
Histories of Dermatan Sulfate Epimerase and Dermatan 4-O-Sulfotransferase from Discovery of Their Enzymes and Genes to Musculocontractural Ehlers-Danlos Syndrome

Abstract
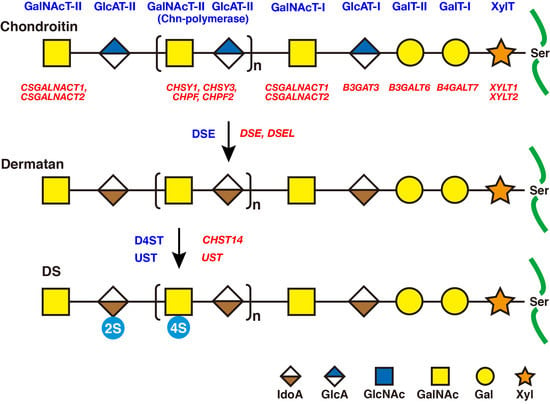
:
1. Introduction
2. Biosynthesis of DS Side Chains on PGs
2.1. GAG-Protein Linker Region
2.2. Repeating Disaccharide Region of DS
3. Roles of DSE and D4ST in Biosynthesis of DS
3.1. DSE
3.2. DSEL/DSE2
3.3. CHST14/D4ST1
3.3.1. Identification of CHST14/D4ST1 Gene
3.3.2. Knockout Mice of Chst14/D4st1
3.3.3. Human Genetic Disorders Caused by Mutations in CHST14/D4ST1
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fransson, L.-A.; Cheng, F.; Yoshida, K.; Heinegård, D.; Malmström, A.; Schmidtchen, A. Patterns of epimerization and sulphation in dermatan sulphate chains. In Dermatan Sulphate Proteoglycans: Chemistry, Biology, Chemical Pathology; Scott, J.E., Ed.; Portland Press: London, UK, 1993; pp. 11–25. [Google Scholar]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117R–125R. [Google Scholar] [CrossRef] [PubMed]
- Thelin, M.A.; Bartolini, B.; Axelsson, J.; Gustafsson, R.; Tykesson, E.; Pera, E.; Oldberg, Å.; Maccarana, M.; Malmström, A. Biological functions of iduronic acid in chondroitin/dermatan sulfate. FEBS J. 2013, 280, 2431–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumoto, S.; Yamada, S. The specific role of dermatan sulfate as an instructive glycosaminoglycan in tissue development. Int. J. Mol. Sci. 2022, 23, 7485. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Kosho, T.; Yamada, S.; Sugahara, K. Pathophysiological significance of dermatan sulfate proteoglycans revealed by human genetic disorders. Pharmaceuticals 2017, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Karl, M.; Chaffee, E. The mucopolysaccharides of skin. J. Biol. Chem. 1941, 138, 491–499. [Google Scholar]
- Anderson, J.C. Isolation of a glycoprotein and proteodermatan sulphate from bovine achilles tendon by affinity chromatography on concanavalin A-Sepharose. Biochim. Biophys. Acta 1975, 379, 444–455. [Google Scholar] [CrossRef]
- Cöster, L.; Fransson, L.A. Isolation and characterization of dermatan sulphate proteoglycans from bovine sclera. Biochem. J. 1981, 193, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Damle, S.P.; Cöster, L.; Gregory, J.D. Proteodermatan sulfate isolated from pig skin. J. Biol. Chem. 1982, 257, 5523–5527. [Google Scholar] [CrossRef]
- Fujii, N.; Nagai, Y. Isolation and characterization of a proteodermatan sulfate from calf skin. J. Biochem. 1981, 90, 1249–1258. [Google Scholar] [CrossRef]
- Pearson, C.H.; Gibson, G.J. Proteoglycans of bovine periodontal ligament and skin. Occurrence of different hybrid-sulphated galactosaminoglycans in distinct proteoglycans. Biochem. J. 1982, 201, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, L.C.; Choi, H.U.; Tang, L.H.; Johnson, T.L.; Pal, S.; Webber, C.; Reiner, A.; Poole, A.R. Isolation of dermatan sulfate proteoglycans from mature bovine articular cartilages. J. Biol. Chem. 1985, 260, 6304–63013. [Google Scholar] [CrossRef]
- Krusius, T.; Ruoslahti, E. Primary structure of an extracellular matrix proteoglycan core protein deduced from cloned cDNA. Proc. Natl. Acad. Sci. USA 1986, 83, 7683–7687. [Google Scholar] [CrossRef] [Green Version]
- Day, A.A.; McQuillan, C.I.; Termine, J.D.; Young, M.R. Molecular cloning and sequence analysis of the cDNA for small proteoglycan II of bovine bone. Biochem. J. 1987, 248, 801–805. [Google Scholar] [CrossRef]
- Li, W.; Vergnes, J.P.; Cornuet, P.K.; Hassell, J.R. cDNA clone to chick corneal chondroitin/dermatan sulfate proteoglycan reveals identity to decorin. Arch. Biochem. Biophys. 1992, 296, 190–197. [Google Scholar] [CrossRef]
- Abramson, S.R.; Woessner, J.F., Jr. cDNA sequence for rat dermatan sulfate proteoglycan-II (decorin). Biochim. Biophys. Acta 1992, 1132, 225–227. [Google Scholar] [CrossRef]
- Fisher, L.W.; Termine, J.D.; Young, M.F. Deduced protein sequence of bone small proteoglycan I (biglycan) shows homology with proteoglycan II (decorin) and several nonconnective tissue proteins in a variety of species. J. Biol. Chem. 1989, 264, 4571–4576. [Google Scholar] [CrossRef]
- Neame, P.J.; Choi, H.U.; Rosenberg, L.C. The primary structure of the core protein of the small, leucine-rich proteoglycan (PG I) from bovine articular cartilage. J. Biol. Chem. 1989, 264, 8653–86561. [Google Scholar] [CrossRef]
- Shinomura, T.; Kimata, K. Proteoglycan-Lb, a small dermatan sulfate proteoglycan expressed in embryonic chick epiphyseal cartilage, is structurally related to osteoinductive factor. J. Biol. Chem. 1992, 267, 1265–1270. [Google Scholar] [CrossRef]
- Lassalle, P.; Molet, S.; Janin, A.; Heyden, J.V.; Tavernier, J.; Fiers, W.; Devos, R.; Tonnel, A.B. ESM-1 is a novel human endothelial cell-specific molecule expressed in lung and regulated by cytokines. J. Biol. Chem. 1996, 271, 20458–20464. [Google Scholar] [CrossRef] [Green Version]
- Béchard, D.; Gentina, T.; Delehedde, M.; Scherpereel, A.; Lyon, M.; Aumercier, M.; Vazeux, R.; Richet, C.; Degand, P.; Jude, B.; et al. Endocan is a novel chondroitin sulfate/dermatan sulfate proteoglycan that promotes hepatocyte growth factor/scatter factor mitogenic activity. J. Biol. Chem. 2001, 276, 48341–48349. [Google Scholar] [CrossRef] [Green Version]
- Bella, A., Jr.; Danishefsky, I. The dermatan sulfate-protein linkage region. J. Biol. Chem. 1968, 243, 2660–2664. [Google Scholar] [CrossRef] [PubMed]
- Fransson, L.A. Structure of dermatan sulfate. IV. Glycopeptides from the carbohydrate-protein linkage region of pig skin dermatan sulfate. Biochim. Biophys. Acta 1968, 156, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.L.; Lindahl, B.; Rodén, L. The linkage of dermatan sulfate to protein. II. Monosaccharide sequence of the linkage region. J. Biol. Chem. 1971, 246, 5707–5715. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Ohkita, Y.; Shibata, Y.; Yoshida, K.; Ikegami, A. Structural studies on the hexasaccharide alditols isolated from the carbohydrate-protein linkage region of dermatan sulfate proteoglycans of bovine aorta: Demonstration of iduronic acid-containing components. J. Biol. Chem. 1995, 270, 7204–7212. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, U.; Rodén, L. The chondroitin 4-sulfate-protein linkage. J. Biol. Chem. 1966, 241, 2113–2119. [Google Scholar] [CrossRef]
- Lindahl, U.; Rodén, L. Carbohydrate-protein linkages in proteoglycans of animal, plant and bacterial origin. In Glycoproteins: Their Composition, Structure and Function; Gottschalk, A., Ed.; Elsevier: Amsterdam, The Netherlands, 1972; pp. 491–517. [Google Scholar]
- Kjellén, L.; Lindahl, U. Proteoglycans: Structures and interactions. Annu. Rev. Biochem. 1991, 60, 443–475. [Google Scholar] [CrossRef]
- Iozzo, R.V. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef] [Green Version]
- Karamanos, N.K.; Piperigkou, Z.; Theocharis, A.D.; Watanabe, H.; Franchi, M.; Baud, S.; Brézillon, S.; Götte, M.; Passi, A.; Vigetti, D.; et al. Proteoglycan chemical diversity drives multifunctional cell regulation and therapeutics. Chem. Rev. 2018, 118, 9152–9232. [Google Scholar] [CrossRef]
- Diehl, V.; Huber, L.S.; Trebicka, J.; Wygrecka, M.; Iozzo, R.V.; Schaefer, L. The role of decorin and biglycan signaling in tumorigenesis. Front. Oncol. 2021, 11, 801801. [Google Scholar] [CrossRef]
- Sugahara, K.; Mikami, T.; Uyama, T.; Mizuguchi, S.; Nomura, K.; Kitagawa, H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 2003, 13, 612–620. [Google Scholar] [CrossRef]
- Sugahara, K.; Mikami, T. Chondroitin/dermatan sulfate in the central nervous system. Curr. Opin. Struct. Biol. 2007, 17, 536–545. [Google Scholar] [CrossRef]
- Mizumoto, S.; Yamada, S.; Sugahara, K. Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 2015, 34, 35–42. [Google Scholar] [CrossRef]
- Hayes, A.J.; Melrose, J. Neural tissue homeostasis and repair is regulated via CS and DS proteoglycan motifs. Front. Cell Dev. Biol. 2021, 9, 696640. [Google Scholar] [CrossRef]
- Grebner, E.E.; Hall, C.W.; Neufeld, E.F. Glycosylation of serine residues by a uridine diphosphate-xylose: Protein xylosyltransferase from mouse mastocytoma. Arch. Biochem. Biophys. 1966, 116, 391–398. [Google Scholar] [CrossRef]
- Robinson, H.C.; Telser, A.; Dorfman, A. Studies on biosynthesis of the linkage region of chondroitin sulfate-protein complex. Proc. Natl. Acad. Sci. USA 1966, 56, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, N.B.; Rodén, L. Biosynthesis of chondroitin sulfate. Purification of UDP-D-xylose:core protein β-D-xylosyltransferase by affinity chromatography. Carbohydr. Res. 1974, 37, 167–180. [Google Scholar] [CrossRef]
- Schwartz, N.B.; Dorfman, A. Purification of rat chondrosarcoma xylosyltransferase. Arch. Biochem. Biophys. 1975, 171, 136–144. [Google Scholar] [CrossRef]
- Kuhn, J.; Götting, C.; Schnölzer, M.; Kempf, T.; Brinkmann, T.; Kleesiek, K. First isolation of human UDP-D-xylose: Proteoglycan core protein β-D-xylosyltransferase secreted from cultured JAR choriocarcinoma cells. J. Biol. Chem. 2001, 276, 4940–4947. [Google Scholar] [CrossRef] [Green Version]
- Götting, C.; Kuhn, J.; Zahn, R.; Brinkmann, T.; Kleesiek, K. Molecular cloning and expression of human UDP-D-Xylose:proteoglycan core protein β-D-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 2000, 304, 517–528. [Google Scholar] [CrossRef]
- Pönighaus, C.; Ambrosius, M.; Casanova, J.C.; Prante, C.; Kuhn, J.; Esko, J.D.; Kleesiek, K.; Götting, C. Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. J. Biol. Chem. 2007, 282, 5201–5206. [Google Scholar] [CrossRef] [Green Version]
- Voglmeir, J.; Voglauer, R.; Wilson, I.B. XT-II, the second isoform of human peptide-O-xylosyltransferase, displays enzymatic activity. J. Biol. Chem. 2007, 282, 5984–5990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schön, S.; Prante, C.; Bahr, C.; Kuhn, J.; Kleesiek, K.; Götting, C. Cloning and recombinant expression of active full-length xylosyltransferase I (XT-I) and characterization of subcellular localization of XT-I and XT-II. J. Biol. Chem. 2006, 281, 14224–14231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helting, T.; Rodén, L. Biosynthesis of chondroitin sulfate. I. Galactosyl transfer in the formation of the carbohydrate-protein linkage region. J. Biol. Chem. 1969, 244, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.B.; Rodén, L. Biosynthesis of chondroitin sulfate. Solubilization of chondroitin sulfate glycosyltransferases and partial purification of uridine diphosphate-D-galactose:D-xylose galactosyltrans. J. Biol. Chem. 1975, 250, 5200–5207. [Google Scholar] [CrossRef] [PubMed]
- Okajima, T.; Yoshida, K.; Kondo, T.; Furukawa, K. Human homolog of Caenorhabditis elegans sqv-3 gene is galactosyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1999, 274, 22915–22918. [Google Scholar] [CrossRef] [Green Version]
- Almeida, R.; Levery, S.B.; Mandel, U.; Kresse, H.; Schwientek, T.; Bennett, E.P.; Clausen, H. Cloning and expression of a proteoglycan UDP-galactose:β-xylose beta1,4-galactosyltransferase I. A seventh member of the human beta4-galactosyltransferase gene family. J. Biol. Chem. 1999, 274, 26165–26171. [Google Scholar] [CrossRef] [Green Version]
- Okajima, T.; Fukumoto, S.; Furukawa, K.; Urano, T. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999, 274, 28841–28844. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, N.B.; Rodén, L.; Dorfman, A. Biosynthesis of chondroitin sulfate: Interaction between xylosyltransferase and galactosyltransferase. Biochem. Biophys. Res. Commun. 1974, 56, 717–724. [Google Scholar] [CrossRef]
- Schwartz, N.B. Biosynthesis of chondroitin sulfate: Immunoprecipitation of interacting xylosyltransferase and galactosyltransferase. FEBS Lett. 1975, 49, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Zhou, D.; Brown, J.R.; Crawford, B.E.; Hennet, T.; Esko, J.D. Biosynthesis of the linkage region of glycosaminoglycans: Cloning and activity of galactosyltransferase II, the sixth member of the β1,3-galactosyltransferase family (beta3GalT6). J. Biol. Chem. 2001, 276, 48189–48195. [Google Scholar] [CrossRef] [Green Version]
- Helting, T.; Rodén, L. Biosynthesis of chondroitin sulfate. II. Glucuronosyl transfer in the formation of the carbohydrate-protein linkage region. J. Biol. Chem. 1969, 244, 2799–2805. [Google Scholar] [CrossRef]
- Brandt, A.E.; Distler, J.; Jourdian, G.W. Biosynthesis of the chondroitin sulfate-protein linkage region: Purification and properties of a glucuronosyltransferase from embryonic chick brain. Proc. Natl. Acad. Sci. USA 1969, 64, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Helting, T. Biosynthesis of heparin. Solubilization and partial purification of uridine diphosphate glucuronic acid: Acceptor glucuronosyltransferase from mouse mastocytoma. J. Biol. Chem. 1972, 247, 4327–4332. [Google Scholar] [CrossRef]
- Kitagawa, H.; Tone, Y.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Oka, S.; Kawasaki, T.; Sugahara, K. Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1998, 273, 6615–6618. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Bai, X.; Sarkar, A.K.; Esko, J.D. Formation of HNK-1 determinants and the glycosaminoglycan tetrasaccharide linkage region by UDP-GlcUA:Galactose beta1, 3-glucuronosyltransferases. J. Biol. Chem. 1999, 274, 7857–7864. [Google Scholar] [CrossRef] [Green Version]
- Rohrmann, K.; Niemann, R.; Buddecke, E. Two N-acetylgalactosaminyltransferase are involved in the biosynthesis of chondroitin sulfate. Eur. J. Biochem. 1985, 148, 463–469. [Google Scholar] [CrossRef]
- Lidholt, K.; Fjelstad, M.; Lindahl, U.; Goto, F.; Ogawa, T.; Kitagawa, H.; Sugahara, K. Assessment of glycosaminoglycan-protein linkage tetrasaccharides as acceptors for GalNAc- and GlcNAc-transferases from mouse mastocytoma. Glycoconj. J. 1997, 14, 737–742. [Google Scholar] [CrossRef]
- Nadanaka, S.; Kitagawa, H.; Goto, F.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Sugahara, K. Involvement of the core protein in the first β-N-acetylgalactosamine transfer to the glycosaminoglycan-protein linkage-region tetrasaccharide and in the subsequent polymerization: The critical determining step for chondroitin sulphate biosynthesis. Biochem. J. 1999, 340, 353–357. [Google Scholar] [CrossRef]
- Uyama, T.; Kitagawa, H.; Tamura, J.; Sugahara, K. Molecular cloning and expression of human chondroitin N-acetylgalactosaminyltransferase: The key enzyme for chain initiation and elongation of chondroitin/dermatan sulfate on the protein linkage region tetrasaccharide shared by heparin/heparan sulfate. J. Biol. Chem. 2002, 277, 8841–8846. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, M.; Sato, T.; Akashima, T.; Iwasaki, H.; Kameyama, A.; Mochizuki, H.; Yada, T.; Inaba, N.; Zhang, Y.; Kikuchi, N.; et al. Enzymatic synthesis of chondroitin with a novel chondroitin sulfate N-acetylgalactosaminyltransferase that transfers N-acetylgalactosamine to glucuronic acid in initiation and elongation of chondroitin sulfate synthesis. J. Biol. Chem. 2002, 277, 38189–38196. [Google Scholar] [CrossRef] [Green Version]
- Uyama, T.; Kitagawa, H.; Tanaka, J.; Tamura, J.; Ogawa, T.; Sugahara, K. Molecular cloning and expression of a second chondroitin N-acetylgalactosaminyltransferase involved in the initiation and elongation of chondroitin/dermatan sulfate. J. Biol. Chem. 2003, 278, 3072–3078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Gotoh, M.; Kiyohara, K.; Akashima, T.; Iwasaki, H.; Kameyama, A.; Mochizuki, H.; Yada, T.; Inaba, N.; Togayachi, A.; et al. Differential roles of two N-acetylgalactosaminyltransferases, CSGalNAcT-1, and a novel enzyme, CSGalNAcT-2. Initiation and elongation in synthesis of chondroitin sulfate. J. Biol. Chem. 2003, 278, 3063–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, R.L.; Telser, A.; Dorfman, A. The biosynthesis of chondroitin sulfate by a cell-free preparation. J. Biol. Chem. 1964, 239, 3623–3629. [Google Scholar] [CrossRef] [PubMed]
- Silbert, J.E. Incorporation of 14C and 3H from labeled nucleotide sugars into a polysaccharide in the presence of a cell-free preparation from cartilage. J. Biol. Chem. 1964, 239, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Telser, A.; Robinson, H.C.; Dorfman, A. The biosynthesis of chondroitin sulfate. Arch. Biochem. Biophys. 1966, 116, 458–465. [Google Scholar] [CrossRef]
- Dorfman, A. Adventures in viscous solutions. Mol. Cell. Biochem. 1974, 4, 45–74. [Google Scholar] [CrossRef]
- Silbert, J.E.; Reppucci, A.C., Jr. Biosynthesis of chondroitin sulfate. Independent addition of glucuronic acid and N-acetylgalactosamine to oligosaccharides. J. Biol. Chem. 1976, 251, 3942–3947. [Google Scholar] [CrossRef]
- Kitagawa, H.; Ujikawa, M.; Tsutsumi, K.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Sugahara, K. Characterization of serum β-glucuronyltransferase involved in chondroitin sulfate biosynthesis. Glycobiology 1997, 7, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, H.; Tsuchida, K.; Ujikawa, M.; Sugahara, K. Detection and characterization of UDP-GalNAc: Chondroitin N-acetylgalactosaminyltransferase in bovine serum using a simple assay method. J. Biochem. 1995, 117, 1083–1087. [Google Scholar] [CrossRef]
- Kitagawa, H.; Tsutsumi, K.; Ujikawa, M.; Goto, F.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Sugahara, K. Regulation of chondroitin sulfate biosynthesis by specific sulfation: Acceptor specificity of serum β-GalNAc transferase revealed by structurally defined oligosaccharides. Glycobiology 1997, 7, 531–537. [Google Scholar] [CrossRef]
- Kitagawa, H.; Uyama, T.; Sugahara, K. Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 2001, 276, 38721–38726. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, H.; Izumikawa, T.; Uyama, T.; Sugahara, K. Molecular cloning of a chondroitin polymerizing factor that cooperates with chondroitin synthase for chondroitin polymerization. J. Biol. Chem. 2003, 278, 23666–23671. [Google Scholar] [CrossRef] [Green Version]
- Yada, T.; Gotoh, M.; Sato, T.; Shionyu, M.; Go, M.; Kaseyama, H.; Iwasaki, H.; Kikuchi, N.; Kwon, Y.D.; Togayachi, A.; et al. Chondroitin sulfate synthase-2. Molecular cloning and characterization of a novel human glycosyltransferase homologous to chondroitin sulfate glucuronyltransferase, which has dual enzymatic activities. J. Biol. Chem. 2003, 278, 30235–30247. [Google Scholar] [CrossRef] [Green Version]
- Izumikawa, T.; Uyama, T.; Okuura, Y.; Sugahara, K.; Kitagawa, H. Involvement of chondroitin sulfate synthase-3 (chondroitin synthase-2) in chondroitin polymerization through its interaction with chondroitin synthase-1 or chondroitin polymerizing factor. Biochem. J. 2007, 403, 545–552. [Google Scholar] [CrossRef]
- Yada, T.; Sato, T.; Kaseyama, H.; Gotoh, M.; Iwasaki, H.; Kikuchi, N.; Kwon, Y.D.; Togayachi, A.; Kudo, T.; Watanabe, H.; et al. Chondroitin sulfate synthase-3. Molecular cloning and characterization. J. Biol. Chem. 2003, 278, 39711–39725. [Google Scholar] [CrossRef] [Green Version]
- Izumikawa, T.; Koike, T.; Shiozawa, S.; Sugahara, K.; Tamura, J.; Kitagawa, H. Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization: Chondroitin polymerization is achieved by multiple enzyme complexes consisting of chondroitin synthase family members. J. Biol. Chem. 2008, 283, 11396–11406. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, M.; Yada, T.; Sato, T.; Akashima, T.; Iwasaki, H.; Mochizuki, H.; Inaba, N.; Togayachi, A.; Kudo, T.; Watanabe, H.; et al. Molecular cloning and characterization of a novel chondroitin sulfate glucuronyltransferase that transfers glucuronic acid to N-acetylgalactosamine. J. Biol. Chem. 2002, 277, 38179–38188. [Google Scholar] [CrossRef] [Green Version]
- Malmström, A.; Fransson, L.A. Biosynthesis of dermatan sulfate. I. Formation of L-iduronic acid residues. J. Biol. Chem. 1975, 250, 3419–3425. [Google Scholar] [CrossRef]
- Malmström, A. Biosynthesis of dermatan sulphate. Loss of C-5 hydrogen during conversion of D-glucuronate to L-iduronate. Biochem. J. 1981, 198, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Malmström, A.; Aberg, L. Biosynthesis of dermatan sulphate. Assay and properties of the uronosyl C-5 epimerase. Biochem. J. 1982, 201, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Malmström, A. Biosynthesis of dermatan sulfate. II. Substrate specificity of the C-5 uronosyl epimerase. J. Biol. Chem. 1984, 259, 161–165. [Google Scholar] [CrossRef]
- Campbell, P.; Feingold, D.S.; Jensen, J.W.; Malmström, A.; Rodén, L. New assay for uronosyl 5-epimerases. Anal. Biochem. 1983, 131, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Cöster, L.; Hernnäs, J.; Malmström, A. Biosynthesis of dermatan sulphate proteoglycans. The effect of β-D-xyloside addition on the polymer-modification process in fibroblast cultures. Biochem. J. 1991, 276, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Hannesson, H.H.; Hagner-McWhirter, A.; Tiedemann, K.; Lindahl, U.; Malmström, A. Biosynthesis of dermatan sulphate. Defructosylated Escherichia coli K4 capsular polysaccharide as a substrate for the D-glucuronyl C-5 epimerase, and an indication of a two-base reaction mechanism. Biochem. J. 1996, 313, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedemann, K.; Larsson, T.; Heinegård, D.; Malmström, A. The glucuronyl C5-epimerase activity is the limiting factor in the dermatan sulfate biosynthesis. Arch. Biochem. Biophys. 2001, 391, 65–71. [Google Scholar] [CrossRef]
- Maccarana, M.; Olander, B.; Malmström, J.; Tiedemann, K.; Aebersold, R.; Lindahl, U.; Li, J.P.; Malmström, A. Biosynthesis of dermatan sulfate: Chondroitin-glucuronate C5-epimerase is identical to SART2. J. Biol. Chem. 2006, 281, 11560–11568. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, B.; Maccarana, M.; Goodlett, D.R.; Malmström, A.; Malmström, L. Identification of the active site of DS-epimerase 1 and requirement of N-glycosylation for enzyme function. J. Biol. Chem. 2009, 284, 1741–1747. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, B.; Malmström, A.; Maccarana, M. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 2009, 284, 9788–9795. [Google Scholar] [CrossRef] [Green Version]
- Nakao, M.; Shichijo, S.; Imaizumi, T.; Inoue, Y.; Matsunaga, K.; Yamada, A.; Kikuchi, M.; Tsuda, N.; Ohta, K.; Takamori, S.; et al. Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J. Immunol. 2000, 164, 2565–2574. [Google Scholar] [CrossRef] [Green Version]
- Goossens, D.; Van Gestel, S.; Claes, S.; De Rijk, P.; Souery, D.; Massat, I.; Van den Bossche, D.; Backhovens, H.; Mendlewicz, J.; Van Broeckhoven, C.; et al. A novel CpG-associated brain-expressed candidate gene for chromosome 18q-linked bipolar disorder. Mol. Psychiatry 2003, 8, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Eklund, E.; Rodén, L.; Malmström, M.; Malmström, A. Dermatan is a better substrate for 4-O-sulfation than chondroitin: Implications in the generation of 4-O-sulfated, L-iduronate-rich galactosaminoglycans. Arch. Biochem. Biophys. 2000, 383, 171–177. [Google Scholar] [CrossRef]
- Evers, M.R.; Xia, G.; Kang, H.G.; Schachner, M.; Baenziger, J.U. Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 2001, 276, 36344–36353. [Google Scholar] [CrossRef] [Green Version]
- Mikami, T.; Mizumoto, S.; Kago, N.; Kitagawa, H.; Sugahara, K. Specificities of three distinct human chondroitin/dermatan N-acetylgalactosamine 4-O-sulfotransferases demonstrated using partially desulfated dermatan sulfate as an acceptor: Implication of differential roles in dermatan sulfate biosynthesis. J. Biol. Chem. 2003, 278, 36115–36127. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, B.; Maccarana, M.; Malmström, A. Dermatan 4-O-sulfotransferase 1 is pivotal in the formation of iduronic acid blocks in dermatan sulfate. Glycobiology 2009, 19, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Sugumaran, G.; Liu, J.; Shworak, N.W.; Silbert, J.E.; Rosenberg, R.D. Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan/chondroitin sulfate. J. Biol. Chem. 1999, 274, 10474–10480. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, S.; Kimata, K.; Habuchi, O. Recognition of sulfation pattern of chondroitin sulfate by uronosyl 2-O-sulfotransferase. J. Biol. Chem. 2005, 280, 39115–39123. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Van Die, I.; Van den Eijnden, D.H.; Yokota, A.; Kitagawa, H.; Sugahara, K. Demonstration of glycosaminoglycans in Caenorhabditis elegans. FEBS Lett. 1999, 459, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Kinoshita-Toyoda, A.; Selleck, S.B. Structural analysis of glycosaminoglycans in Drosophila and Caenorhabditis elegans and demonstration that tout-velu, a Drosophila gene related to EXT tumor suppressors, affects heparan sulfate in vivo. J. Biol. Chem. 2000, 275, 2269–2275. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Okada, Y.; Ueno, M.; Iwata, S.; Deepa, S.S.; Nishimura, S.; Fujita, M.; Van Die, I.; Hirabayashi, Y.; Sugahara, K. Determination of the glycosaminoglycan-protein linkage region oligosaccharide structures of proteoglycans from Drosophila melanogaster and Caenorhabditis elegans. J. Biol. Chem. 2002, 277, 31877–31886. [Google Scholar] [CrossRef] [Green Version]
- Maccarana, M.; Kalamajski, S.; Kongsgaard, M.; Magnusson, S.P.; Oldberg, A.; Malmström, A. Dermatan sulfate epimerase 1-deficient mice have reduced content and changed distribution of iduronic acids in dermatan sulfate and an altered collagen structure in skin. Mol. Cell. Biol. 2009, 29, 5517–5528. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, B.; Thelin, M.A.; Svensson, L.; Ghiselli, G.; van Kuppevelt, T.H.; Malmström, A.; Maccarana, M. Iduronic acid in chondroitin/dermatan sulfate affects directional migration of aortic smooth muscle cells. PLoS ONE 2013, 8, e66704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, R.; Stachtea, X.; Maccarana, M.; Grottling, E.; Eklund, E.; Malmström, A.; Oldberg, A. Dermatan sulfate epimerase 1 deficient mice as a model for human abdominal wall defects. Birth Defects Res. A Clin. Mol. Teratol. 2014, 100, 712–720. [Google Scholar] [CrossRef] [Green Version]
- Nadafi, R.; Koning, J.J.; Veninga, H.; Stachtea, X.N.; Konijn, T.; Zwiers, A.; Malmström, A.; den Haan, J.M.M.; Mebius, R.E.; Maccarana, M.; et al. Dendritic cell migration to skin-draining lymph nodes is controlled by dermatan sulfate and determines adaptive immunity magnitude. Front. Immunol. 2018, 9, 206. [Google Scholar] [CrossRef] [Green Version]
- Gouignard, N.; Maccarana, M.; Strate, I.; von Stedingk, K.; Malmström, A.; Pera, E.M. Musculocontractural Ehlers-Danlos syndrome and neurocristopathies: Dermatan sulfate is required for Xenopus neural crest cells to migrate and adhere to fibronectin. Dis. Model Mech. 2016, 9, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Dam, G.B.; Yamada, S.; Kobayashi, F.; Purushothaman, A.; van de Westerlo, E.M.; Bulten, J.; Malmström, A.; Sugahara, K.; Massuger, L.F.; van Kuppevelt, T.H. Dermatan sulfate domains defined by the novel antibody GD3A12, in normal tissues and ovarian adenocarcinomas. Histochem. Cell. Biol. 2009, 132, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Thelin, M.A.; Svensson, K.J.; Shi, X.; Bagher, M.; Axelsson, J.; Isinger-Ekstrand, A.; van Kuppevelt, T.H.; Johansson, J.; Nilbert, M.; Zaia, J.; et al. Dermatan sulfate is involved in the tumorigenic properties of esophagus squamous cell carcinoma. Cancer Res. 2012, 72, 1943–1952. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Mizumoto, S.; Suresh, I.; Komatsu, Y.; Vodopiutz, J.; Dundar, M.; Straub, V.; Lingenhel, A.; Melmer, A.; Lechner, S.; et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum. Mol. Genet. 2013, 22, 3761–3772. [Google Scholar] [CrossRef] [Green Version]
- Syx, D.; Van Damme, T.; Symoens, S.; Maiburg, M.C.; van de Laar, I.; Morton, J.; Suri, M.; Del Campo, M.; Hausser, I.; Hermanns-Lê, T.; et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 2015, 36, 535–547. [Google Scholar] [CrossRef]
- Ranza, E.; Huber, C.; Levin, N.; Baujat, G.; Bole-Feysot, C.; Nitschke, P.; Masson, C.; Alanay, Y.; Al-Gazali, L.; Bitoun, P.; et al. Chondrodysplasia with multiple dislocations: Comprehensive study of a series of 30 cases. Clin. Genet. 2017, 91, 868–880. [Google Scholar] [CrossRef]
- Schirwani, S.; Metcalfe, K.; Wagner, B.; Berry, I.; Sobey, G.; Jewell, R. DSE associated musculocontractural EDS, a milder phenotype or phenotypic variability. Eur. J. Med. Genet. 2020, 63, 103798. [Google Scholar] [CrossRef]
- Lautrup, C.K.; Teik, K.W.; Unzaki, A.; Mizumoto, S.; Syx, D.; Sin, H.H.; Nielsen, I.K.; Markholt, S.; Yamada, S.; Malfait, F.; et al. Delineation of musculocontractural Ehlers-Danlos syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genom. Med. 2020, 8, e1197. [Google Scholar] [CrossRef] [Green Version]
- Akatsu, C.; Mizumoto, S.; Kaneiwa, T.; Maccarana, M.; Malmström, A.; Yamada, S.; Sugahara, K. Dermatan sulfate epimerase 2 is the predominant isozyme in the formation of the chondroitin sulfate/dermatan sulfate hybrid structure in postnatal developing mouse brain. Glycobiology 2011, 21, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Nishimura, S.; Mikami, T.; Yamada, S.; Itoh, N.; Sugahara, K. Chondroitin sulfate/dermatan sulfate hybrid chains from embryonic pig brain, which contain a higher proportion of L-iduronic acid than those from adult pig brain, exhibit neuritogenic and growth factor binding activities. J. Biol. Chem. 2004, 279, 9765–9776. [Google Scholar] [CrossRef] [Green Version]
- Mitsunaga, C.; Mikami, T.; Mizumoto, S.; Fukuda, J.; Sugahara, K. Chondroitin sulfate/dermatan sulfate hybrid chains in the development of cerebellum. Spatiotemporal regulation of the expression of critical disulfated disaccharides by specific sulfotransferases. J. Biol. Chem. 2006, 281, 18942–18952. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, B.; Thelin, M.A.; Rauch, U.; Feinstein, R.; Oldberg, A.; Malmström, A.; Maccarana, M. Mouse development is not obviously affected by the absence of dermatan sulfate epimerase 2 in spite of a modified brain dermatan sulfate composition. Glycobiology 2021, 22, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Stachtea, X.N.; Tykesson, E.; van Kuppevelt, T.H.; Feinstein, R.; Malmström, A.; Reijmers, R.M.; Maccarana, M. Dermatan sulfate-free mice display embryological defects and are neonatal lethal despite normal lymphoid and non-lymphoid organogenesis. PLoS ONE 2015, 10, e0140279. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Potash, J.B.; Knowles, J.A.; Weissman, M.M.; Coryell, W.; Scheftner, W.A.; Lawson, W.B.; DePaulo, J.R., Jr.; Gejman, P.V.; Sanders, A.R.; et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiatry 2011, 16, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Zayed, H.; Chao, R.; Moshrefi, A.; Lopezjimenez, N.; Delaney, A.; Chen, J.; Shaw, G.M.; Slavotinek, A.M. A maternally inherited chromosome 18q22.1 deletion in a male with late-presenting diaphragmatic hernia and microphthalmia-evaluation of DSEL as a candidate gene for the diaphragmatic defect. Am. J. Med. Genet. 2010, 152A, 916–923. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, S.; Mita, S.; Matsubara, T.; Fukuta, M.; Habuchi, H.; Kimata, K.; Habuchi, O. Molecular cloning and expression of chondroitin 4-sulfotransferase. J. Biol. Chem. 2000, 275, 8975–8981. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, N.; Nakagawa, H.; Ong, E.; Akama, T.O.; Fukuda, M.N.; Fukuda, M. Molecular cloning and expression of two distinct human chondroitin 4-O-sulfotransferases that belong to the HNK-1 sulfotransferase gene family. J. Biol. Chem. 2000, 75, 20188–20196. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.G.; Evers, M.R.; Xia, G.; Baenziger, J.U.; Schachner, M. Molecular cloning and characterization of chondroitin-4-O-sulfotransferase-3. A novel member of the HNK-1 family of sulfotransferases. J. Biol. Chem. 2002, 277, 34766–34772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, G.; Evers, M.R.; Kang, H.G.; Schachner, M.; Baenziger, J.U. Molecular cloning and expression of the pituitary glycoprotein hormone N-acetylgalactosamine-4-O-sulfotransferase. J. Biol. Chem. 2000, 275, 38402–38409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.G.; Evers, M.R.; Xia, G.; Baenziger, J.U.; Schachner, M. Molecular cloning and expression of an N-acetylgalactosamine-4-O-sulfotransferase that transfers sulfate to terminal and Non-terminal β 1,4-linked N-acetylgalactosamine. J. Biol. Chem. 2001, 276, 10861–10869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tykesson, E.; Hassinen, A.; Zielinska, K.; Thelin, M.A.; Frati, G.; Ellervik, U.; Westergren-Thorsson, G.; Malmström, A.; Kellokumpu, S.; Maccarana, M. Dermatan sulfate epimerase 1 and dermatan 4-O-sulfotransferase 1 form complexes that generate long epimerized 4-O-sulfated blocks. J. Biol. Chem. 2018, 293, 13725–13735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Akyüz, N.; Bernreuther, C.; Loers, G.; Laczynska, E.; Jakovcevski, I.; Schachner, M. Dermatan sulfotransferase Chst14/D4st1, but not chondroitin sulfotransferase Chst11/C4st1, regulates proliferation and neurogenesis of neural progenitor cells. J. Cell Sci. 2011, 124, 4051–4063. [Google Scholar] [CrossRef] [Green Version]
- Clement, A.M.; Nadanaka, S.; Masayama, K.; Mandl, C.; Sugahara, K.; Faissner, A. The DSD-1 carbohydrate epitope depends on sulfation, correlates with chondroitin sulfate D motifs, and is sufficient to promote neurite outgrowth. J. Biol. Chem. 1998, 273, 28444–28453. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Hikino, M.; Yajima, Y.; Mikami, T.; Sirko, S.; von Holst, A.; Faissner, A.; Fukui, S.; Sugahara, K. Structural characterization of the epitopes of the monoclonal antibodies 473HD, CS-56, and MO-225 specific for chondroitin sulfate D-type using the oligosaccharide library. Glycobiology 2005, 15, 593–603. [Google Scholar] [CrossRef]
- von Holst, A.; Sirko, S.; Faissner, A. The unique 473HD-Chondroitinsulfate epitope is expressed by radial glia and involved in neural precursor cell proliferation. J. Neurosci. 2006, 26, 4082–4094. [Google Scholar] [CrossRef] [Green Version]
- Akyüz, N.; Rost, S.; Mehanna, A.; Bian, S.; Loers, G.; Oezen, I.; Mishra, B.; Hoffmann, K.; Guseva, D.; Laczynska, E.; et al. Dermatan 4-O-sulfotransferase1 ablation accelerates peripheral nerve regeneration. Exp. Neurol. 2013, 247, 517–530. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Mizumoto, S.; Takahashi, Y.; Shimada, S.; Sugahara, K.; Nakayama, J.; Takeda, S.; Nomura, Y.; Nitahara-Kasahara, Y.; Okada, T.; et al. Vascular abnormalities in the placenta of Chst14-/- fetuses: Implications in the pathophysiology of perinatal lethality of the murine model and vascular lesions in human CHST14/D4ST1 deficiency. Glycobiology 2018, 28, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Dündar, M.; Müller, T.; Zhang, Q.; Pan, J.; Steinmann, B.; Vodopiutz, J.; Gruber, R.; Sonoda, T.; Krabichler, B.; Utermann, G.; et al. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009, 85, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Mizumoto, S.; Hashimoto, A.; Takahashi, Y.; Yoshizawa, T.; Nitahara-Kasahara, Y.; Takahashi, N.; Nakayama, J.; Takehana, K.; Okada, T.; et al. Systematic investigation of the skin in Chst14-/- mice: A model for skin fragility in musculocontractural Ehlers-Danlos syndrome caused by CHST14 variants (mcEDS-CHST14). Glycobiology 2021, 31, 137–150. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Mizumoto, S.; Inoue, Y.U.; Saka, S.; Posadas-Herrera, G.; Nakamura-Takahashi, A.; Takahashi, Y.; Hashimoto, A.; Konishi, K.; Miyata, S.; et al. Muscle pathophysiology in mouse models of musculocontractural Ehlers-Danlos syndrome due to CHST14 mutations (mcEDS-CHST14), generated through CRISPR/Cas9-mediated genomic editing. Dis. Model. Mech. 2021, 14, dmm048963. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Posadas-Herrera, G.; Mizumoto, S.; Nakamura-Takahashi, A.; Inoue, Y.U.; Inoue, T.; Nomura, Y.; Takeda, S.; Yamada, S.; Kosho, T.; et al. Myopathy associated with dermatan sulfate-deficient decorin and myostatin in musculocontractural Ehlers-Danlos syndrome: A mouse model investigation. Front. Cell Dev. Biol. 2021, 9, 695021. [Google Scholar] [CrossRef]
- Dundar, M.; Demiryilmaz, F.; Demiryilmaz, I.; Kumandas, S.; Erkilic, K.; Kendirci, M.; Tuncel, M.; Ozyazgan, I.; Tolmie, J.L. An autosomal recessive adducted thumb-club foot syndrome observed in Turkish cousins. Clin. Genet. 1997, 51, 61–64. [Google Scholar] [CrossRef]
- Kosho, T.; Takahashi, J.; Ohashi, H.; Nishimura, G.; Kato, H.; Fukushima, Y. Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. A 2005, 138A, 282–287. [Google Scholar] [CrossRef]
- Kosho, T.; Miyake, N.; Hatamochi, A.; Takahashi, J.; Kato, H.; Miyahara, T.; Igawa, Y.; Yasui, H.; Ishida, T.; Ono, K.; et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. 2010, 152A, 1333–1346. [Google Scholar] [CrossRef]
- Miyake, N.; Kosho, T.; Mizumoto, S.; Furuichi, T.; Hatamochi, A.; Nagashima, Y.; Arai, E.; Takahashi, K.; Kawamura, R.; Wakui, K.; et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010, 31, 966–974. [Google Scholar] [CrossRef]
- Hirose, T.; Takahashi, N.; Tangkawattana, P.; Minaguchi, J.; Mizumoto, S.; Yamada, S.; Miyake, N.; Hayashi, S.; Hatamochi, A.; Nakayama, J.; et al. Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 623–631. [Google Scholar] [CrossRef]
- Mizumoto, S.; Kosho, T.; Hatamochi, A.; Honda, T.; Yamaguchi, T.; Okamoto, N.; Miyake, N.; Yamada, S.; Sugahara, K. Defect in dermatan sulfate in urine of patients with Ehlers-Danlos syndrome caused by a CHST14/D4ST1 deficiency. Clin. Biochem. 2017, 50, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Malfait, F.; Syx, D.; Vlummens, P.; Symoens, S.; Nampoothiri, S.; Hermanns-Lê, T.; Van Laer, L.; De Paepe, A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010, 31, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Okamoto, N.; Miyake, N.; Taira, K.; Sato, Y.; Matsuda, K.; Akimaru, N.; Ohashi, H.; Wakui, K.; Fukushima, Y.; et al. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: Observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. A 2011, 155A, 1949–1958. [Google Scholar] [CrossRef]
- Voermans, N.C.; Kempers, M.; Lammens, M.; van Alfen, N.; Janssen, M.C.; Bönnemann, C.; van Engelen, B.G.; Hamel, B.C. Myopathy in a 20-year-old female patient with D4ST-1 deficient Ehlers-Danlos syndrome due to a homozygous CHST14 mutation. Am. J. Med. Genet. A 2012, 158A, 850–855. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Londono, R.; Chitayat, D.; Kahr, W.H.; Hinek, A.; Blaser, S.; Dupuis, L.; Goh, E.; Badilla-Porras, R.; Howard, A.; Mittaz, L.; et al. Extracellular matrix and platelet function in patients with musculocontractural Ehlers-Danlos syndrome caused by mutations in the CHST14 gene. Am. J. Med. Genet. A 2012, 158A, 1344–1354. [Google Scholar] [CrossRef]
- Winters, K.A.; Jiang, Z.; Xu, W.; Li, S.; Ammous, Z.; Jayakar, P.; Wierenga, K.J. Re-assigned diagnosis of D4ST1-deficient Ehlers-Danlos syndrome (adducted thumb-clubfoot syndrome) after initial diagnosis of Marden-Walker syndrome. Am. J. Med. Genet. A 2012, 158A, 2935–2940. [Google Scholar] [CrossRef]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170A, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef]
- Sandal, S.; Kaur, A.; Panigrahi, I. Novel mutation in the CHST14 gene causing musculocontractural type of Ehlers-Danlos syndrome. BMJ Case Rep. 2018, 2018, bcr2018226165. [Google Scholar] [CrossRef]
- Uehara, M.; Kosho, T.; Yamamoto, N.; Takahashi, H.E.; Shimakura, T.; Nakayama, J.; Kato, H.; Takahashi, J. Spinal manifestations in 12 patients with musculocontractural Ehlers-Danlos syndrome caused by CHST14/D4ST1 deficiency (mcEDS-CHST14). Am. J. Med. Genet. A 2018, 176, 2331–2341. [Google Scholar] [CrossRef]
- Uehara, M.; Oba, H.; Hatakenaka, T.; Ikegami, S.; Kuraishi, S.; Takizawa, T.; Munakata, R.; Mimura, T.; Yamaguchi, T.; Kosho, T.; et al. Posterior spinal fusion for severe spinal deformities in musculocontractural Ehlers-Danlos syndrome: Detailed observation of a novel case and review of 2 reported cases. World Neurosurg. 2020, 143, 454–461. [Google Scholar] [CrossRef]
- Minatogawa, M.; Unzaki, A.; Morisaki, H.; Syx, D.; Sonoda, T.; Janecke, A.R.; Slavotinek, A.; Voermans, N.C.; Lacassie, Y.; Mendoza-Londono, R.; et al. Clinical and molecular features of 66 patients with musculocontractural Ehlers-Danlos syndrome caused by pathogenic variants in CHST14 (mcEDS-CHST14). J. Med. Genet. 2022, 59, 865–877. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Du, Y.F.; Lu, Q.; Zhai, X.Z.; Shi, M.F.; Chen, D.Y.; Liu, S.R.; Zhong, Y. Case Report: A novel mutation identified in CHST14 gene in a fetus with structural abnormalities. Front. Genet. 2022, 13, 853907. [Google Scholar] [CrossRef]
- Qian, H.; Zhou, T.; Zheng, N.; Lu, Q.; Han, Y. Case report: Multiple gastrointestinal perforations in a rare musculocontractural Ehlers-Danlos syndrome with multiple organ dysfunction. Front. Genet. 2022, 13, 846529. [Google Scholar] [CrossRef]
- Kosho, T.; Mizumoto, S.; Watanabe, T.; Yoshizawa, T.; Miyake, N.; Yamada, S. Recent advances in the pathophysiology of musculocontractural Ehlers-Danlos syndrome. Genes 2019, 11, 43. [Google Scholar] [CrossRef] [Green Version]
- Miyake, N.; Kosho, T.; Matsumoto, N. Ehlers Danlos syndrome with glycosaminoglycan abnormalities. Adv. Exp. Med. Biol. 2021, 1348, 235–249. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| DS-PGs | Coding Genes | Chromosomal Mapping | Gene ID a | MIM No. b | GAG Type (Number) | Human Genetic Disorders |
|---|---|---|---|---|---|---|
| Decorin | DCN | 12q21.33 | 1634 | 125255 610048 | DS/CS (1) | Congenital stromal corneal dystrophy |
| Biglycan | BGN | Xq28 | 633 | 300106 300989 301870 | DS/CS (2) | Spondyloepimetaphyseal dysplasia, X-linked Meester–Loeys syndrome |
| Epiphycan | EPYC | 12q21.33 | 1833 | 601657 | DS/CS (2–3) | — |
| Endocan | ESM1 | 5q11.2 | 11082 | 601521 | DS/CS (1) | — |
| Enzyme | Coding Gene | Chromosomal Mapping | Gene ID | MIM No. | Human Genetic Disorders |
|---|---|---|---|---|---|
| DSE | DSE | 6q22.1 | 29940 | 605942 615539 | Ehlers-Danlos syndrome musculocontractural type 2 |
| DSEL | DSEL | 18q22.1 | 92126 | 611125 | Bipolar disorder |
| D4ST | CHST14 | 15q15.1 | 113189 | 601776 608429 | Ehlers-Danlos syndrome musculocontractural type 1; adducted thumb–clubfoot syndrome |
| UST | UST | 6q25.1 | 10090 | 610752 | Multiple congenital anomalies of the heart and central nervous system |
| Coding Gene | Human Genetic Disorders | Variants |
|---|---|---|
| DSE | Ehlers-Danlos syndrome musculocontractural type 2 | p.Ser268Leu, p.Arg267Gly, p.Tyr320*, p.Val333Cysfs*4, p.Pro384Trpfs*9, p.Tyr867*, and p.Val938Asp |
| DSEL | Bipolar disorder | p.Val287Ile, p.Pro673Ser, p.Tyr730Cys, p.Pro942Ser, and p.Ile1113Met |
| Diaphragmatic defect | p.Met14Ile, p.Asn276Ser, p.Pro683Ser, p.Tyr740Cys, p.Thr842Ser, and p.Asp991Asn | |
| CHST14 | Ehlers-Danlos syndrome musculocontractural type 1; adducted thumb–clubfoot syndrome | p.Arg29Glyfs*113, p.Val49*, p.Lys69*, p.Gln133Argfs*14, p.Arg135Gly, p.Leu137Gln, p.Phe209Ser, p.Arg213Pro, p.Arg218Ser, p.Lys226Alafs*16, p.Arg274Pro, p.Met280Leu, p.Pro281Leu, p.Cys289Ser, p.Tyr293Cys, and p.Glu334Glyfs*107 |
| UST | Multiple congenital anomalies of the heart and central nervous system | 0.63 Mb deletion in 6q25.1, which includes TAB2, LATS1, and UST |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizumoto, S.; Yamada, S. Histories of Dermatan Sulfate Epimerase and Dermatan 4-O-Sulfotransferase from Discovery of Their Enzymes and Genes to Musculocontractural Ehlers-Danlos Syndrome. Genes 2023, 14, 509. https://doi.org/10.3390/genes14020509
Mizumoto S, Yamada S. Histories of Dermatan Sulfate Epimerase and Dermatan 4-O-Sulfotransferase from Discovery of Their Enzymes and Genes to Musculocontractural Ehlers-Danlos Syndrome. Genes. 2023; 14(2):509. https://doi.org/10.3390/genes14020509
Chicago/Turabian StyleMizumoto, Shuji, and Shuhei Yamada. 2023. "Histories of Dermatan Sulfate Epimerase and Dermatan 4-O-Sulfotransferase from Discovery of Their Enzymes and Genes to Musculocontractural Ehlers-Danlos Syndrome" Genes 14, no. 2: 509. https://doi.org/10.3390/genes14020509