
The Reversion of the Epigenetic Signature of Coronary Heart Disease in Response to Smoking Cessation
by
 ,
,
Robert Philibert
1,2,3,* ,
,
Joanna Moody
1,
Willem Philibert
1,
Meeshanthini V. Dogan
2,3 and
Eric A. Hoffman
3,4 1
Department of Psychiatry, University of Iowa, Iowa City, IA 52242, USA
2
Cardio Diagnostics Inc., Chicago, IL 60642, USA
3
Department of Biomedical Engineering, University of Iowa, Iowa City, IA 52242, USA
4
Department of Radiology, University of Iowa, Iowa City, IA 52242, USA
*
Author to whom correspondence should be addressed.
Genes 2023, 14(6), 1233; https://doi.org/10.3390/genes14061233
Submission received: 15 May 2023
/
Revised: 31 May 2023
/
Accepted: 5 June 2023
/
Published: 8 June 2023
(This article belongs to the Section Molecular Genetics and Genomics)
Abstract
:Coronary heart disease (CHD) is the leading cause of death worldwide. However, current diagnostic tools for CHD, such as coronary computed tomography angiography (CCTA), are poorly suited for monitoring treatment response. Recently, we have introduced an artificial-intelligence-guided integrated genetic–epigenetic test for CHD whose core consists of six assays that determine methylation in pathways known to moderate the pathogenesis of CHD. However, whether methylation at these six loci is sufficiently dynamic to guide CHD treatment response is unknown. To test that hypothesis, we examined the relationship of changes in these six loci to changes in cg05575921, a generally accepted marker of smoking intensity, using DNA from a cohort of 39 subjects undergoing a 90-day smoking cessation intervention and methylation-sensitive digital PCR (MSdPCR). We found that changes in epigenetic smoking intensity were significantly associated with reversion of the CHD-associated methylation signature at five of the six MSdPCR predictor sites: cg03725309, cg12586707, cg04988978, cg17901584, and cg21161138. We conclude that methylation-based approaches could be a scalable method for assessing the clinical effectiveness of CHD interventions, and that further studies to understand the responsiveness of these epigenetic measures to other forms of CHD treatment are in order.
1. Introduction
Coronary heart disease (CHD) is the leading cause of death in the world, with over 9 million individuals dying annually from myocardial infarctions [1]. Up to 90% of these deaths may be preventable [2]. However, to achieve this goal, more scalable approaches for both recognizing and monitoring those at risk of CHD for initial preventive intervention and those with current CHD for more intensive therapy are necessary.
Unfortunately, with respect to the latter, there are significant limitations in utilizing contemporary CHD diagnostic approaches for monitoring the effectiveness of interventions, both lifestyle and therapeutic. The currently accepted methods for diagnosing stable CHD from the American Heart Association/American College of Cardiology include exercise stress tests with electrocardiograms (ECG), exercise or pharmacological stress tests with echocardiography, myocardial perfusion imaging, coronary computed tomographic angiography (CCTA), and coronary angiography [3]. The choice of which test is employed is dependent on a number of factors, including the degree of clinical suspicion for CHD, the ability to exercise, the likelihood of experiencing high-risk events, and the availability of testing resources [4]. Unfortunately, each of those modalities has significant limitations that affect their clinical implementation. For example, the exercise ECG is the most commonly used and least invasive method, but has only 58% sensitivity and 62% specificity [3]. Perfusion imaging has better sensitivity (~87%) and specificity (70%), but requires expensive infrastructure, specialized interpretation, and requires considerable radiation exposure (15–35 millisieverts) [5,6]. The final method is coronary angiography, which is considered by many to be the “gold standard” for the assessment of CHD. However, it is invasive, entails considerable exposure to radiation and iodinated contrast dyes, and is insensitive to non-obstructive forms of CHD. More importantly, these diagnostic tools fall short in providing actionable information to personalize patient care and lack the ability to measure the effectiveness of interventions. Hence, there is considerable need for less costly and invasive methods for assessing CHD status that also provide actionable information that can be leveraged by healthcare providers to tailor, refine, and enhance treatment plans for each patient more effectively.
Recently, Cardio Diagnostics has introduced an artificial intelligence-guided genetic–epigenetic test (PrecisionCHD™) for assessing current CHD [7]. The test uses a machine learning model to interpret the genetically contextual signal of six methylation-sensitive digital PCR (MSdPCR) assays to determine CHD status, with overall area under the curve, sensitivity, and specificity values of 82%, 79% and 76%, respectively. The test uses DNA from whole blood and a relatively inexpensive PCR-based laboratory method; therefore, unlike many of the current diagnostic tests, this new CHD testing approach is inherently more scalable and compatible with the operations of most clinical laboratories. In addition, it presents two distinct potential advantages as compared with current methods for assessing CHD. First, because the MSdPCR assays each measure DNA methylation in pathways involved in the pathogenesis of CHD, it allows clinicians to gather patient specific insight into potentially targetable lifestyle or physiological factors. Second, because DNA methylation is dynamic, it may also be possible to use DNA methylation to serve as a proxy for the success of CHD therapy.
Devising more scalable methods for assessing the success of CHD therapy could have a high clinical impact. The introduction of the hemoglobin A1c (HbA1c) metric for both assessing disease status and monitoring the effects of diabetic therapy revolutionized the treatment of diabetes [8]. However, unlike diabetes, there is not a single pathway whose status can be measured to determine the success of therapy for CHD. As a result, clinicians are relegated to aggregating separate measurements of individual risk factors, such as high blood pressure, elevated cholesterol, or HbA1c status itself, to determine the effectiveness of therapy for CHD. However, many of those measures, such as blood pressure and cholesterol levels, are relatively imprecise, can vary considerably from day to day, and are not sufficient to fully address CHD [9]. Therefore, a more robust test, such as the HbA1c assay, which be used to directly and more precisely assess CHD status and the effectiveness of treatment, would be a significant advancement.
Previously, we have shown that one of the three MSdPCR measures contained in an integrated genetic–epigenetic test for assessing the 3-year risk for CHD significantly changed as a function of treatment for smoking [10]. Since smoking is a key driver of CHD [11,12] and smoking cessation therapy can markedly improve survival in smokers with CHD, this suggests that methylation at one of more of the six loci assessed in the PrecisionCHD test may also respond to smoking therapy. Therefore, in this communication, we assess DNA methylation at each of the six loci in the new CHD test in DNA samples from 39 research subjects collected before and after undergoing 90 days of smoking cessation therapy [13].
2. Materials and Methods
The methods and procedures used in this smoking cessation study have been described previously. All methods were approved by the University of Iowa Institutional Review Board (IRB#201706713).
The 39 subjects who participated in this study were part of a 90-day incentive-based smoking cessation program (National Clinical Trials NCT02682147) whose overarching purpose was to understand the relationship between smoking cessation and pulmonary inflammation [14]. Each potential participant, who was alerted to the study by a series of advertisements to patients and staff of the University of Iowa, had to complete a brief online survey in which they reported smoking at least 10 cigarettes per day with a total of five or more pack-years of lifetime consumption in order to qualify for the study. After providing written informed consent, each participant was enrolled in a protocol that included an intake visit and three subsequent monthly follow-up visits at 30-, 60-, and 90 days post intake. At each of these visits, each of the subjects were asked about their current levels of smoking and were phlebotomized to provide DNA for the epigenetic analyses. Due to concerns that medications for smoking cessation may interfere with the pulmonary imaging studies, subjects were instructed not to use medications to quit smoking. However, they were offered USD 400 for having biochemically verified smoking cessation at each of the three follow-up visits. Smoking cessation can take a variable course and self-reports of smoking can be unreliable [15,16]; therefore, smoking intensity at each visit was assessed using digital measures of cg05575921, a generally accepted measure of smoking intensity and smoking cessation [17,18]. At the time of the preparation of the sample, a total of 75 subjects had enrolled in the study, with 39 successfully attending all four clinic visits.
DNA for the epigenetic studies was prepared from whole blood using the method developed by Lahiri and Nurnberger [19]. The MSdPCR assays and bisulfite-sensitive methylation standards used in this study were provided by Cardio Diagnostics Inc. (Chicago, IL, USA) or Behavioral Diagnostics LLC (cg05575921), then used according to the protocols described elsewhere [7,20,21]. In brief, determination of DNA methylation at cg05575921 and the six loci in the PrecisionCHD test (cg03725309, cg12586707, cg04988978, cg17901584, cg21161138, and cg12655112) was conducted using universal droplet digital PCR reagents and equipment obtained from Bio-Rad (Hercules, CA, USA). In brief, 1 µg of DNA was bisulfite-converted using a Qiagen EpiTect Bisulfite kit (Hilden, Germany), according to manufacturer’s directions, with the modified DNA then being eluted in 70 µL of tris buffer. Fourteen cycles of high-stringency PCR amplification of the target region were then performed on a 3 µL aliquot of each bisulfite-converted DNA sample using a set of amplicon-specific proprietary primers. Then, an aliquot of the enriched amplicon target solution was diluted 1:1500, mixed together with primer and probes specific for the target loci and droplet digital PCR reagents, partitioned into droplets with a Bio-Rad droplet generator, and then PCR amplified. Fractional methylation (methylated CpG/(methylated + unmethylation CpG)) of each sample was then determined using a Bio-Rad QX-200 reader and its accompanying Bio-Rad QuantaSoft™ software. The results from all bisulfite methylation testing standards were within their calibrated ranges.
The data were analyzed using the suite of general linear model analytic algorithms embedded in JMP Version 17 (SAS Institute, Cary, SC, USA). Linear regression was used to assess the relationship between each of the six DNA methylation markers in the PrecisionCHD test and the cg05575921 DNA methylation marker for smoking [22]. Comparisons between groups were conducted using Student’s t-tests [22]. Bonferroni correction was used to adjust for multiple comparisons [23].
3. Results
The clinical and demographic characteristics of the 39 subjects who successfully completed the protocol are given in Table 1. In general, the subjects tended to be White, in their early 40s, and male (22 of 39, or 56%). The subjects tended to be heavy smokers, with the average subject smoking 17 cigarettes per day over the month prior to study intake and having previously had 28 pack-years of total cigarette consumption. The average serum cotinine at intake was 278 ng/mL, with an average Fagerstrom Test for Nicotine Dependence score of 3.7 ± 9.7. Notably, the average cg05575921 methylation value, a generally accepted measure of smoking intensity, was 52.3% at intake.
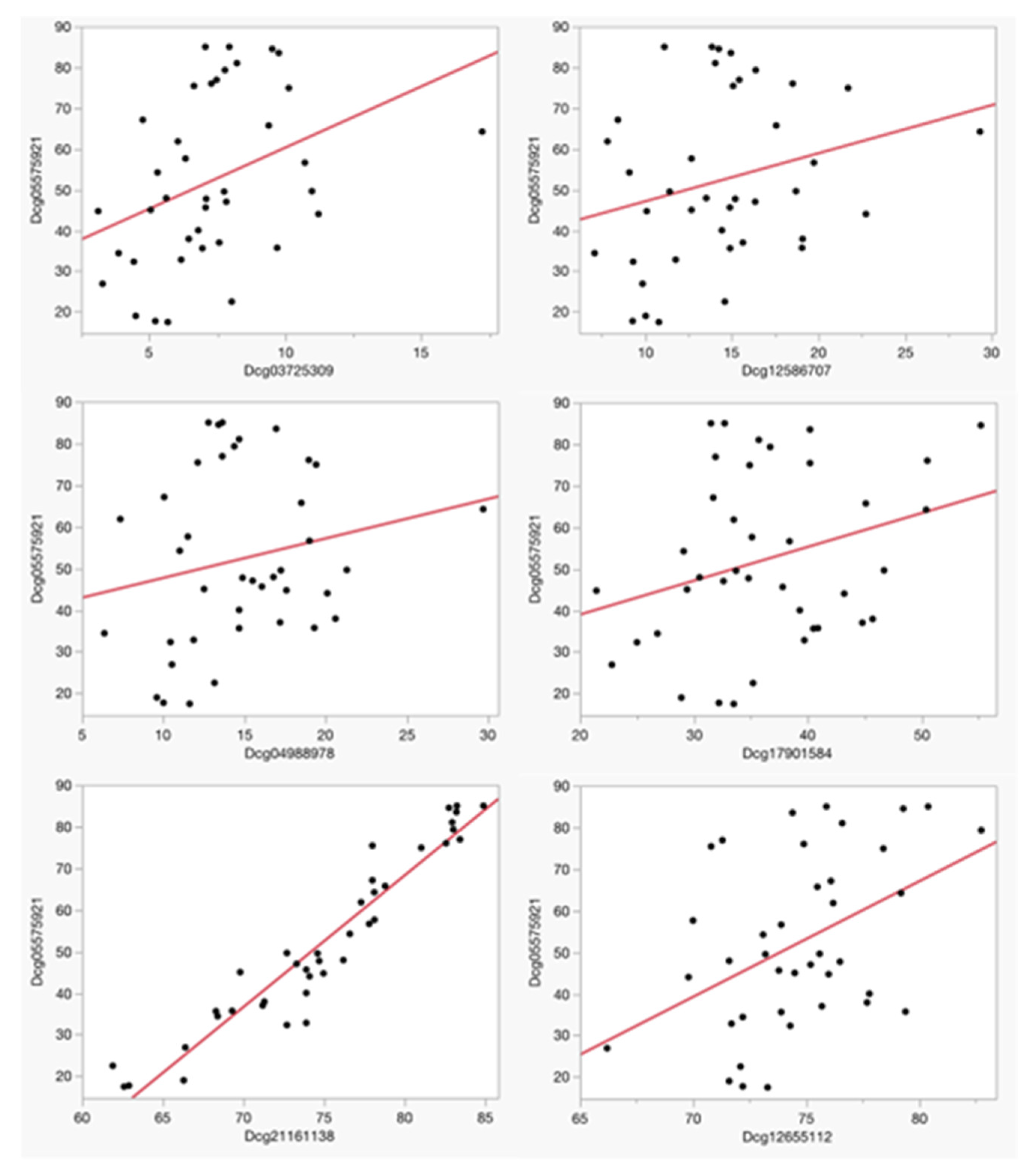
Table 2 details the identity and chromosomal location of the six CpG sites assessed in the PrecisionCHD test for current CHD assessment, while both Table 2 and Figure 1 delineate the strengths of the linear relationships between methylation at cg05575921 and methylation values at the each of the six CpG loci used in PrecisionCHD at study intake. After correction for multiple comparisons, cg05575921 values were significantly associated with both cg21161138 (Adj R2 = 0.91, p < 0.0001) and cg12655112 (Adj R2 = 0.20, p < 0.0048), with a nominal association being further noted with cg03725309 (Adj R2 = 0.15, p < 0.0162). There was no significant relationship between methylation at cg05575921 and methylation at cg04988978 (Adj R2 = 0.04, p < 0.21), cg17901584 (Adj R2 = 0.09, p < 0.07) or cg12586707 (Adj R2 = 0.07, p < 0.11).
Over the course of the 90-day treatment paradigm, 22 subjects exhibited undetectable levels of cotinine at all three follow-up visits, while the remaining 17 subjects exhibited a positive cotinine value at one or more follow-up visits. Overall, those with negative cotinine levels at all three visits reduced smoking intensity more than those with one or more positive cotinine tests (∆cg05575921; −7.1% ± 6.2 vs. −2.5% ± 5.4; p < 0.02), but both groups had decreases in self-reported intake and increases in overall cg05575921 methylation.
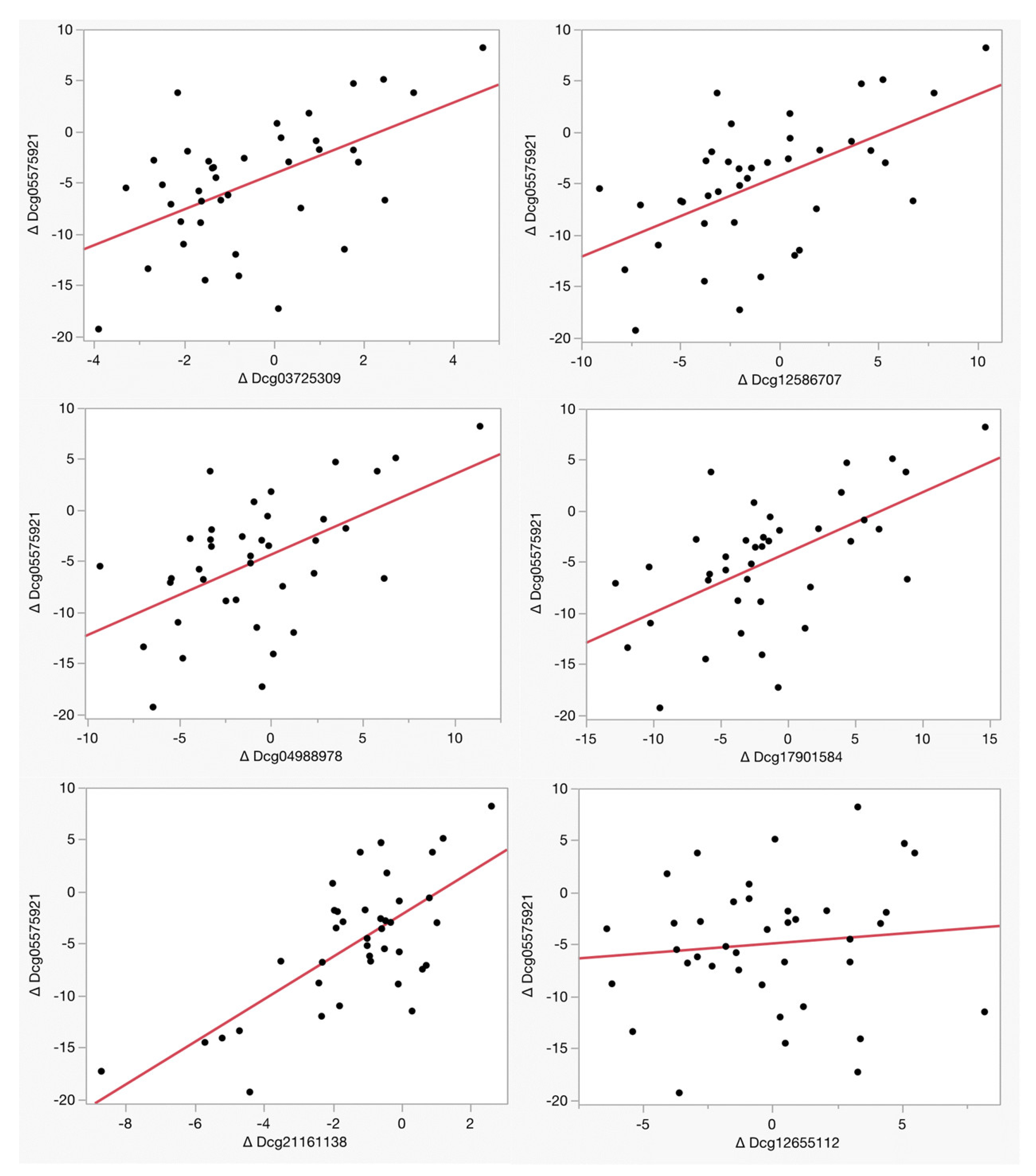
Table 3 and Figure 2 illustrate the relationship between the changes in methylation at each of the six loci used in the PrecisionCHD test as a function of cg05575921 methylation. Remarkably, five of the six loci, cg03725309, cg12586707, cg04988978, cg17901584, and cg21161138, exhibited a significant reversion of CHD-associated methylation as a function of cg05575921 which indicated a smoking intensity reduction over the 90-day treatment period. In contrast, there was no significant reversion at cg12655112 (Adj R2 = 0.01, p < 0.53) at the EH Domain Containing 4 (EHD4) gene CpG site, whose baseline status is associated with smoking intensity.
4. Discussion
Creating better, more scalable methods to assess the effectiveness of CHD interventions may be an important step to increasing the effectiveness of CHD treatment. In that regard, our demonstration that five of the six loci of the PrecisionCHD test showing significant changes in response to a 90-day course of smoking cessation is a promising first step. One of the limitations of this study is the relatively small sample size and the cohort being almost exclusively White. Furthermore, we only analyzed one CHD-related intervention: smoking cessation.
The complex dynamics of the methylomic response to successful CHD therapy detailed in this manuscript reflect the complexity of the pathways being interrogated by the test [7]. The six CpG loci targeted by the PrecisionCHD test assess six different molecular pathways moderating vulnerability to CHD. The CpG site in the first pathway, which is assessed by the cg03725309 MSdPCR assay, maps to a candidate cis regulatory element in intron 1 of the seryl-tRNA synthetase 1 gene (SARS1). Demethylation at this locus is associated with obesity, coronary artery calcification, and cardiometabolic syndrome [24,25,26]. The CpG site in the second pathway, measured by the cg12586707 MSdPCR assay, is found in a candidate cis regulatory element approximately 1.5 kb downstream of the 5′ UTR of the CXCL1 gene [27]. CXCL1 is a key member of a group of chemotactic messengers involved in the pathogenesis of a number of inflammatory disorders, such as CHD, and has an important role in the regulation of angiogenesis and cardiac remodeling [27,28]. The CpG site in the third pathway, which is assessed by the cg04988978 MSdPCR assay, maps to the upstream promoter area of myeloperoxidase (MPO), which has been shown to contribute to atherosclerosis by the oxidation of LDL [29]. The CpG site in the fourth pathway, which is assessed by the cg17901584 MSdPCR assay, is found in an intron of 24-Dehydrocholesterol Reductase Divergent Transcript (DHCR24-DT), a long non-coding RNA (lncRNA) gene that is in a divergent (head-to-head) configuration with DHCR24, a key gene in cholesterol biosynthesis [30]. The CpG targeted by the cg21161138 MSdPCR assay maps to the aryl hydrocarbon receptor repressor (AHRR) 267 kb distal to cg05575921 [31]. Similarly to cg05575921, this marker assesses activity in the xenobiotic pathway that is essential for detoxifying the polyaromatic hydrocarbons found in smoke [32]. Finally, the sixth assay, cg12655112, measures methylation at a regulatory region of the first intron of EH Domain Containing 4 (EHD4) gene. EHD4 methylation at this site has been negatively associated with serum glucose levels, while EHD4 expression predicts the success of pancreatic islet transplants [33,34,35].
Both cg05575921 and cg21161138 map to the AHRR gene and both demethylate in response to smoking; therefore, the strong correlation observed between cg05575921 with this new predictor for CHD is to be expected [31]. However, it is interesting to note that the other outright (cg12655112) and nominal (cg03725309) associations with smoking intensity at study baseline were with the two CpG sites whose methylation alterations were previously associated with diabetes [33,36,37]. Smoking is associated with increased risk for diabetes; as such, this makes intuitive sense [6,38]. At the same time, smoking has also been repeatedly implicated as a major cause of inflammation [39]. Thus, the absence of a robust association of smoking intensity with DNA methylation in the inflammatory pathway interrogated by cg12586707 (CXCL1) is somewhat surprising. Then again, it is important to note that the use of the term “inflammation” is often broad, and not all inflammatory processes may be affected by smoking.
To clinicians familiar with the psychosocial processes involved in smoking cessation, the finding that methylation at five of the six pathways interrogated by the MSdPCR assays begin to revert to baseline after only 90 days of therapy is not surprising. In order to cut down or quit smoking, patients often make radical changes to their lifestyle in order to avoid stimuli, such as bars or friends who smoke, that can trigger the urge to smoke. Often, these changes are accompanied with improvements in diet and exercise patterns and decreases in alcohol consumption [40]. Therefore, it should not be surprising that methylation at most, but not all, of the CpG loci targeted in this new diagnostic test were reverted as a function of the reversion of cg05575921 methylation (i.e., successful smoking cessation).
Interestingly, the one locus that did not manifest significant reversion in response to smoking cessation was the EDH4 locus, whose methylation status was associated with baseline cg05575921 methylation. This lack of a significant finding may be secondary to the short time scale of this study. There have been several epigenome-wide studies of the time dependency of methylation reversion in those undergoing smoking cessation [18,41,42]. These studies have demonstrated that the reversion of smoking-associated methylation changes in the thousands of CpG sites affected can be divided into three categories. The first is those whose sites whose methylation status changes quickly, such as cg05575921. The second class is those which revert more slowly, while the third class is those sites which revert very slowly, if at all. Therefore, it should not be surprising to find variation in the time scale of responsiveness at these loci which predict CHD status as well. Given the relationship of cg12655112 (EHD4) methylation with diabetes, it would be interesting to understand if there are any changes in the hemoglobin A1c values of the subjects as a function of smoking cessation in longer-term studies.
Although they are remarkably significant from a statistical point of view, these findings should only be regarded as an initial foray into the epigenetics of effective CHD therapy. Effective treatment of CHD requires targeting the unique lifestyle or physiological factors driving the pathology. For some patients, this means targeting weight; for others, diabetes; for others still, cholesterol levels. However, since successfully addressing these causative factors also often entails broad changes in other clinical drivers of CHD, investigators should be encouraged to use broad measures to fully capture the impact of changes that occur in patients’ lives as a function of successful CHD therapy. Crucially, this was only a 90-day study. Since patients often relapse into unhealthy habits, it is essential to gather longer-term information so that clinicians and public health policy experts can balance the cost of interventions with their long-term impact on CHD outcomes.
Critical to any success to the extension of these findings will be the ability to accurately quantitate changes in the CHD factor being studied. To a large extent, one reason for the success of this study is secondary to the availability of the cg05575921 MSdPCR assay that can precisely measure changes in smoking intensity. Therefore, given the ease of availability of hemoglobin A1c, a precise marker of diabetes status, it would be relatively easy to design a study to test for the reversion of CHD-related signals as a function of changes in hemoglobin A1c values. However, for studies of clinical factors for which less precise steady-state measures are available, such as serum lipid levels, clinicians may need to use innovative strategies or large sample sizes to detect significant changes in methylation. In contrast, the use of self-report variables, such as history of smoking or FTND, which is based solely on self-report and requires a mild degree of introspection, should be used only as necessary because they can be much less reliable.
Avenues through which these findings can be improved upon include the obvious extensions, such as enlarging the sample size and diversity, and more nuanced extensions, such as understanding the role in co-morbid clinical and genetic factors, in moderating reversion. Increases in the size and diversity of the sample are absolutely necessary if this approach is to find clinical application. It is essential to understand how variables such as age, sex, and ethnicity affect reversion before using this to guide clinical care. Furthermore, the subjects in this study were relatively healthy. Understanding the effect of co-morbid illnesses such as obesity or diabetes on clinical response is similarly essential. These examinations should be performed in conjunction with genetic analyses, such as those afforded by polygenic risk scores, which can help parse the acquired effects of any co-morbidity with the inherited genetic factors that contribute to those illnesses.
It may well be that quantifying and understanding these potentially confounding effects is difficult. In this regard, this may be an excellent opportunity for artificial intelligence (AI) to further its role in the derivation and implementation of this testing technology. AI technologies for aggregating and analyzing methylation and genetic data are increasingly commonplace [43,44]. These tools will need to be integrated with tools for parsing clinical laboratory or text data from the electronic medical records in order to create a more complete understanding of the performance and implications of these technologies in the real world [45].
If these trials are adequately powered and treatment compliance is carefully monitored, we believe that additional studies of the methylomics for other types of CHD therapy have a good chance of succeeding. As evidence of that, we note that in a naturalistic longitudinal study of statin therapy, Qin et al. used a mixed linear effects modeling in a sample of 535 Danish subjects to show changes in DNA methylation at three previously identified CpG sites (cg10177197, cg17901584, and cg27243685) as a function of treatment with a variety of statins. Furthermore, using a cross-sectional epigenome-wide approach and data from two large cohorts of subjects, Schrader et al. showed that statin therapy was associated with DNA methylation at one of the sites interrogated by the PrecisionCHD test (cg17901584), as well as two other novel sites (cg27243685 and cg05119988) [46]. Unfortunately, in each of these studies, treatment compliance was not assessed, and both the time and type of statin used varied. However, as evidence of proof of principle, these studies more than demonstrate the potential for precision epigenetic approaches for guiding the treatment of CHD.
In summary, we show significant changes in DNA methylation at five of the six loci measured in the recently described PrecisionCHD test for the assessment of current CHD. We suggest that further studies to understand the methylomic response to CHD treatment may lead to improved methods for guiding the treatment of CHD.
Author Contributions
W.P., R.P. and J.M. performed the methylation assessments and conducted the initial methylation determination analyses. M.V.D. helped write the first draft and contributed to the editing and interpretation of data. E.A.H. and R.P. obtained the funding for the project. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by 5R01HL130883 and S10OD026960 (Eric Hoffman, PI) and by R44CA213507 (Robert Philibert, PI). Additional support for reagents and publications costs was provided by Cardio Diagnostics Inc.
Institutional Review Board Statement
All protocols and procedures used in this study were conducted according to the guidelines of the Declaration of Helsinki and approved by the University of Iowa Institutional Review Board (IRB201706713). Written informed consent was obtained from all subjects.
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The datasets used during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
Robert Philibert and Meeshanthini Dogan are officers, directors, and stockholders of Cardio Diagnostics Inc. (www.cardiodiagnosticsinc.com accessed on 1 of May 2023). The use of DNA methylation and gene–methylation interaction effects for the assessment, diagnosis, and monitoring of cardiovascular disease is covered by United States Patent 11,414,704, European Patent EP3472344B1, and other both granted and pending intellectual property claims elsewhere. The University of Iowa Research Foundation may be entitled to royalties from the use of this technology. Philibert is also the Chief Executive Officer of Behavioral Diagnostics and inventor on a number of granted and pending patent applications with respect to tobacco consumption related to the material discussed herein. The use of cg05575921 status to determine smoking status is protected by US Patents 8,637,652 and 9,273,358. The other authors have no conflicts to disclose.
References
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the global burden of disease study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruff, C.T.; Braunwald, E. The evolving epidemiology of acute coronary syndromes. Nat. Rev. Cardiol. 2011, 8, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.; De Lemos, J. Stable ischemic heart disease. In Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 12th ed.; Elsevier: Philadelphia, PA, USA, 2022. [Google Scholar]
- Fihn, S.D.; Gardin, J.M.; Abrams, J.; Berra, K.; Blankenship, J.C.; Dallas, A.P.; Douglas, P.S.; Foody, J.M.; Gerber, T.C.; Hinderliter, A.L. 2012 accf/aha/acp/aats/pcna/scai/sts guideline for the diagnosis and management of patients with stable ischemic heart disease: A report of the american college of cardiology foundation/american heart association task force on practice guidelines, and the american college of physicians, american association for thoracic surgery, preventive cardiovascular nurses association, society for cardiovascular angiography and interventions, and society of thoracic surgeons. J. Am. Coll. Cardiol. 2012, 60, e44–e164. [Google Scholar] [PubMed] [Green Version]
- De Gonzalez, A.B.; Kim, K.-P.; Smith-Bindman, R.; McAreavey, D. Myocardial Perfusion Scans. Circulation 2010, 122, 2403–2410. [Google Scholar] [CrossRef]
- Willi, C.; Bodenmann, P.; Ghali, W.A.; Faris, P.D.; Cornuz, J. Active smoking and the risk of type 2 diabetes: A systematic review and meta-analysis. JAMA 2007, 298, 2654–2664. [Google Scholar] [CrossRef]
- Philibert, R.; Dogan, T.; Knight, S.; Ahmad, F.; Lau, S.; Miles, G.; Knowlton, K.; Dogan, M.V. The validation of an integrated genetic-epigenetic test for the assessment of coronary heart disease. J. Am. Heart Assoc. 2023. [Google Scholar]
- Ang, S.H.; Thevarajah, M.; Alias, Y.; Khor, S.M. Current aspects in hemoglobin A1c detection: A review. Clin. Chim. Acta 2015, 439, 202–211. [Google Scholar] [CrossRef]
- Bookstein, L.; Gidding, S.S.; Donovan, M.; Smith, F.A. Day-to-day variability of serum cholesterol, triglyceride, and high-density lipoprotein cholesterol levels. Arch. Intern. Med. 1990, 150, 1653–1657. [Google Scholar] [CrossRef]
- Philibert, W.; Andersen, A.M.; Hoffman, E.A.; Philibert, R.; Dogan, M. The Reversion of DNA Methylation at Coronary Heart Disease Risk Loci in Response to Prevention Therapy. Processes 2021, 9, 699. [Google Scholar] [CrossRef]
- Jacobs, D.R., Jr.; Adachi, H.; Mulder, I.; Kromhout, D.; Menotti, A.; Nissinen, A.; Blackburn, H. Cigarette Smoking and Mortality Risk: Twenty-five–Year Follow-up of the Seven Countries Study. Arch. Intern. Med. 1999, 159, 733–740. [Google Scholar] [CrossRef]
- Breitling, L.P.; Salzmann, K.; Rothenbacher, D.; Burwinkel, B.; Brenner, H. Smoking, F2RL3 methylation, and prognosis in stable coronary heart disease. Eur. Heart J. 2012, 33, 2841–2848. [Google Scholar] [CrossRef] [Green Version]
- Critchley, J.A.; Capewell, S. Smoking cessation for the secondary prevention of coronary heart disease. Cochrane Database Syst. Rev. 2003, CD003041. [Google Scholar] [CrossRef]
- Liu, Y.; Comellas, A.; Kaczka, D.; Motahari, A.; Lee, C.; Gerke, A.; Wilson, J.; Salisbury, S.; O’Connel-Moore, D.; Philibert, R. An incentive-based program coupled with sildenafil provides enhanced success of smoking cessation associated with an accelerated loss of ct assessed smoking-associated lung density (inflammation) and improved dlco. In D76. Copd: Clinical Studies; American Thoracic Society: New York, NY, USA, 2020; p. A7556. [Google Scholar]
- Caraballo, R.S.; Asman, K. Epidemiology of menthol cigarette use in the United States. Tob. Induc. Dis. 2011, 9, S1. [Google Scholar] [CrossRef] [Green Version]
- Caraballo, R.S.; Giovino, G.A.; Pechacek, T.F.; Mowery, P.D. Factors Associated with Discrepancies between Self-Reports on Cigarette Smoking and Measured Serum Cotinine Levels among Persons Aged 17 Years or Older: Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol. 2001, 153, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Gelernter, J.; Polimanti, R. Genetics of substance use disorders in the era of big data. Nat. Rev. Genet. 2021, 22, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Andersen, A.M.; Philibert, R.; Hancock, D.B. Epigenetic biomarkers for smoking cessation. Addict. Neurosci. 2023, 6, 100079. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Nurnberger, J.I., Jr. A rapid non-enzymatic method for the preparation of hmw DNA from blood for rflp studies. Nucleic Acids Res. 1991, 19, 5444. [Google Scholar] [CrossRef]
- Philibert, R.; Dogan, M.; Beach, S.R.H.; Mills, J.A.; Long, J.D. AHRR methylation predicts smoking status and smoking intensity in both saliva and blood DNA. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2020, 183, 51–60 . [Google Scholar] [CrossRef]
- Dawes, K.; Andersen, A.; Reimer, R.; Mills, J.A.; Hoffman, E.; Long, J.D.; Miller, S.; Philibert, R. The relationship of smoking to cg05575921 methylation in blood and saliva DNA samples from several studies. Sci. Rep. 2021, 11, 21627. [Google Scholar] [CrossRef]
- Fleiss, J.L. Statistical Methods for Rates and Proportions, 2nd ed.; John Wiley & Sons Inc.: New York, NY, USA, 1981. [Google Scholar]
- Dunn, O.J. Multiple comparisons among means. J. Am. Stat. Assoc. 1961, 56, 52–64. [Google Scholar] [CrossRef]
- Zheng, Y.; Joyce, B.T.; Hwang, S.-J.; Ma, J.; Liu, L.; Allen, N.B.; Krefman, A.E.; Wang, J.; Gao, T.; Nannini, D.R.; et al. Association of Cardiovascular Health Through Young Adulthood with Genome-Wide DNA Methylation Patterns in Midlife: The CARDIA Study. Circulation 2022, 146, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, M.M.; Marioni, R.E.; Joehanes, R.; Liu, C.; Hedman, K.; Aslibekyan, S.; Demerath, E.W.; Guan, W.; Zhi, D.; Yao, C.; et al. Association of Body Mass Index with DNA Methylation and Gene Expression in Blood Cells and Relations to Cardiometabolic Disease: A Mendelian Randomization Approach. PLoS Med. 2017, 14, e1002215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.-C.; Ried, J.S.; Zhang, W.; Yang, Y. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Barczak, K.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Cxcl1: Gene, promoter, regulation of expression, mrna stability, regulation of activity in the intercellular space. Int. J. Mol. Sci. 2022, 23, 792. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.-L.; Lin, Q.-Y.; Liu, Y.; Guan, X.-M.; Ma, X.-L.; Cao, H.-J.; Liu, Y.; Bai, J.; Xia, Y.-L.; et al. CXCL1–CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 2018, 39, 1818–1831. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Muret, K.; Désert, C.; Lagoutte, L.; Boutin, M.; Gondret, F.; Zerjal, T.; Lagarrigue, S. Long noncoding RNAs in lipid metabolism: Literature review and conservation analysis across species. BMC Genom. 2019, 20, 882. [Google Scholar] [CrossRef] [Green Version]
- Philibert, R.A.; Beach, S.R.H.; Lei, M.K.; Brody, G.H. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin. Epigenet. 2013, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Esser, C. Biology and function of the aryl hydrocarbon receptor: Report of an international and interdisciplinary conference. Arch. Toxicol. 2012, 86, 1323–1329. [Google Scholar] [CrossRef]
- Meeks, K.A.C.; Henneman, P.; Venema, A.; Addo, J.; Bahendeka, S.; Burr, T.; Danquah, I.; Galbete, C.; Mannens, M.M.A.M.; Mockenhaupt, F.P.; et al. Epigenome-wide association study in whole blood on type 2 diabetes among sub-Saharan African individuals: Findings from the RODAM study. Leuk. Res. 2018, 48, 58–70. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, W.; Wang, B.; Cao, W.; Lv, J.; Yu, C.; Pang, Z.; Cong, L.; Wang, H.; Wu, X. Correlation between fasting plasma glucose, hba1c and DNA methylation in adult twins. Beijing Da Xue Xue Bao. Yi Xue Ban = J. Peking Univ. Health Sci. 2020, 52, 425–431. [Google Scholar]
- Kurian, S.M.; Ferreri, K.; Wang, C.-H.; Todorov, I.; Al-Abdullah, I.H.; Rawson, J.; Mullen, Y.; Salomon, D.R.; Kandeel, F. Gene expression signature predicts human islet integrity and transplant functionality in diabetic mice. PLoS ONE 2017, 12, e0185331. [Google Scholar] [CrossRef] [Green Version]
- Walaszczyk, E. DNA Methylation in Type 2 Diabetes and Metabolic Health. Ph.D. Thesis, University of Groningen, Groningen, The Netherlands, 2021. [Google Scholar]
- Hong, X.; Wu, Z.; Cao, W.; Lv, J.; Yu, C.; Huang, T.; Sun, D.; Liao, C.; Pang, Y.; Pang, Z.; et al. Longitudinal Association of DNA Methylation with Type 2 Diabetes and Glycemic Traits: A 5-Year Cross-Lagged Twin Study. Diabetes 2022, 71, 2804–2817. [Google Scholar] [CrossRef] [PubMed]
- Rimm, E.B.; Manson, J.E.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C.; Rosner, B.; Hennekens, C.H.; Speizer, F.E. Cigarette smoking and the risk of diabetes in women. Am. J. Public Health 1993, 83, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibuakuu, M.; Kamimura, D.; Kianoush, S.; DeFilippis, A.P.; Al Rifai, M.; Reynolds, L.M.; White, W.B.; Butler, K.R.; Mosley, T.H.; Turner, S.T.; et al. The association between cigarette smoking and inflammation: The Genetic Epidemiology Network of Arteriopathy (GENOA) study. PLoS ONE 2017, 12, e0184914. [Google Scholar] [CrossRef] [PubMed]
- Vogel, E.A.; Ramo, D.E. Smoking cessation, metabolic risk behaviors, and stress management over time in a sample of young adult smokers. Transl. Behav. Med. 2019, 11, 189–197. [Google Scholar] [CrossRef] [PubMed]
- McCartney, D.L.; Stevenson, A.J.; Hillary, R.F.; Walker, R.M.; Bermingham, M.L.; Morris, S.W.; Clarke, T.-K.; Campbell, A.; Murray, A.D.; Whalley, H.C.; et al. Epigenetic signatures of starting and stopping smoking. Ebiomedicine 2018, 37, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, F.; Sandanger, T.M.; Castagne, R.; Campanella, G.; Polidoro, S.; Palli, D.; Krogh, V.; Tumino, R.; Sacerdote, C.; Panico, S.; et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet. 2015, 24, 2349–2359. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; He, D.; Cheng, Y.; Yu, L.; Qi, D.; Li, B.; Zheng, F. Identification of DNA methylation-regulated genes as potential biomarkers for coronary heart disease via machine learning in the Framingham Heart Study. Clin. Epigenet. 2022, 14, 122. [Google Scholar] [CrossRef]
- Dogan, M.V.; Grumbach, I.M.; Michaelson, J.J.; Philibert, R.A. Integrated genetic and epigenetic prediction of coronary heart disease in the Framingham Heart Study. PLoS ONE 2018, 13, e0190549. [Google Scholar] [CrossRef] [Green Version]
- Lauritsen, S.M.; Kristensen, M.; Olsen, M.V.; Larsen, M.S.; Lauritsen, K.M.; Jørgensen, M.J.; Lange, J.; Thiesson, B. Explainable artificial intelligence model to predict acute critical illness from electronic health records. Nat. Commun. 2020, 11, 3852. [Google Scholar] [CrossRef] [PubMed]
- Schrader, S.; Perfilyev, A.; Martinell, M.; García-Calzón, S.; Ling, C. Statin therapy is associated with epigenetic modifications in individuals with Type 2 diabetes. Epigenomics 2021, 13, 919–925. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The relationship between baseline Dcg05575921 and the six loci used to assess CHD status. Methylation values are expressed as a percentage. The adjusted R2 and p-values of the linear associations of each of these comparisons are listed in Table 2.
Figure 1.
The relationship between baseline Dcg05575921 and the six loci used to assess CHD status. Methylation values are expressed as a percentage. The adjusted R2 and p-values of the linear associations of each of these comparisons are listed in Table 2.

Figure 2.
The relationship of the change in smoking intensity with the change in the measure of CHD status. Methylation values for each assay are expressed as percentages. The adjusted R2 and p-values of the linear associations of each of these comparisons are given in Table 3.
Figure 2.
The relationship of the change in smoking intensity with the change in the measure of CHD status. Methylation values for each assay are expressed as percentages. The adjusted R2 and p-values of the linear associations of each of these comparisons are given in Table 3.


{kind=link}
{kind=link}
Table 1.
Demographic and clinical characteristics of the subjects.
| N | 39 |
| Age | 42.7 ± 10.4 years |
| Gender | |
| Male | 22 |
| Female | 17 |
| Ethnicity | |
| White | 37 |
| African American | 1 |
| Other | 1 |
| Smoking Variables | |
| Pack-Year Consumption | 28 ± 20 |
| Cigarettes per day | 17 ± 9.7 |
| FTND | 3.7 ± 9.7 |
| Cotinine (ng/mL) | 278 ± 135 |
FTND, Fagerstrom Test for Nicotine Dependence score.
Table 2.
Methylation values and association with smoking intensity at intake.
| Smoking Intensity | ||||
|---|---|---|---|---|
| Assay | Gene Localization | Average Value (%) | ||
| Dcg05575921 | AHRR | 52.3 ± 20.6 | ||
| Coronary Heart Disease (PrecisionCHD) | Association with cg05575921 | |||
| Adj R2 | Nominal p-value | |||
| Dcg03725309 | SARS1 | 7.3 ± 2.6 | 0.12 | p < 0.02 |
| Dcg12586707 | CXCL1 | 14.4 ± 4.6 | 0.04 | p < 0.11 |
| Dcg04988978 | MPO | 14.8 ± 4.4 | 0.04 | p < 0.22 |
| Dcg17901584 | DHCR24-DT | 36.4 ± 7.6 | 0.09 | p < 0.07 |
| Dcg21161138 * | AHRR | 72.0 ± 6.2 | 0.91 | p < 0.0001 |
| Dcg12655112 | EHD4 | 74.7 ± 3.3 | 0.19 | p < 0.005 |
* p-value for significance after multiple corrections is p < 0.0083; Dcg is the MSdPCR assay for its respective cg methylation marker.
Table 3.
Linear associations with 90-day changes in cg05575921 methylation.
| Smoking Intensity | |||
|---|---|---|---|
| Assay | Average Change (%) | ||
| Dcg05575921 | −4.9 ± 6.2 | ||
| Coronary Heart Disease (PrecisionCHD) | Association with ∆cg05575921 | ||
| Adj R2 | Nominal p-value | ||
| Dcg03725309 | −0.5 ± 1.9 | 0.29 | p < 0.0004 |
| Dcg12586707 | −0.9 ± 4.5 | 0.32 | p < 0.0002 |
| Dcg04988978 | −0.7 ± 4.2 | 0.29 | p < 0.0004 |
| Dcg17901584 | −1.4 ± 6.1 | 0.33 | p < 0.0001 |
| Dcg21161138 | −1.3 ± 2.1 | 0.50 | p < 0.0001 |
| Dcg12655112 | −0.1 ± 3.4 | 0.01 | p < 0.53 |
p-value for significance after multiple corrections is p < 0.0083; Dcg is the MSdPCR assay for its respective cg methylation marker.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Philibert, R.; Moody, J.; Philibert, W.; Dogan, M.V.; Hoffman, E.A. The Reversion of the Epigenetic Signature of Coronary Heart Disease in Response to Smoking Cessation. Genes 2023, 14, 1233. https://doi.org/10.3390/genes14061233
AMA Style
Philibert R, Moody J, Philibert W, Dogan MV, Hoffman EA. The Reversion of the Epigenetic Signature of Coronary Heart Disease in Response to Smoking Cessation. Genes. 2023; 14(6):1233. https://doi.org/10.3390/genes14061233
Chicago/Turabian StylePhilibert, Robert, Joanna Moody, Willem Philibert, Meeshanthini V. Dogan, and Eric A. Hoffman. 2023. "The Reversion of the Epigenetic Signature of Coronary Heart Disease in Response to Smoking Cessation" Genes 14, no. 6: 1233. https://doi.org/10.3390/genes14061233
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.