horse dqa gene. Immunogenetics 1998, 47, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Klumplerova, M.; Vychodilova, L.; Bobrova, O.; Cvanova, M.; Futas, J.; Janova, E.; Vyskocil, M.; Vrtkova, I.; Putnova, L.; Dusek, L.; et al. Major histocompatibility complex and other allergy-related candidate genes associated with insect bite hypersensitivity in icelandic horses. Mol. Biol. Rep. 2013, 40, 3333–3340. [Google Scholar] [CrossRef]
- McGuire, T.C.; Leib, S.R.; Mealey, R.H.; Fraser, D.G.; Prieur, D.J. Presentation and binding affinity of equine infectious anemia virus ctl envelope and matrix protein epitopes by an expressed equine classical mhc class i molecule. J. Immunol. 2003, 171, 1984–1993. [Google Scholar] [CrossRef]
- Ramsay, J.D.; Leib, S.R.; Orfe, L.; Call, D.R.; Tallmadge, R.L.; Fraser, D.G.; Mealey, R.H. Development of a DNA microarray for detection of expressed equine classical mhc class i sequences in a defined population. Immunogenetics 2010, 62, 633–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Kim, E.; Igarashi, M.; Ito, K.; Hasebe, R.; Fukushi, H.; Sawa, H.; Kimura, T. Single amino acid residue in the a2 domain of major histocompatibility complex class i is involved in the efficiency of equine herpesvirus-1 entry. J. Biol. Chem. 2011, 286, 39370–39378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalai, G.; Antczak, D.F.; Gerber, H.; Lazary, S. Molecular cloning and characterization of horse dqa cdna. Immunogenetics 1994, 40, 457. [Google Scholar] [CrossRef]
- Szalai, G.; Antczak, D.F.; Gerber, H.; Lazary, S. Molecular cloning and characterization of horse dqb cdna. Immunogenetics 1994, 40, 458. [Google Scholar] [CrossRef]
- Szalai, G.; Bailey, E.; Gerber, H.; Lazary, S. DNA sequence analysis of serologically detected ela class ii haplotypes at the equine dq beta locus. Anim. Genet. 1993, 24, 187–190. [Google Scholar] [CrossRef]
- Tallmadge, R.L.; Campbell, J.A.; Miller, D.C.; Antczak, D.F. Analysis of mhc class i genes across horse mhc haplotypes. Immunogenetics 2010, 62, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Egana, V.; Sanders, H.; Chitnis, N. New challenges, new opportunities: Next generation sequencing and its place in the advancement of hla typing. Hum. Immunol. 2021, 82, 478–487. [Google Scholar] [CrossRef]
- Profaizer, T.; Kumanovics, A. Human leukocyte antigen typing by next-generation sequencing. Clin. Lab. Med. 2018, 38, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Pereira, S.; Jaramillo, A.; Stoll, S.T.; Khan, F.M.; Berka, N.; Mostafa, A.A.; Pando, M.J.; Usenko, C.Y.; Bettinotti, M.P.; et al. Comparison of sequence-specific oligonucleotide probe vs next generation sequencing for hla-a, b, c, drb1, drb3/b4/b5, dqa1, dqb1, dpa1, and dpb1 typing: Toward single-pass high-resolution hla typing in support of solid organ and hematopoietic cell transplant programs. HLA 2019, 94, 296–306. [Google Scholar] [PubMed]
- Morgan, R.A.; Karl, J.A.; Bussan, H.E.; Heimbruch, K.E.; O’Connor, D.H.; Dudley, D.M. Restricted mhc class i a locus diversity in olive and hybrid olive/yellow baboons from the southwest national primate research center. Immunogenetics 2018, 70, 449–458. [Google Scholar] [CrossRef]
- Razali, H.; O’Connor, E.; Drews, A.; Burke, T.; Westerdahl, H. A quantitative and qualitative comparison of illumina miseq and 454 amplicon sequencing for genotyping the highly polymorphic major histocompatibility complex (mhc) in a non-model species. BMC Res. Notes 2017, 10, 346. [Google Scholar] [CrossRef]
- Sundaram, A.Y.M.; Garseth, A.H.; Maccari, G.; Grimholt, U. An illumina approach to mhc typing of atlantic salmon. Immunogenetics 2020, 72, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Vasoya, D.; Law, A.; Motta, P.; Yu, M.; Muwonge, A.; Cook, E.; Li, X.; Bryson, K.; MacCallam, A.; Sitt, T.; et al. Rapid identification of bovine mhci haplotypes in genetically divergent cattle populations using next-generation sequencing. Immunogenetics 2016, 68, 765–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasoya, D.; Oliveira, P.S.; Muriel, L.A.; Tzelos, T.; Vrettou, C.; Morrison, W.I.; de Miranda Santos, I.K.F.; Connelley, T. High throughput analysis of mhc-i and mhc-dr diversity of brazilian cattle populations. HLA 2021, 98, 93–113. [Google Scholar] [CrossRef]
- Silwamba, I.; Vasoya, D.; Simuunza, M.; Tzelos, T.; Nalubamba, K.S.; Simulundu, E.; Vrettou, C.; Mainda, G.; Watson, M.; Muma, J.B.; et al. High throughput analysis of mhc class i and class ii diversity of zambian indigenous cattle populations. HLA 2023, 101, 458–483. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 19 December 2022).
- Maccari, G.; Robinson, J.; Bontrop, R.E.; Otting, N.; de Groot, N.G.; Ho, C.S.; Ballingall, K.T.; Marsh, S.G.E.; Hammond, J.A. IPD-MHC: Nomenclature requirements for the non-human major histocompatibility complex in the next-generation sequencing era. Immunogenetics 2018, 70, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Fuselli, S.; Baptista, R.P.; Panziera, A.; Magi, A.; Guglielmi, S.; Tonin, R.; Benazzo, A.; Bauzer, L.G.; Mazzoni, C.J.; Bertorelle, G. A new hybrid approach for mhc genotyping: High-throughput ngs and long read minion nanopore sequencing, with application to the non-model vertebrate alpine chamois (rupicapra rupicapra). Heredity 2018, 121, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Pardal, S.; Drews, A.; Alves, J.A.; Ramos, J.A.; Westerdahl, H. Characterization of mhc class i in a long distance migratory wader, the icelandic black-tailed godwit. Immunogenetics 2017, 69, 463–478. [Google Scholar] [CrossRef] [Green Version]
- McGivney, B.A.; Han, H.; Corduff, L.R.; Katz, L.M.; Tozaki, T.; MacHugh, D.E.; Hill, E.W. Genomic inbreeding trends, influential sire lines and selection in the global thoroughbred horse population. Sci. Rep. 2020, 10, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, E.T.; Ho, S.Y.W.; Thomson, P.C.; Ang, R.A.; Velie, B.D.; Hamilton, N.A. Founder-specific inbreeding depression affects racing performance in thoroughbred horses. Sci. Rep. 2018, 8, 6167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef] [Green Version]
- Petersen, J.L.; Mickelson, J.R.; Cothran, E.G.; Andersson, L.S.; Axelsson, J.; Bailey, E.; Bannasch, D.; Binns, M.M.; Borges, A.S.; Brama, P.; et al. Genetic diversity in the modern horse illustrated from genome-wide snp data. PLoS ONE 2013, 8, e54997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, M.; Jonsson, H.; Chang, D.; Der Sarkissian, C.; Ermini, L.; Ginolhac, A.; Albrechtsen, A.; Dupanloup, I.; Foucal, A.; Petersen, B.; et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc. Natl. Acad. Sci. USA 2014, 111, E5661–E5669. [Google Scholar] [CrossRef] [PubMed]
- Wallner, B.; Palmieri, N.; Vogl, C.; Rigler, D.; Bozlak, E.; Druml, T.; Jagannathan, V.; Leeb, T.; Fries, R.; Tetens, J.; et al. Y chromosome uncovers the recent oriental origin of modern stallions. Curr. Biol. 2017, 27, 2029–2035.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, C.M.; Violette, N.; Miller, D.; Wagner, B.; Svansson, V.; Antczak, D.F. Mhc haplotype diversity in icelandic horses determined by polymorphic microsatellites. Genes Immun. 2019, 20, 660–670. [Google Scholar] [CrossRef]
- Jaworska, J.; Ropka-Molik, K.; Woclawek-Potocka, I.; Siemieniuch, M. Inter- and intrabreed diversity of the major histocompatibility complex (mhc) in primitive and draft horse breeds. PLoS ONE 2020, 15, e0228658. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lei, H.; Ran, X.; Wang, J. Genetic variation and selection in the major histocompatibility complex class ii gene in the guizhou pony. PeerJ 2020, 8, e9889. [Google Scholar] [CrossRef]
- Sadeghi, R.; Moradi-Shahrbabak, M.; Miraei Ashtiani, S.R.; Miller, D.C.; Antczak, D.F. Mhc haplotype diversity in persian arabian horses determined using polymorphic microsatellites. Immunogenetics 2018, 70, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, G.; Zhao, S.; Li, K.; Zhang, D.; Liu, S.; Hu, D. Major histocompatibility complex (mhc) diversity of the reintroduction populations of endangered przewalski’s horse. Genes 2022, 13, 928. [Google Scholar] [CrossRef]
- Mealey, R.H.; Lee, J.H.; Leib, S.R.; Littke, M.H.; McGuire, T.C. A single amino acid difference within the alpha-2 domain of two naturally occurring equine mhc class i molecules alters the recognition of gag and rev epitopes by equine infectious anemia virus-specific ctl. J. Immunol. 2006, 177, 7377–7390. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C.; Ellis, S.A. Evolutionary history of mhc class i genes in the mammalian order perissodactyla. J. Mol. Evol. 1999, 49, 316–324. [Google Scholar] [CrossRef]
- Allen, R.L. Non-classical immunology. Genome Biol. 2001, 2, REPORTS4004. [Google Scholar] [CrossRef]
- Bergmann, T.; Lindvall, M.; Moore, E.; Moore, E.; Sidney, J.; Miller, D.; Tallmadge, R.L.; Myers, P.T.; Malaker, S.A.; Shabanowitz, J.; et al. Peptide-binding motifs of two common equine class i mhc molecules in thoroughbred horses. Immunogenetics 2017, 69, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, T.; Moore, C.; Sidney, J.; Miller, D.; Tallmadge, R.; Harman, R.M.; Oseroff, C.; Wriston, A.; Shabanowitz, J.; Hunt, D.F.; et al. The common equine class i molecule eqca-1*00101 (ela-a3.1) is characterized by narrow peptide binding and t cell epitope repertoires. Immunogenetics 2015, 67, 675–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeg, C.A.; Marti, E.; Gaillard, C.; Kaspers, B. Equine recurrent uveitis is strongly associated with the mhc class i haplotype ela-a9. Equine Vet. J. 2004, 36, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Curik, I.; Fraser, D.; Eder, C.; Achmann, R.; Swinburne, J.; Crameri, R.; Brem, G.; Solkner, J.; Marti, E. Association between the mhc gene region and variation of serum ige levels against specific mould allergens in the horse. Genet. Sel. Evol. 2003, 35 (Suppl. S1), S177–S190. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.L.; Kaese, H.J.; Valberg, S.J.; Hendrickson, J.A.; Rendahl, A.K.; Bellone, R.R.; Dynes, K.M.; Wagner, M.L.; Lucio, M.A.; Cuomo, F.M.; et al. Genetic risk factors for insidious equine recurrent uveitis in appaloosa horses. Anim. Genet. 2014, 45, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Schurink, A.; Wolc, A.; Ducro, B.J.; Frankena, K.; Garrick, D.J.; Dekkers, J.C.; van Arendonk, J.A. Genome-wide association study of insect bite hypersensitivity in two horse populations in the netherlands. Genet. Sel. Evol. 2012, 44, 31. [Google Scholar] [CrossRef] [Green Version]
- Andersson, L.S.; Swinburne, J.E.; Meadows, J.R.; Brostrom, H.; Eriksson, S.; Fikse, W.F.; Frey, R.; Sundquist, M.; Tseng, C.T.; Mikko, S.; et al. The same ela class ii risk factors confer equine insect bite hypersensitivity in two distinct populations. Immunogenetics 2012, 64, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Staiger, E.A.; Tseng, C.T.; Miller, D.; Cassano, J.M.; Nasir, L.; Garrick, D.; Brooks, S.A.; Antczak, D.F. Host genetic influence on papillomavirus-induced tumors in the horse. Int. J. Cancer 2016, 139, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeannerat, E.; Marti, E.; Berney, C.; Janett, F.; Bollwein, H.; Sieme, H.; Burger, D.; Wedekind, C. Stallion semen quality depends on major histocompatibility complex matching to teaser mare. Mol. Ecol. 2018, 27, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Burger, D.; Meuwly, C.; Marti, E.; Sieme, H.; Oberthur, M.; Janda, J.; Meinecke-Tillmann, S.; Wedekind, C. Mhc-correlated preferences in diestrous female horses (equus caballus). Theriogenology 2017, 89, 318–323.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, D.; Thomas, S.; Aepli, H.; Dreyer, M.; Fabre, G.; Marti, E.; Sieme, H.; Robinson, M.R.; Wedekind, C. Major histocompatibility complex-linked social signalling affects female fertility. Proc. Biol. Sci. 2017, 284, 20171824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, D.; Dolivo, G.; Marti, E.; Sieme, H.; Wedekind, C. Female major histocompatibility complex type affects male testosterone levels and sperm number in the horse (equus caballus). Proc. Biol. Sci. 2015, 282, 20150407. [Google Scholar] [CrossRef]
- Kydd, J.H.; Case, R.; Winton, C.; MacRae, S.; Sharp, E.; Ricketts, S.L.; Rash, N.; Newton, J.R. Polarisation of equine pregnancy outcome associated with a maternal mhc class i allele: Preliminary evidence. Vet. Microbiol. 2016, 188, 34–40. [Google Scholar] [CrossRef]
- Berglund, A.K.; Schnabel, L.V. Allogeneic major histocompatibility complex-mismatched equine bone marrow-derived mesenchymal stem cells are targeted for death by cyt.totoxic anti-major histocompatibility complex antibodies. Equine Vet. J. 2017, 49, 539–544. [Google Scholar] [CrossRef]
- Barrachina, L.; Cequier, A.; Romero, A.; Vitoria, A.; Zaragoza, P.; Vazquez, F.J.; Rodellar, C. Allo-antibody production after intraarticular administration of mesenchymal stem cells (mscs) in an equine osteoarthritis model: Effect of repeated administration, msc inflammatory stimulation, and equine leukocyte antigen (ela) compatibility. Stem Cell Res. Ther. 2020, 11, 52. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Orientation | Sequence | Amplicon Size (bp) | Mis-Matches |
|---|---|---|---|---|
| DRA | For | CAGCTGTCCTGATGAGCTTT | 360 | |
| Rev | AGCCACGTGACATCGATCAC | |||
| DRB | For | GAGGCTCCTGGATGGCAGCT | 341 | A not G@6 for Eqca-DRB2*006:01 |
| Rev | GTCTTTGCAGGATACACAGT | |||
| DQA | For | GATCCTAAACAGAGCTCTGA | 370 | |
| Rev | AAGACAGATGAGGGTGTTGG | |||
| DQB | For | GGCCTTTGGACAKYAGCT | 351 | |
| Rev | RGATGGGGAGAYGGTCAC | |||
| MHCI | For1 | GTYGGCTAYGTGGACGAC | 378 | T not A@11 and C not T@12 for Eqca-1*005:02, 1*008:01, 1*009:01, 18*002:01 and 18*003:01 |
| Rev2.2 | SCCMTCYAGGTAGKYCCT | A not T@8 for Eqca-4*001:01 T not C@13 For Eqca-18*003:01 | ||
| MHCI | For3.2 | GGGCCGSARTATTGGGA | 318 | A not G@12 for Eqca-17*002:02 |
| Rev1 | CTCCAGGTRTCTGMGGAGC | A not C@5 for Eqca-6*001:01 C not T@11 for Eqca-4*001:01 A not T@12 for Eqca-N*001:01 |
| Haplotype. | Number of Occurrences in Cohort | Total Number of Expressed Genes | Standard | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of ‘Standard Genes’ Expressed | Allele | Allele | Allele | Allele | Allele | Allele | Allele | Allele | Allele | Allele | Allele | |||
| HP1.1a | 30 | 11 | 5 | 4*001:AA | N*001:01 | MHCI*gb8:02 | 16*001:01 | MHCI*gb6:03 | ||||||
| HP1.1b | 7 | 10 | 5 | MHCI*gb38:01 | N*001:01 | MHCI*gb8:02 | 16*001:AA | MHCI*gb6:04 | ||||||
| HP1.2 | 42 | 12 | 7 | MHCI*gb12:01 | MHCI*gb50:01 | MHCI*gb2:01 | MHCI*gb4:01 | MHCI*gb27:01 | 6*001:01 | MHCI*gb25:01 | ||||
| HP1.3 | 27 | 13 | 8 | MHCI*gb38:01 | MHCI*gb40:01 | 1*002:02 | 16*003:01 | MHCI*gb16:01 | MHCI*gb1:02 | MHCI*gb4:02 | MHCI*gb27:01 | |||
| HP1.4 | 16 | 16 | 10 | MHCI*gb5:01 | N*005:AA | 1*003:AA | MHCI*gb19:01 | MHCI*gb6:01 | MHCI*gb15:01 | MHCI*gb12:02 | MHCI*gb4:02 | 6*001:01 | MHCI*gb25:01 | |
| HP1.5 | 13 | 12 | 7 | MHCI*gb38:01 | MHCI*gb21:01 | MHCI*gb22:01 | 1*005:02 | MHCI*gb23:01 | MHCI*gb15:01 | MHCI*gb1:01 | ||||
| HP1.6a | 34 | 15 | 8 | MHCI*gb31:01 | 4*001:01 | 1*001:01 | 3*001:01 | MHCI*gb6:02 | MHCI*gb18:01 | 6*001:01 | MHCI*gb25:01 | |||
| HP1.6b | 11 | 11 | 5 | 4*001:01 | 1*001:01 | 3*001:01 | MHCI*gb6:02 | MHCI*gb18:01 | ||||||
| HP1.7a | 2 | 12 | 7 | MHCI*gb37:01 | MHCI*gb36:01 | MHCI*gb8:01 | MHCI*gb30:01 | MHCI*gb1:02 | MHCI*gb4:02 | 6*001:01 | ||||
| HP1.8 | 3 | 16 | 10 | MHCI*gb7:03 | 4*001:AA:01 | MHCI*gb29:01 | MHCI*gb3:01 | MHCI*gb5:02 | MHCI*gb1:02 | MHCI*gb4:02 | MHCI*gb27:01 | 6*001:01 | MHCI*gb25:01 | |
| HP1.9 | 2 | 8 | 4 | MHCI*gb38:01 | MHCI*gb7:02 | MHCI*gb34:01 | MHCI*gb35:01 | |||||||
| HP1.10 | 2 | 6 | 2 | MHCI*gb3:02 | MHCI*gb49:01 | |||||||||
| unHP1.11 | 1 | 13 | 7 | MHCI*gb43:01 | MHCI*gb47:01 | MHCI*gb10:01 | MHCI*gb48:01 | MHCI*gb30:01 | MHCI*gb4:02 | MHCI*gb27:01 | ||||
| Haplotype. | Number of Occurrences in Cohort | Total Number of Expressed Genes | Universal | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of ‘Universal Alleles’ Expressed | Allele | Allele | Allele | Allele | Allele | Allele | Allele | |||
| HP1.1a | 30 | 11 | 6 | MHCI*gb11:01:02 | 2*001:AB | MHCI*gb13:01 | 7*001:01 | MHCI*gb9:01 | MHCI*gb26:01 | |
| HP1.1b | 7 | 10 | 5 | MHCI*gb11:02 | 2*001:AB | MHCI*gb13:02 | 7*002:01:AA | MHCI*gb26:01 | ||
| HP1.2 | 42 | 12 | 5 | 2*001:AE | MHCI*gb13:01 | 7*001:01 | MHCI*gb9:01 | MHCI*gb26:01 | ||
| HP1.3 | 27 | 13 | 5 | MHCI*gb11:01:02 | 2*001:03 | MHCI*gb13:01 | 7*001:01 | MHCI*gb26:01 | ||
| HP1.4 | 16 | 16 | 6 | MHCI*gb11:01:01 | 2*001:AD | MHCI*gb13:01 | 7*001:01 | MHCI*gb9:01 | MHCI*gb26:01 | |
| HP1.5 | 13 | 12 | 5 | MHCI*gb11:01:02 | 2*001:AC | MHCI*gb13:01 | 7*002:01:AA | MHCI*gb26:01 | ||
| HP1.6a | 34 | 15 | 7 | MHCI*gb11:01:02 | 2*001:01 | MHCI*gb13:03 | MHCI*gb13:02 | 7*002:01:AA | MHCI*gb9:01 | MHCI*gb26:01 |
| HP1.6b | 11 | 11 | 6 | MHCI*gb11:01:02 | 2*001:01 | MHCI*gb13:01 | 7*001:01 | MHCI*gb9:01 | MHCI*gb26:01 | |
| HP1.7a | 2 | 12 | 5 | MHCI*gb11:01:02 | 2*001:AH | 7*001:01 | MHCI*gb9:02 | MHCI*gb26:01 | ||
| HP1.8 | 3 | 16 | 6 | MHCI*gb11:01:02 | 2*003:01 | MHCI*gb13:01 | 7*001:01 | MHCI*gb9:01 | MHCI*gb26:01 | |
| HP1.9 | 2 | 8 | 4 | MHCI*gb11:01:02 | 2*001:AA | 7*002:01:AA | MHCI*gb26:01 | |||
| HP1.10 | 2 | 6 | 4 | MHCI*gb11:01:01 | MHCI*gb13:01 | MHCI*gb9:01 | MHCI*gb26:01 | |||
| unHP1.11 | 1 | 13 | 6 | MHCI*gb11:01:02 | 2*003:01 | MHCI*gb13:01 | 7*001:01 | MHCI*gb9:02 | MHCI*gb26:01 | |
| Allele | Cluster | Inferred Function | Chr. | Genomic Location—Start | Genomic Location—Stop | Orientation | Query Start | Query End | Length | Score | E-Val | % ID | Automated Gnomon Prediction |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MHCI*gb11:01:02 | 3-5 | Expressed non-classical | 20 | 29901699 | 29901880 | F | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | |
| 20 | 29902124 | 29902353 | F | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | ||||
| MHCI*gb18:01 | 20 | 29958884 | 29958973 | F | 1 | 90 | 90 | 178 | 1 × 10−42 | 100 | Yes | ||
| 20 | 29959206 | 29959437 | F | 87 | 318 | 232 | 459 | 5 × 10−127 | 100 | ||||
| MHCI*gb6:02 | 20 | 30001109 | 30001290 | F | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | |||
| 20 | 30001526 | 30001755 | F | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | ||||
| 4*001:01 | 3-4 | Expressed classical | 20 | 30070552 | 30070733 | F | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | |
| 20 | 30070970 | 30071199 | F | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | ||||
| 3*001:01 | 3-3 | Expressed classical | 20 | 30225873 | 30226054 | F | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | |
| 20 | 30226290 | 30226519 | F | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | ||||
| 2*001:01 | 3-2 | Expressed classical | 20 | 30277079 | 30277260 | F | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | |
| 20 | 30277488 | 30277717 | F | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | ||||
| 1*001:01 | 3-1 | Expressed classical | 20 | 30335949 | 30336130 | F | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | |
| 20 | 30336359 | 30336588 | F | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | ||||
| MHCI*gb13:03 | 20 | 31136277 | 31136506 | R | 89 | 318 | 230 | 455 | 7 × 10−126 | 100 | Yes | ||
| 20 | 31143897 | 31143986 | R | 1 | 90 | 90 | 178 | 1 × 10−42 | 100 | ||||
| MHCI*gb13:02 | 20 | 31256964 | 31257193 | R | 89 | 318 | 230 | 455 | 7 × 10−126 | 100 | Yes | ||
| 20 | 31264867 | 31264956 | R | 1 | 90 | 90 | 178 | 1 × 10−42 | 100 | ||||
| 7*002:AA | 3-7 | Expressed non-classical | 20 | 31524136 | 31524365 | R | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | |
| 20 | 31524606 | 31524787 | R | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | ||||
| MHCI*gb9:01 | 20 | 31884461 | 31884642 | F | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | |||
| 20 | 31884876 | 31885105 | F | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | ||||
| MHCI*gb26:01 | 3-11 | Pseudogene | 20 | 31981174 | 31981403 | R | 89 | 318 | 230 | 455 | 7 × 10−126 | 100 | |
| 20 | 31981615 | 31981704 | R | 1 | 90 | 90 | 178 | 1 × 10−42 | 100 | ||||
| 6*001:01 | 3-6 | Expressed non-classical | 20 | 32015995 | 32016224 | R | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | |
| 20 | 32016463 | 32016644 | R | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | ||||
| MHCI*gb25:01 | 3-10 | Pseudogene | 20 | 32060222 | 32060451 | R | 181 | 410 | 230 | 455 | 1 × 10−125 | 100 | |
| 20 | 32060689 | 32060870 | R | 1 | 182 | 182 | 360 | 4 × 10−97 | 100 | ||||
| MHCI*gb31:01 | 20 | 30336359 | 30336550 | F | 181 | 372 | 192 | 316 | 4 × 10−84 | 95.83 | |||
| 20 | 31823220 | 31823401 | F | 1 | 182 | 182 | 344 | 2 × 10−92 | 98.90 |
| Haplotype | Number of Occurences in Cohort | Number of Expressed Genes | Previousy Defined | DQA1 | DQA2 | DQB1 | DQB2 | DRA | DRB1 | DRB2 | DRB3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HP2.1 | 40 | 8 | A3 | DQA1*001:01 | DQA2*001:01 | DQB*gb2:01 | DQB*gb3:01 | DRA*001:01/ DRA*001:04 | DRB1*001:01 | DRB2*002:01 | DRB3*001:01:02 /DRB3*001:01:01 |
| HP2.2a | 42 | 8 | DQA1*002:03 | DQA*gb1:01 | DQB1*006:01 | DQB2*003:01 | DRA*001:02 | DRB1*003:01 | DRB2*001:01/ DRB2*001:02 | DRB3*001:01:02/ DRB3*001:01:01 | |
| HP2.2b | 2 | 8 | DQA1*002:03 | DQA*gb1:01 | DQB1*006:01 | DQB2*003:01 | DRA*001:02 | DRB1*003:01 | DRB2*002:01 | DRB3*001:01:02/ DRB3*001:01:01 | |
| HP2.2c | 5 | 8 | DQA1*002:03 | DQA*gb1:01 | DQB1*006:01 | DQB2*003:01 | DRA*001:02 | DRB1*003:01 | DRB2*005:01 | DRB3*002:01/ DRB3*002:02 | |
| HP2.3 | 38 | 8 | A2 | DQA1*002:01 | DQA*gb4:01 | DQB1*002:01 | DQB2*002:01 | DRA*001:01/ DRA*001:04 | DRB1*002:01 | DRB2*003:01 | DRB3*001:01:02/ DRB3*001:01:01 |
| HP2.4a | 28 | 7 | DQA1*003:01 | DQA2*002:01 | DQB1*004:01 | DQB2*004:01 | DRA*001:03 | DRB1*004:01 | DRB2*004:01 | ||
| unHP2.4b | 1 | 7 | A9 | DQA1*003:01 | DQA2*002:01 | DQB1*004:01 | DQB2*004:01 | DRA*001:01 /DRA*001:04 | DRB1*004:01 | DRB2*004:01 | |
| HP2.4c | 3 | 7 | DQA1*003:01 | DQA2*002:01 | DQB1*004:01 | DQB2*004:01 | DRA*001:02 | DRB1*003:01 | DRB2*004:01 | ||
| HP2.5 | 21 | 7 | A10 | DQA1*004:01 | DQB1*005:01 | DQB2*005:01 | DRA*001:01/ DRA*001:04 | DRB1*005:01 | DRB2*001:01/ DRB2*001:02 | DRB3*002:01 /DRB3*002:02 | |
| HP2.6a | 3 | 7 | DQA1*002:02 | DQA2*002:01 | DQB1*007:01 | DQB2*004:01 | DRA*001:01/ DRA*001:04 | DRB*gb1:01 | DRB*gb2:01 | DRB3*001:01:02/ DRB3*001:01:01* | |
| HP2.6b | 2 | 8 | DQA1*002:02 | DQA2*002:01 | DQB1*007:01 | DQB2*004:01 | DRA*001:02 | DRB1*003:01 | DRB*gb2:01 | DRB3*001:01:02/ DRB3*001:01:01 | |
| HP2.7 | 3 | 8 | DQA1*002:05 | DQA*gb3:01 | DQB1*003:AA | DQB2*003:01 | DRA*001:01/ DRA*001:04 | DRB1*007:01/ DRB1*007:02 | DRB2*002:01 | DRB3*001:01:02/ DRB3*001:01:01 | |
| HP2.8 | 2 | 7 | DQA*gb2:01 | DQA2*002:AA | DQB*gb1:01 | DQB*gb4:01 | DRA*001:03 | DRB*gb2:01 | DRB3*002:01 /DRB3*002:02 | ||
| unHP2.9a | 1 | 7 | DQA1*005:01 | DQA*gb1:04 | DQB*gb5:01 | DQB2*003:01 | DRA*001:03 | DRB1*008:01 | DRB2*006:01 | DRB3*001:01:02/ DRB3*001:01:01* | |
| unHP2.10 | 1 | 7 | DQA1*002:02 | DQA*gb1:01 | DQB1*003:01 | DQB2*003:01 | DRA*001:03 | DRB*gb3:01 | DRB2*001:01/ DRB2*001:02 | DRB3*001:01:02/ DRB3*001:01:01* |
| Haplotype | Number of Occurences in Icelandic Ponies | Number of Occurences in Norwegian Fjord Horses | Standard | Universal | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of ‘Standard Alleles’ Expressed | Allele | Allele | Allele | Allele | Allele | Allele | Allele | Allele | Allele | |||
| HP1.10 | 6 | 4 | 2 | MHCI*gb3:02 | MHCI*gb49:01 | |||||||
| HP1.12 | 2 | 1 | 7 | 1*001:AA | MHCI*no34:01 | MHCI*gb1:02 | MHCI*no6:01 | MHCI*gb6:05 | 4*001:AB | 3*001:01 | 2*001:01 | |
| HP1.13 | 3 | 0 | 6 | N*006:01 | MHCI*no5:01 | MHCI*no3:02 | MHCI*gb8:03 | MHCI*gb6:05 | 6*001:01 | 2*001:AC | ||
| HP1.14 | 2 | 0 | 7 | MHCI*no25:01 | MHCI*gb34:01 | MHCI*no3:02 | MHCI*gb40:01 | MHCI*gb6:05 | 1*002:AA | MHCI*gb1:02 | 2*001:AA | |
| HP1.15 | 3 | 0 | 5 | MHCI*no17:01 | MHCI*no1:02 | MHCI*gb1:02:02 | MHCI*gb49:02 | 3*001:AA | ||||
| HP1.16 | 8 | 0 | 6 | MHCI*no16:01 | MHCI*no28:01 | MHCI*gb15:01 | MHCI*gb34:02 | MHCI*gb6:03 | MHCI*gb49:04 | |||
| HP1.17 | 0 | 2 | 5 | MHCI*no29:01 | MHCI*no1:02 | MHCI*no3:01 | MHCI*gb8:05 | MHCI*gb6:05 | ||||
| unHP1.18a | 1 | 0 | 4 | MHCI*gb1:01 | MHCI*no2:02 | MHCI*gb34:01 | MHCI*no32:01 | 2*001:AI | ||||
| unHP1.18b | 1 | 0 | 4 | MHCI*gb1:01 | MHCI*no2:01 | MHCI*gb34:01 | MHCI*no32:01 | 2*001:AI | ||||
| HP1.19a | 0 | 16 | 6 | MHCI*no24:01 | MHCI*no9:01 | MHCI*gb2:01 | MHCI*gb4:03 | MHCI*gb15:03 | MHCI*gb10:02 | 2*001:AJ | ||
| HP1.19b | 1 | 19 | 7 | MHCI*no24:01 | MHCI*no9:01 | MHCI*no12:01 | MHCI*gb4:03 | MHCI*gb6:03 | MHCI*gb15:03 | 3*001:AA | 2*001:AJ | |
| HP1.20 | 3 | 3 | 5 | MHCI*gb41:01 | MHCI*no31:01 | MHCI*gb4:02 | MHCI*gb1:01 | MHCI*gb12:03 | 2*003:01 | |||
| HP1.21 | 0 | 9 | 4 | MHCI*no35:01 | MHCI*no19:01 | MHCI*gb1:01 | MHCI*gb10:01 | 2*001:AM | ||||
| HP1.22 | 0 | 5 | 4 | MHCI*gb43:01:01 | MHCI*gb36:02 | MHCI*gb3:01 | MHCI*gb4:02 | 2*003:01 | ||||
| HP1.23 | 3 | 0 | 4 | MHCI*no31:01 | MHCI*no7:01 | MHCI*gb4:02 | MHCI*gb12:03 | 2*003:01 | ||||
| HP1.24 | 19 | 0 | 7 | MHCI*no23:01 | MHCI*is1:01:01 | MHCI*gb4:02 | MHCI*gb15:01 | MHCI*no8:01 | MHCI*gb1:02:02 | 3*001:AC | 2*001:03 | |
| HP1.25 | 3 | 0 | 6 | MHCI*no13:01 | MHCI*no4:01 | MHCI*no22:01 | MHCI*gb7:06 | MHCI*gb1:05 | MHCI*gb1:03 | 2*001:03 | ||
| HP1.26 | 5 | 10 | 7 | MHCI*no1:01 | MHCI*no18:01 | MHCI*gb8:02 | MHCI*gb15:02 | MHCI*gb49:02 | 3*001:AA | MHCI*gb7:04 | 2*001:AB | |
| HP1.27 | 1 | 2 | 5 | N*001:01 | MHCI*gb8:02 | 16*001:AA | 2*001:AB | MHCI*gb27:01 | ||||
| HP1.28 | 5 | 0 | 8 | MHCI*no21:01 | MHCI*no15:01 | MHCI*gb6:01 | MHCI*gb19:01 | MHCI*gb15:02 | 3*001:AD | MHCI*gb1:02 | 6*001:01 | 2*001:03 |
| unHP1.30 | 1 | 0 | 7 | 16*003:01 | 1*002:02 | 3*001:AA:01 | MHCI*gb15:03 | MHCI*gb40:01 | MHCI*gb12:03 | MHCI*gb1:01:01 | 2*001:03 | |
| unHP1.31 | 1 | 0 | 8 | MHCI*gb5:02 | MHCI*gb29:01 | MHCI*gb1:02 | MHCI*gb15:01 | MHCI*gb4:02 | MHCI*gb7:05 | MHCI*gb1:05 | 3*001:AB | |
| unHP1.32 | 0 | 1 | 6 | MHCI*no4:02 | MHCI*no20:01 | MHCI*gb41:02 | MHCI*gb21:01 | MHCI*gb1:01 | 6*001:01 | 2*001:AC | ||
| unHP1.33 | 1 | 0 | 7 | MHCI*no30:01 | MHCI*no33:01 | MHCI*gb15:01 | MHCI*no36:01 | MHCI*gb49:03 | MHCI*gb7:06 | MHCI*no37:01 | 2*001:AL | |
| unHP1.7b | 1 | 0 | 7 | MHCI*gb37:01 | MHCI*gb36:02 | MHCI*gb8:01 | MHCI*gb30:01 | MHCI*gb1:02 | MHCI*gb4:02 | MHCI*gb10:01 | 2*001:AH | |
| Haplotype | Number of Occurences Combined in Icelandic Horses | Number of Occurences in Norwegian Fjord Horses | Number of Expressed Genes | DQA1 | DQA2 | DQA3 | DQB1 | DQB2 | DQB3 | DRA | DRB1 | DRB2 | DRB3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HP2.5 | 0 | 2 | 7 | DQA1*004:01 | DQB1*005:01 | DQB2*005:01 | DRA*001:01/ DRA*001:04 | DRB1*005:01 | DRB2*001:01/ DRB2*001:02 | DRB3*002:01/ DRB3*002:02 | |||
| HP2.9b | 3 | 0 | 8 | DQA1*005:01 | DQA*gb1:04 | DQB*gb5:01 | DQB2*003:01 | DRA*001:01/ DRA*001:04 | DRB1*007:01/ DRB1*007:02 | DRB2*006:01 | DRB3*002:01/ DRB3*002:02 | ||
| HP2.11 | 9 | 2 | 6 | DQA*no1:01 | DQA1*002:01:AA | DQB1*007:AA | DQB*no1:01 | DRA*001:01/ DRA*001:04 | DRB*is10:01 | ||||
| HP2.12 | 3 | 0 | 8 | DQA1*002:03 | DQA*gb1:01 | DQB*gb1:03 | DQB2*003:AA | DRA*001:01/ DRA*001:04 | DRB*is6:01 | DRB2*002:01:AA | DRB3*001:01:02/ DRB3*001:01:01 | ||
| HP2.13 | 2 | 1 | 7 | DQA*gb1:06 | DQB*is3:01 | DQB2*002:AA | DRA*001:03 | DRB*gb3:01 | DRB*gb2:01 | DRB3*002:01/ DRB3*002:02 | |||
| HP2.14 | 4 | 17 | 6 | DQA1*002:02 | DQA*gb1:02 | DQB*no9:01 | DQB2*003:01 | DRA*001:01/ DRA*001:04 | DRB*gb1:01 | ||||
| HP2.15 | 6 | 0 | 7 | DQA*gb2:01 | DQA2*002:AA | DQB*gb1:01 | DQB*gb4:01 | DRA*001:01/ DRA*001:04 | DRB*is6:01 | DRB3*001:01:02/ DRB3*001:01:01 | |||
| HP2.16a | 8 | 0 | 8 | DQA1*002:AA | DQA*gb1:05:04 | DQB1*006:AA | DQB2*002:AB | DRA*001:01/ DRA*001:04 | DRB*is8:01 | DRB2*002:01:AA | DRB3*002:01/ DRB3*002:02 | ||
| unHP2.16b | 1 | 0 | 7 | DQA1*002:AA | DQA*gb1:05:04 | DQB1*006:AA | DQB2*002:AB | DRA*001:01/ DRA*001:04 | DRB*is8:01 | DRB2*002:AA | |||
| HP2.17 | 3 | 0 | 6 | DQA*is4:01 | DQA*gb3:01 | DQB*is4:01 | DRA*001:01/ DRA*001:04 | DRB*is9:01 | DRB2*004:01 | ||||
| HP2.18 | 0 | 4 | 7 | DQA*no1:01 | DQB*no7:01 | DQB*no4:01 | DRA*001:01/ DRA*001:04 | DRB*no7:01 | DRB2*003:01 | DRB3*002:01/ DRB3*002:02 | |||
| HP2.19a | 4 | 0 | 8 | DQA*is5:01 | DQA*gb1:01 | DQA*is3:01 | DQB*is8:01 | DQB*is2:01 | DRA*001:01/ DRA*001:04 | DRB1*002:01 | DRB2*005:01 | ||
| HP2.19b | 2 | 0 | 8 | DQA*is5:01 | DQA*gb1:01 | DQA*is3:01 | DQB*is8:01 | DQB*is2:01 | DRA*001:01/ DRA*001:04 | DRB1*001:01 | DRB2*002:01 | ||
| HP2.20 | 4 | 0 | 8 | DQA1*002:01 | DQA*gb1:02 | DQB1*007:AB | DQB2*003:01 | DRA*001:01/ DRA*001:04 | DRB*no7:01 | DRB2*001:01/ DRB2*001:02 | DRB3*002:01/ DRB3*002:02 | ||
| HP2.21 | 1 | 3 | 7 | DQA*no3:01 | DQA2*002:01 | DQB*no5:01 | DQB2*004:01 | DRA*001:01/ DRA*001:04 | DRB*no8:01 | DRB2*002:01 | |||
| HP2.22a | 0 | 2 | 9 | DQA*no2:01 | DQA*gb3:01 | DQA*no5:01 | DQB*no8:01 | DQB*no10:01 | DRA*001:01/ DRA*001:04 | DRB*no1:01:01 | DRB*no6:01 | DRB3*002:01/ DRB3*002:02 | |
| unHP2.22b | 0 | 1 | 9 | DQA*no2:01 | DQA*gb3:01 | DQA*no5:01 | DQB*no8:01 | DQB*no10:01 | DRA*001:01/ DRA*001:04 | DRB*no1:03 | DRB*no6:01 | DRB3*002:01/ DRB3*002:02 | |
| unHP2.22c | 0 | 1 | 9 | DQA*no2:01 | DQA*gb3:01 | DQA*no5:01 | DQB*no8:01 | DQB*no10:01 | DRA*001:01/ DRA*001:04 | DRB*no1:02 | DRB*no6:01 | DRB3*002:01/ DRB3*002:02 | |
| HP2.23a | 0 | 17 | 6 | DQA*no1:01 | DQB*no1:01 | DQB*no6:01 | DRA*001:01/ DRA*001:04 | DRB1*002:01 | DRB3*002:01/ DRB3*002:02 | ||||
| unHP2.23b | 0 | 1 | 7 | DQA*no1:01 | DQB*no1:01 | DQB*no6:01 | DRA*001:01/ DRA*001:04 | DRB1*002:01 | DRB2*002:01 | DRB3*001:01:02/ DRB3*001:01:01 | |||
| HP2.23c | 0 | 2 | 7 | DQA*no1:01 | DQB*no1:01 | DQB*no6:01 | DRA*001:01/ DRA*001:04 | DRB1*002:01 | DRB2*002:01 | DRB3*002:AA | |||
| unHP2.23d | 1 | 0 | 7 | DQA*no1:01 | DQB*no1:03 | DQB*no6:01 | DRA*001:01/ DRA*001:04 | DRB1*002:01 | DRB2*002:01 | DRB3*001:01:02/ DRB3*001:01:01 | |||
| HP2.24 | 0 | 5 | 8 | DQA1*002:05 | DQA2*002:AA | DQB1*003:01 | DQB*no1:02 | DRA*001:03 | DRB1*008:01 | DRB2*002:01 | DRB3*002:01/ DRB3*002:02 | ||
| HP2.25a | 0 | 11 | 7 | DQA*gb1:05:03 | DQA*no4:01 | DQB*no11:01 | DQB*is7:01 | DQB*is1:03 | DRA*001:AA | DRB2*004:01 | |||
| HP2.25b | 4 | 0 | 7 | DQA*gb1:05:03 | DQA*no4:01 | DQB*no11:01 | DQB*is7:01 | DQB*is1:02 | DRA*001:AA | DRB2*005:01 | |||
| unHP2.25c | 1 | 0 | 6 | DQA*gb1:05:03 | DQA*no4:01 | DQB*no11:01 | DQB*is8:01 | DRA*001:AA | DRB2*005:01 | ||||
| pHP2.26 | 0 | 2 | 3 | DQB*no7:01 | DQB*no4:01 | DRA*001:01/ DRA*001:04 | DRB*no7:01 | ||||||
| unHP2.27 | 1 | 0 | 10 | DQA*is5:01 | DQA*gb1:01 | DQA*is3:01 | DQB*no11:01 | DQB*is7:01 | DQB*is1:02 | DRA*001:01/ DRA*001:04 | DRB1*001:01 | DRB2*002:01 | DRB3*002:01/ DRB3*002:02 |
| unHP2.28a | 1 | 0 | 5 | DQA*gb2:02 | DQB*gb1:02 | DRA*001:AA | DRB*is10:01 | DRB2*002:AA | |||||
| unHP2.28b | 1 | 0 | 4 | DQA*gb2:02 | DQB*gb1:02 | DRA*001:01/ DRA*001:04 | DRB*is12:01 | DRB2*002:AA | |||||
| unHP2.29 | 1 | 0 | 8 | DQA*no1:01 | DQA1*002:01:AA | DQB*no7:01 | DQB*no4:01 | DRA*001:01/ DRA*001:04 | DRB1*001:01 | DRB2*002:AA | DRB3*002:01/ DRB3*002:02 | ||
| unHP2.30 | 1 | 0 | 7 | DQA*no2:01 | DQA*gb1:01 | DQB*is6:01 | DQB*is7:01 | DQB*is1:01 | DRA*001:AA | DRB2*002:AA | |||
| unHP2.31 | 1 | 0 | 7 | DQA*gb1:05:01 | DQB*no11:01 | DQB*is7:01 | DRB2*002:01 | DRA*001:AA | DRB*no2:01:02 | DRB3*002:01/ DRB3*002:02 | |||
| unHP2.32 | 1 | 0 | 7 | DQA*gb1:05:02 | DQA*is2:01 | DQB*is5:01 | DQB2*002:AA | DRA*001:AA | DRB1*001:01 | DRB*gb2:02 | |||
| unHP2.33a | 1 | 0 | 8 | DQA*gb2:02 | DQA1*002:01:AA | DQA*gb1:03 | DQB*gb1:02 | DQB2*003:01 | DRA*001:01/ DRA*001:04 | DRB*is12:01 | DRB2*002:AA | ||
| unHP2.33b | 1 | 0 | 8 | DQA*gb2:02 | DQA*gb2:01 | DQA*gb1:02 | DQB*gb1:02 | DQB2*003:01 | DRA*001:01/ DRA*001:04 | DRB*is12:01 | DRB2*002:AA | ||
| unHP2.34 | 1 | 0 | 7 | DQA*gb2:03 | DQA2*002:AA | DQB1*003:AA | DQB*no1:02 | DRA*001:01/ DRA*001:04 | DRB1*002:01 | DRB2*002:01 | |||
| unHP2.35 | 1 | 0 | 6 | DQA*gb3:01 | DQB1*003:AA | DRA*001:03 | DRB1*008:01 | DRB2*002:01 | DRB3*001:01:02/ DRB3*001:01:01 | ||||
| unHP2.36 | 1 | 0 | 6 | DQA*is4:01 | DQB*is4:01 | DQB*is9:01 | DRA*001:01/ DRA*001:04 | DRB*is9:01 | DRB3*002:AA | ||||
| unHP2.37 | 1 | 0 | 7 | DQA1*002:01 | DQA*gb1:02 | DQB*no9:01 | DQB2*003:01 | DRA*001:01/ DRA*001:04 | DRB*no7:01 | DRB2*005:01 | |||
| unHP2.38 | 0 | 1 | 7 | DQA1*002:02 | DQA2*002:01 | DQB2*004:01:AA | DQB1*004:AA | DRA*001:01/ DRA*001:04 | DRB*is8:01 | DRB2*002:01 | DRB3*002:01/ DRB3*002:02 | ||
| unHP2.39 | 0 | 1 | 8 | DQA*gb1:05:03 | DQA*no4:01 | DQB*no7:01 | DQB*no4:01 | DRA*001:01/ DRA*001:04 | DRB*no7:01 | DRB2*003:01 | DRB3*002:01/ DRB3*002:02 | ||
| unpHP2.40 | 1 | 0 | 2 | DRA*001:03 | DRB*is11:01 | ||||||||
| unpHP2.41 | 0 | 1 | 4 | DQB*no7:01 | DRA*001:01/ DRA*001:04 | DRB*no7:01 | DRB2*003:01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasoya, D.; Tzelos, T.; Benedictus, L.; Karagianni, A.E.; Pirie, S.; Marr, C.; Oddsdóttir, C.; Fintl, C.; Connelley, T. High-Resolution Genotyping of Expressed Equine MHC Reveals a Highly Complex MHC Structure. Genes 2023, 14, 1422. https://doi.org/10.3390/genes14071422
Vasoya D, Tzelos T, Benedictus L, Karagianni AE, Pirie S, Marr C, Oddsdóttir C, Fintl C, Connelley T. High-Resolution Genotyping of Expressed Equine MHC Reveals a Highly Complex MHC Structure. Genes. 2023; 14(7):1422. https://doi.org/10.3390/genes14071422
Chicago/Turabian StyleVasoya, Deepali, Thomas Tzelos, Lindert Benedictus, Anna Eleonora Karagianni, Scott Pirie, Celia Marr, Charlotta Oddsdóttir, Constanze Fintl, and Timothy Connelley. 2023. "High-Resolution Genotyping of Expressed Equine MHC Reveals a Highly Complex MHC Structure" Genes 14, no. 7: 1422. https://doi.org/10.3390/genes14071422