

Whole Exome Sequencing Study Suggests an Impact of FANCA, CDH1 and VEGFA Genes on Diffuse Gastric Cancer Development
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Methods
2.2.1. Whole Exome Sequencing and Data Analysis
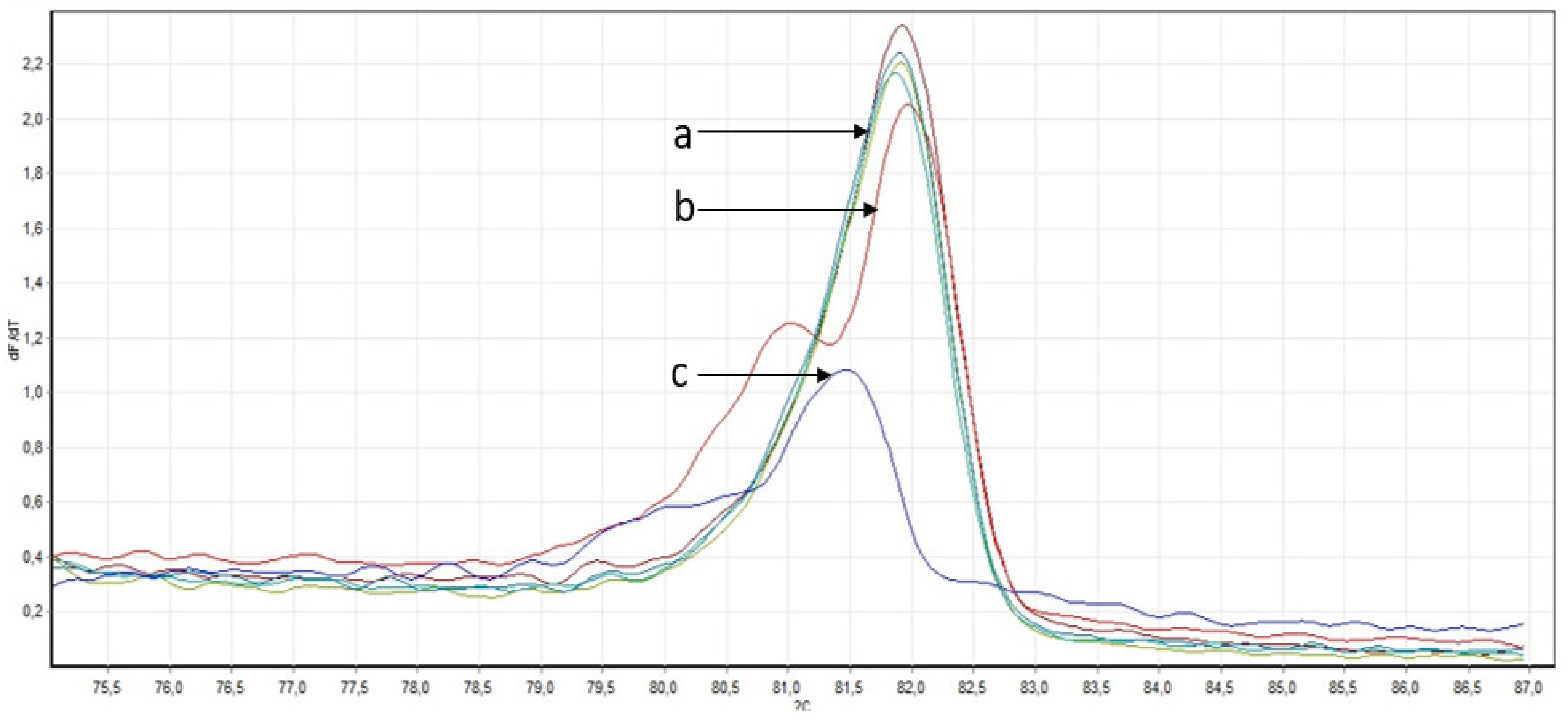
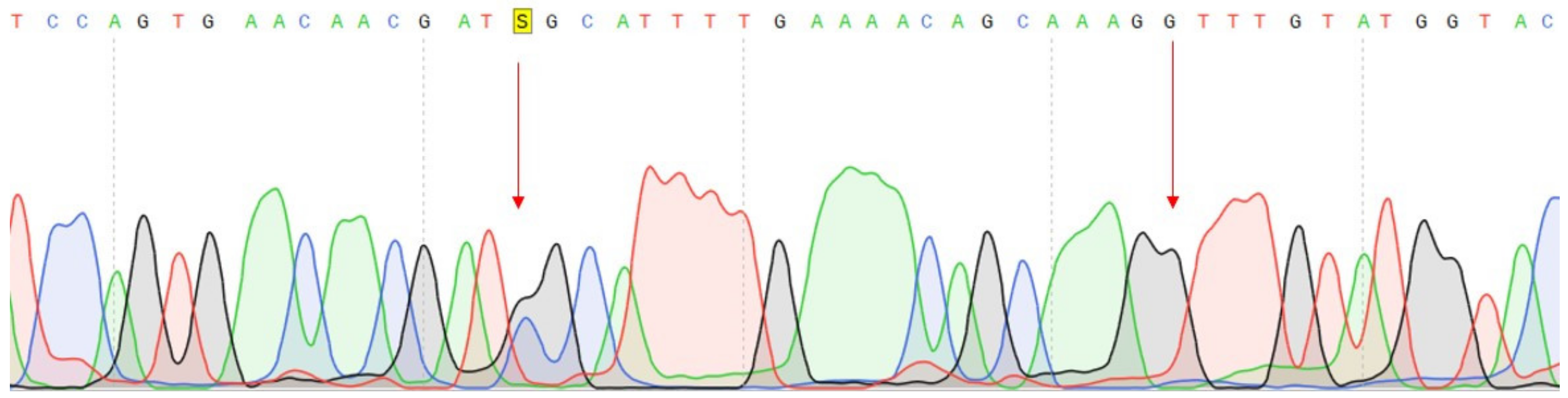
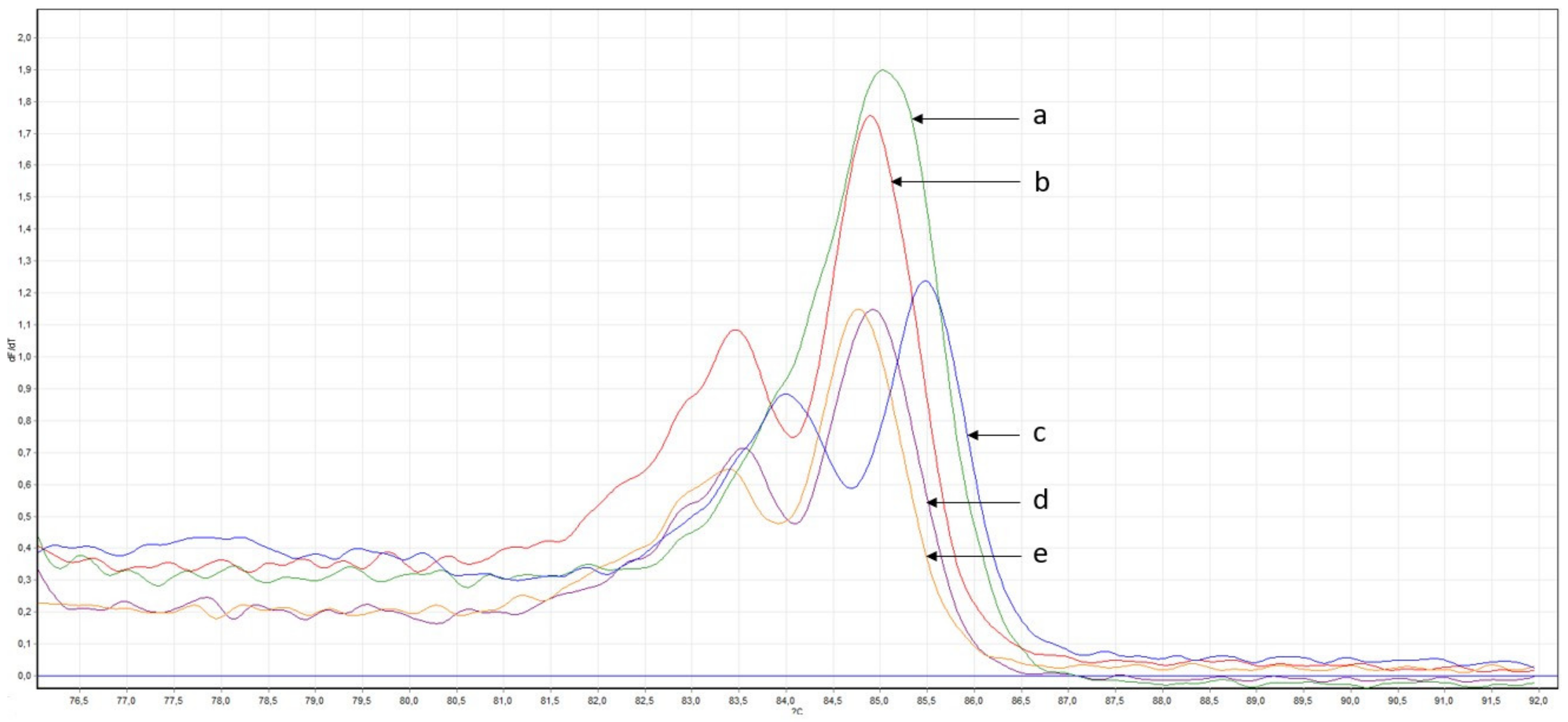
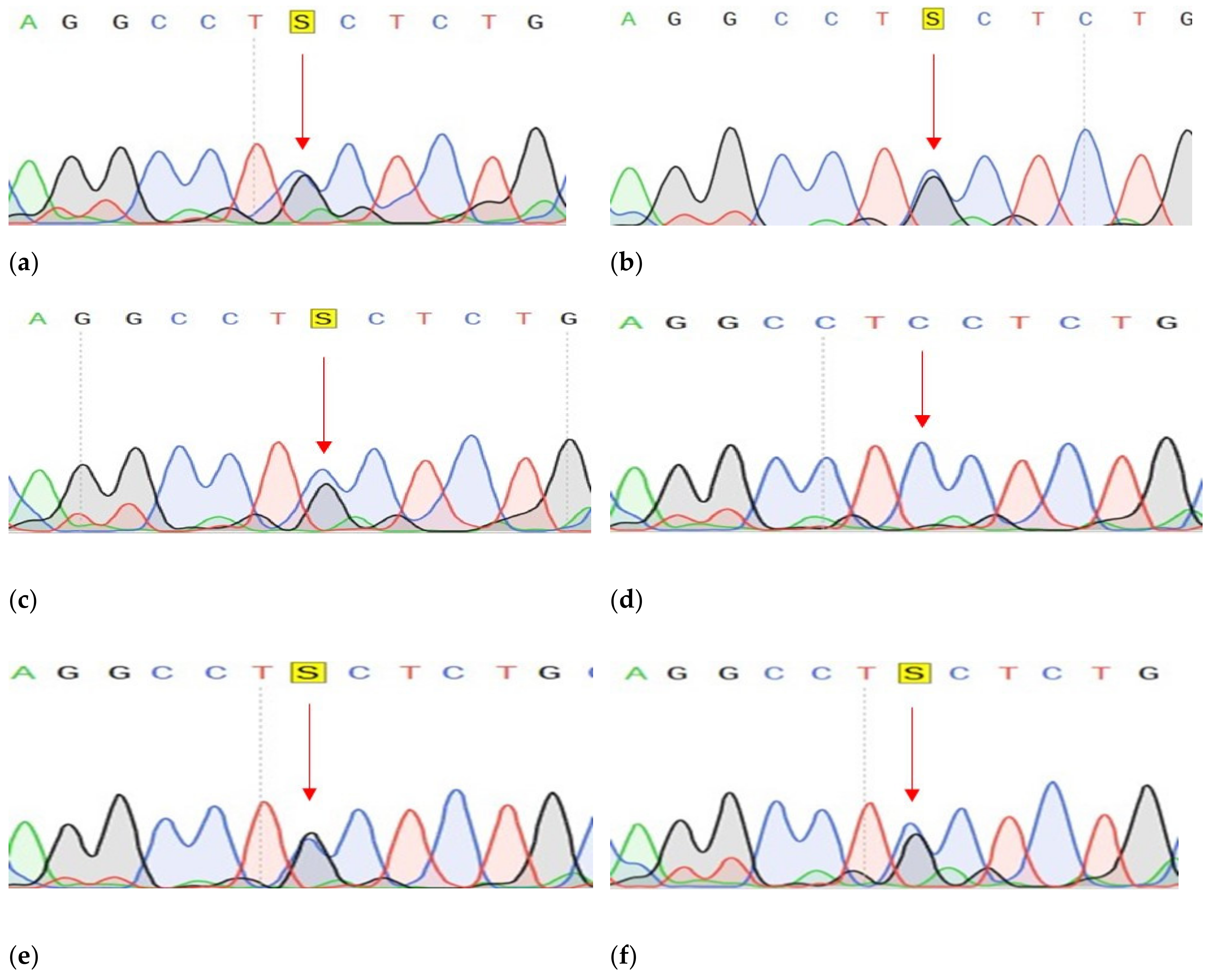
2.2.2. Confirmation of Mutations
2.2.3. Search for Described Variants in a Larger Cohort of Samples
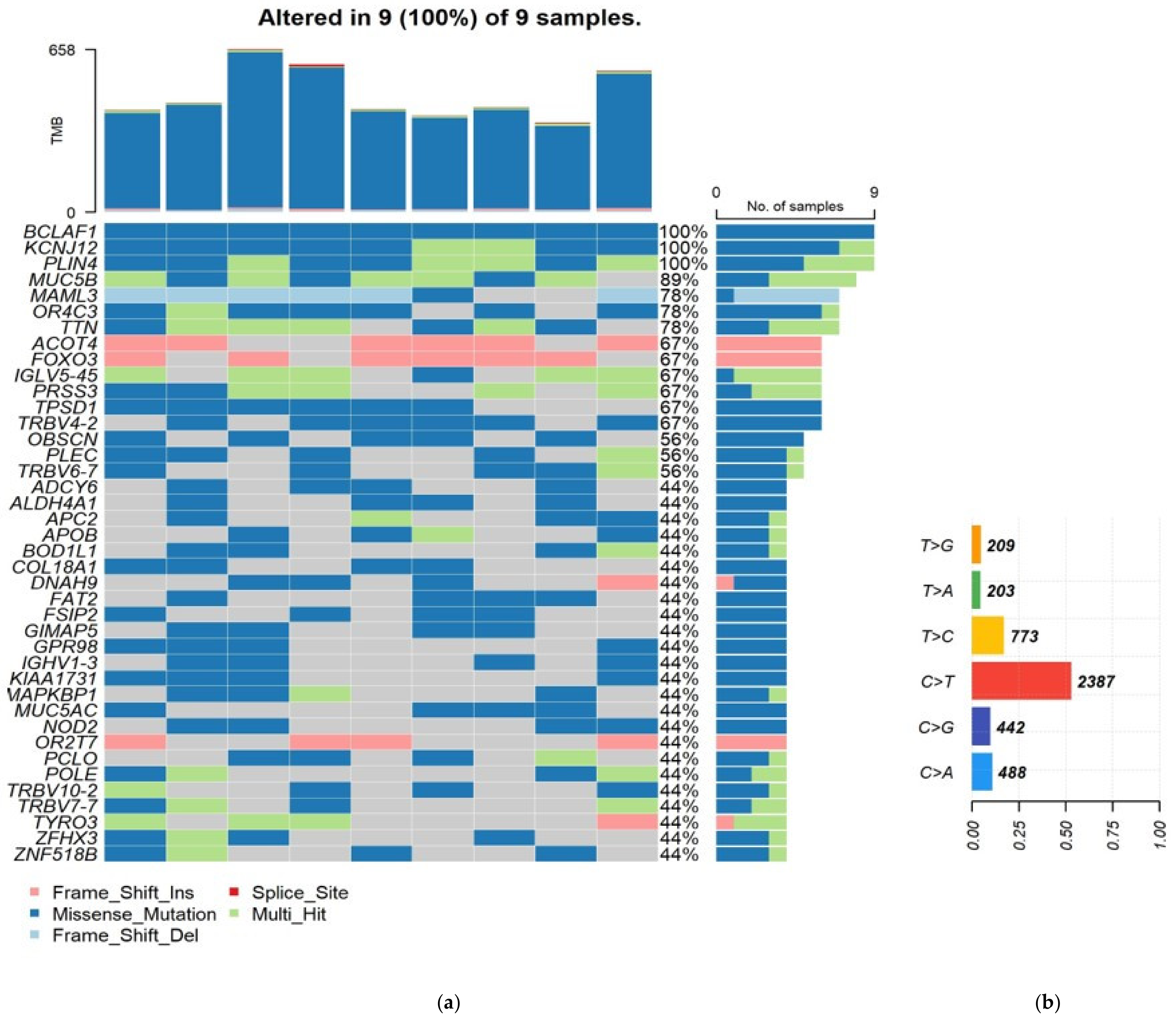
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaprin, A.D. Malignant Neoplasms in Russia in 2020 (Morbidity and Mortality); Kaprina, H.E.L.L., Starinsky, V.V., Shakhzadova, A.O., Eds.; M.: MNIOI them. P.A. Herzen-Branch of the Federal State Budgetary Institution “NMITs Radiology” of the Ministry of Health of Russia: Saint Petersburg, Russia, 2021; 250p.
- Baniak, N.; Senger, J.L.; Ahmed, S.; Kanthan, S.C.; Kanthan, R. Gastric biomarkers: A global review. World J. Surg. Oncol. 2016, 14, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusupova, L.F.; Nurgaliyeva, A.K.; Gilyazova, I.R.; Prokofyeva, D.S.; Munasypov, F.R.; Khusnutdinov, S.M.; Rakhimov, R.R.; Abdeev, R.R.; Sakaeva, D.D.; Khusnutdinova, E.K. The role of allelic variants of a number of cytokine genes in the development of gastric cancer. Genetics 2019, 55, 348–358. [Google Scholar]
- Ng, S.B.; Turner, E.H.; Robertson, P.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.Y.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Hwang, W.C.; Do, I.-G.; Wang, K.; Kang, S.Y.; Lee, J.; Park, S.H.; Park, J.O.; Kang, W.K.; Jang, J.; et al. Exome sequencing identifies early gastric carcinoma as an early stage of advanced gastric cancer. PLoS ONE 2013, 8, e82770. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Kim, S.Y.; Jung, E.S.; Yoo, J.; Kim, T.-M. Mutation heterogeneity between primary gastric cancers and their matched lymph node metastases. Gastric Cancer 2019, 22, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Shi, C.; Huang, X.; Zheng, J.; Zhu, Z.; Li, Q.; Qiu, S.; Huang, Z.; Zhuang, Z.; Wu, R.; et al. Molecular Profiles and Metastasis Markers in Chinese Patients with Gastric Carcinoma. Sci. Rep. 2019, 9, 13995. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Chugh, S.; Gnanapragassam, V.S.; Jain, M.; Rachagani, S.; Ponnusamy, M.P.; Batra, S.K. Pathobiological implications of mucin glycans in cancer: Sweet poison and novel targets. Biochim. Biophys. Acta. 2015, 1856, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Fang, D.; Liu, W.; Luo, Y. Aberrant Expression of MUC2 and MUC3 Genes in Gastric Carcinoma and Its Significance. Chin. Med. J. 2000, 113, 502–507. [Google Scholar]
- Cui, J.; Yin, Y.; Ma, Q.; Wang, G.; Olman, V.; Zhang, Y.; Chou, W.-C.; Hong, C.; Zhang, C.; Cao, S.; et al. Comprehensive characterization of the genomic alterations in human gastric cancer. Int. J. Cancer 2015, 137, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Chung, Y.S.; Takatsuka, S.; Ogawa, Y.; Sawada, T.; Yamashita, Y.; Onoda, N.; Kato, Y.; Nitta, A.; Arimoto, Y.; et al. Tumor angiogenesis as a predictor of recurrence in gastric carcinoma. J. Clin. Oncol. 1995, 13, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar] [PubMed]
- Pepper, M.S.; Ferrara, N.; Orci, L.; Montesano, R. Vascular endothelial growth factor (VEGF) induces plasminogen activators and plasminogen activator inhibitor-1 in microvascular endothelial cells. Biochem. Biophys. Res. Commun. 1991, 181, 902–906. [Google Scholar] [CrossRef]
- Unemori, E.N.; Ferrara, N.; Bauer, E.A.; Amento, E.P. Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J. Cell. Physiol. 1992, 153, 557–562. [Google Scholar] [CrossRef]
- Gerber, H.P.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 1998, 273, 13313–13316. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Tamma, R.; Annese, T.; Ruggieri, S.; Marzullo, A.; Nico, B.; Ribatti, D. VEGFA and VEGFR2 RNAscope Determination in Gastric Cancer. J. Mol. Histol. 2018, 49, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, A.; Gasparini, P.; Lonardi, S.; Corallo, S.; Fornaro, L.; Laterza, M.M.; Di Salvatore, M.; Giommoni, E.; Lotesoriere, C.; Murgioni, S.; et al. Vascular endothelial growth factor A (VEGF-A) amplification and long-term response to ramucirumab (ram) in metastatic gastric cancer (mGC): The VERA study. J. Clin. Oncol. 2019, 37, 3143. [Google Scholar] [CrossRef]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent Gain-Of-Function Mutations of RHOA in Diffuse-Type Gastric Carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, Y.; Yamashita, S.; Niwa, T.; Ushijima, T.; Nagatsuma, A.; Kuwata, T.; Yoshino, T.; Doi, T.; Ochiai, A.; Ohtsu, A. Comprehensive analyses using next-generation sequencing and immunohistochemistry enable precise treatment in advanced gastric cancer. Ann. Oncol. 2016, 27, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Levy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef] [Green Version]
- Wen, K.W.; Grenert, J.P.; Joseph, N.M.; Shafizadeh, N.; Huang, A.; Hosseini, M.; Kakar, S. Genomic profile of appendiceal goblet cell carcinoid is distinct compared to appendiceal neuroendocrine tumor and conventional adenocarcinoma. Hum. Pathol. 2018, 77, 166–174. [Google Scholar] [CrossRef]
- Ghoumid, J.; Stichelbout, M.; Jourdain, A.S.; Frenois, F.; Lejeune-Dumoulin, S.; Alex-Cordier, M.P.; Lebrun, M.; Guerreschi, P.; Duquennoy-Martinot, V.; Vinchon, M. Blepharocheilodontic syndrome is a CDH1 pathway-related disorder due to mutations in CDH1 and CTNND1. Genet. Med. 2017, 19, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Kievit, A.; Tessadori, F.; Douben, H.; Jordens, I.; Maurice, M.; Hoogeboom, J.; Hennekam, R.; Nampoothiri, S.; Kayserili, H.; Castori, M.; et al. Variants in members of the cadherin–catenin complex, CDH1 and CTNND1, cause blepharocheilodontic syndrome. Eur. J. Hum. Genet. 2018, 26, 210–219. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Age | Sex | Cancer Stage | Histological Examination of the Tumor |
|---|---|---|---|---|
| 1 GC | 59 | m | 3 | Undifferentiated adenogenic gastric carcinoma with invasion of the muscularis |
| 2 GC | 69 | f | 3 | Undifferentiated adenogenic gastric cancer with invasion of the muscular layer |
| 3 GC | 77 | f | 3 | Poorly differentiated adenocarcinoma of a scirrhous structure, germinating all layers of the stomach wall with the presence of perineural invasion |
| 4 GC | 54 | m | 3 | Surface ulcerated malignant tumor |
| 5 GC | 61 | m | 3 | Moderately differentiated adenocarcinoma invading all layers of the stomach wall |
| 6 GC | 45 | m | 3 | In the submucosal layer, a poorly differentiated adenocarcinoma of the stomach, growing into the muscle layer |
| 7 GC | 65 | m | 3 | Undifferentiated adenogenic gastric cancer growing into subserosis |
| 8 GC | 67 | m | 3 | Ulcerated on the surface, low-grade adenocarcinoma of the stomach with germination of the muscular membrane and the presence of cancer emboli in the vessels |
| 9 GC | 74 | m | 3 | Moderately differentiated adenocarcinoma of the stomach, sprouting all layers of its wall with the presence of perinephric invasion |
| Sample | 1 GC | 2 GC | 3 GC | 4 GC | 5 GC | 6 GC | 7 GC | 8 GC | 9 GC |
| Tumor tissue | |||||||||
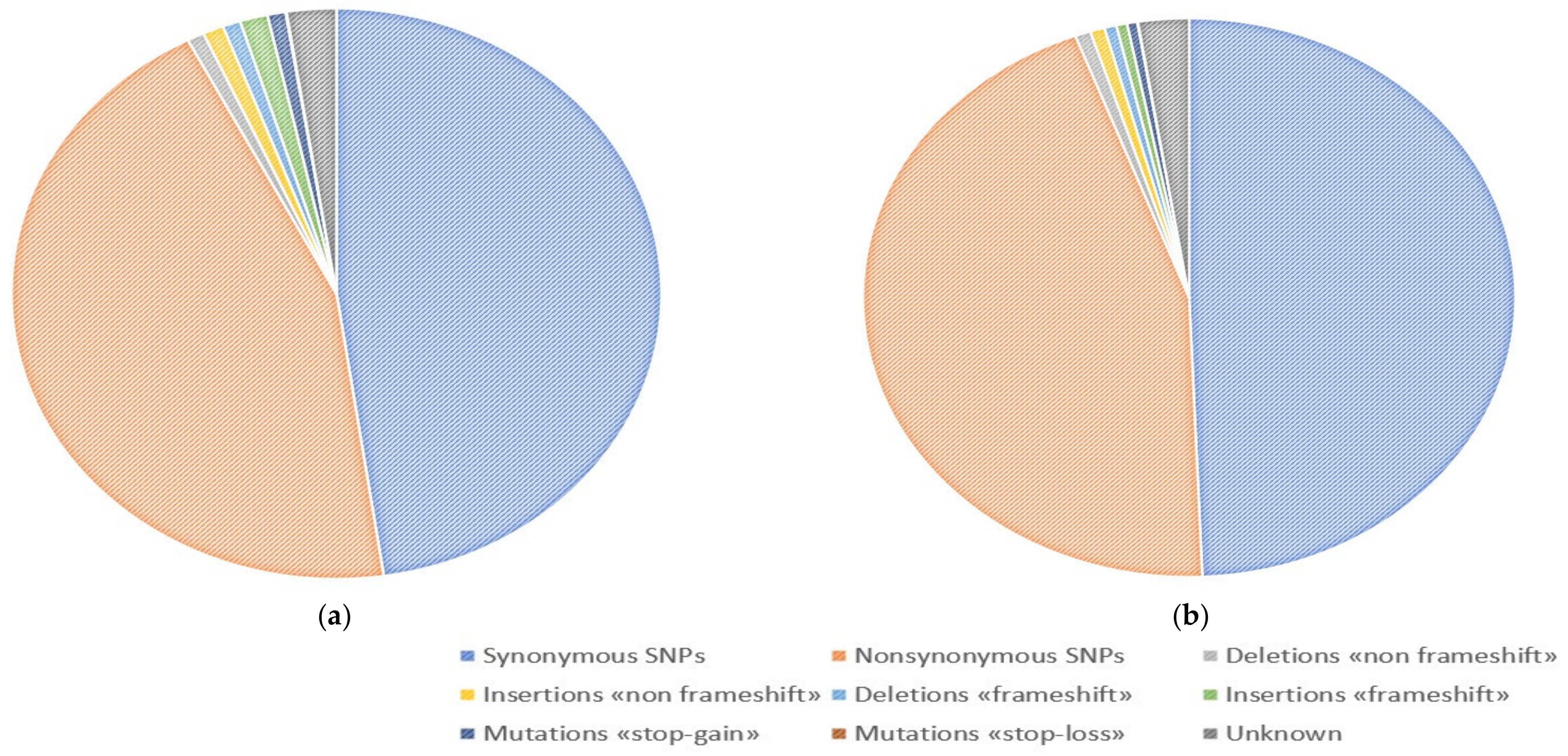
| Total number of variants | 19,179 | 19,154 | 14,829 | 11,850 | 11,527 | 12,213 | 14,975 | 20,788 | 10,275 |
| Synonymous SNPs | 9584 | 9530 | 7377 | 5563 | 5755 | 5694 | 7519 | 8293 | 4939 |
| Nonsynonymous SNPs | 8623 | 8670 | 6650 | 5493 | 5103 | 5547 | 6662 | 9000 | 4573 |
| Deletions «non frameshift» | 136 | 127 | 116 | 106 | 111 | 99 | 119 | 237 | 93 |
| Insertions «non frameshift» | 118 | 114 | 105 | 98 | 88 | 137 | 107 | 552 | 84 |
| Deletions «frameshift» | 88 | 92 | 89 | 87 | 60 | 155 | 65 | 489 | 79 |
| Insertions «frameshift» | 68 | 65 | 48 | 84 | 65 | 163 | 63 | 1217 | 95 |
| Mutations «stop-gain» | 68 | 80 | 55 | 121 | 79 | 112 | 58 | 550 | 92 |
| Mutations «stop-loss» | 7 | 9 | 5 | 3 | 6 | 6 | 6 | 15 | 2 |
| Unknown | 487 | 467 | 384 | 295 | 260 | 300 | 376 | 435 | 318 |
| Sample | 1 GC | 2 GC | 3 GC | 4 GC | 5 GC | 6 GC | 7 GC | 8 GC | 9 GC |
| Normal tissue | |||||||||
| Total number of variants | 20,200 | 21,068 | 12,140 | 12,576 | 15,478 | 14,848 | 9169 | 10,323 | 12,031 |
| Synonymous SNPs | 10,115 | 10,517 | 5967 | 6145 | 7758 | 7280 | 4349 | 5024 | 5930 |
| Nonsynonymous SNPs | 9087 | 9508 | 5443 | 5714 | 6929 | 6722 | 4135 | 4640 | 5309 |
| Deletions «non frameshift» | 133 | 143 | 108 | 99 | 117 | 89 | 94 | 99 | 99 |
| Insertions «non frameshift» | 124 | 140 | 91 | 92 | 99 | 96 | 92 | 92 | 99 |
| Deletions «frameshift» | 91 | 86 | 68 | 68 | 94 | 110 | 69 | 70 | 106 |
| Insertions «frameshift» | 93 | 95 | 59 | 59 | 65 | 65 | 94 | 52 | 105 |
| Mutations «stop-gain» | 71 | 84 | 71 | 75 | 54 | 84 | 81 | 53 | 77 |
| Mutations «stop-loss» | 7 | 9 | 2 | 5 | 4 | 5 | 2 | 2 | 3 |
| Unknown | 479 | 486 | 331 | 319 | 358 | 397 | 253 | 291 | 303 |
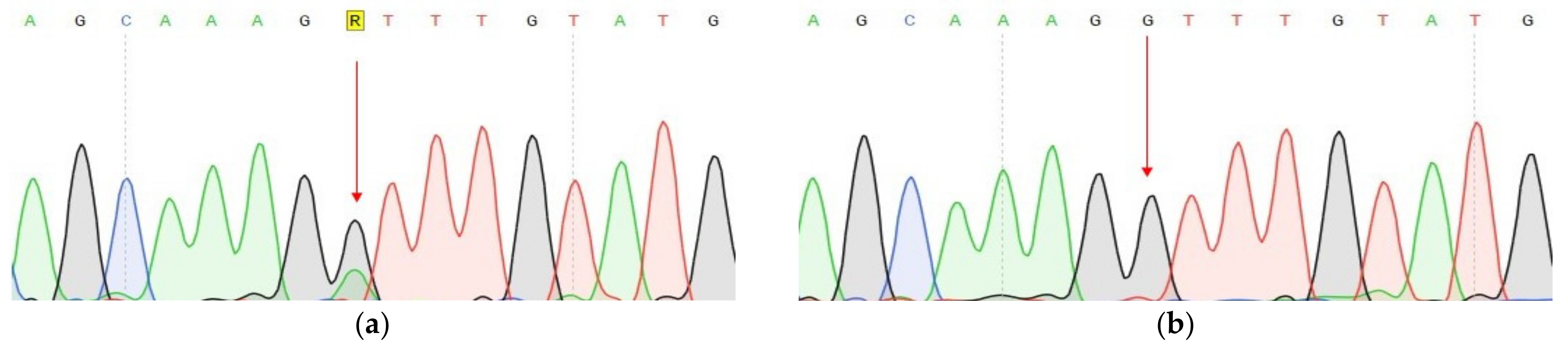
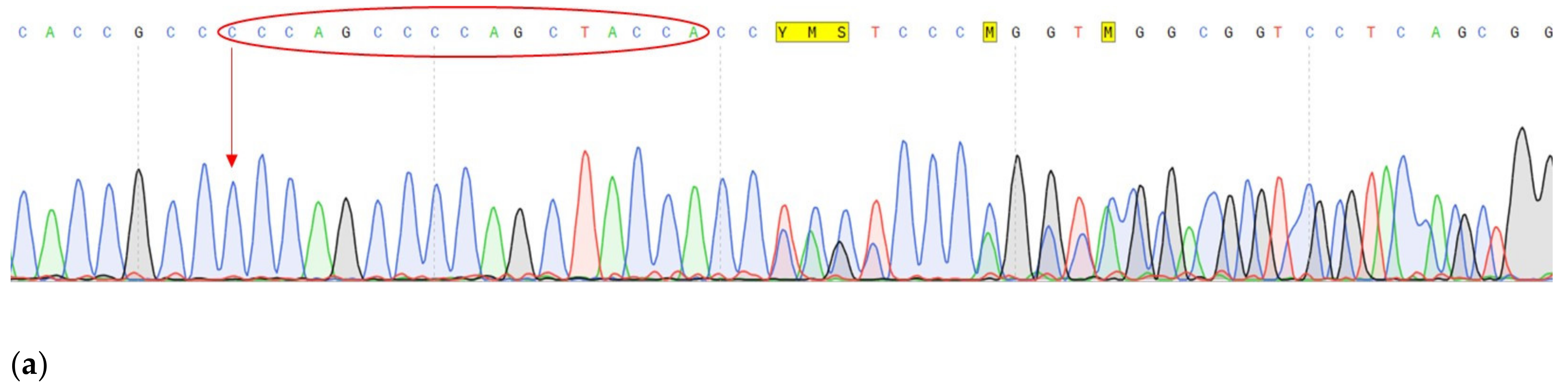
| Gen | CDH1 | VEGFA | FANCA |
|---|---|---|---|
| Tissue | tumor | tumor | tumor, normal |
| Chromosome | Chr16 | Chr6 | Chr16 |
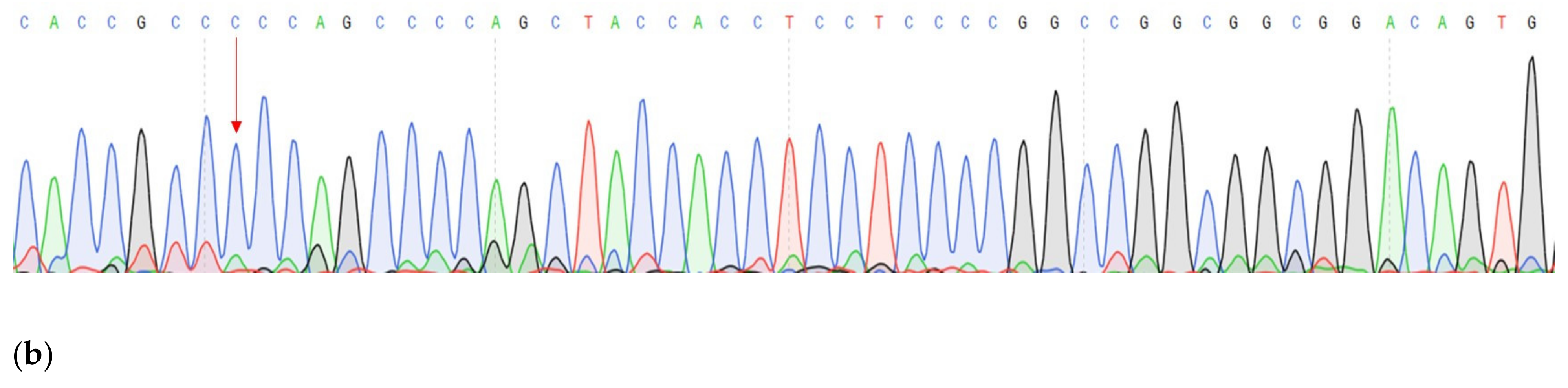
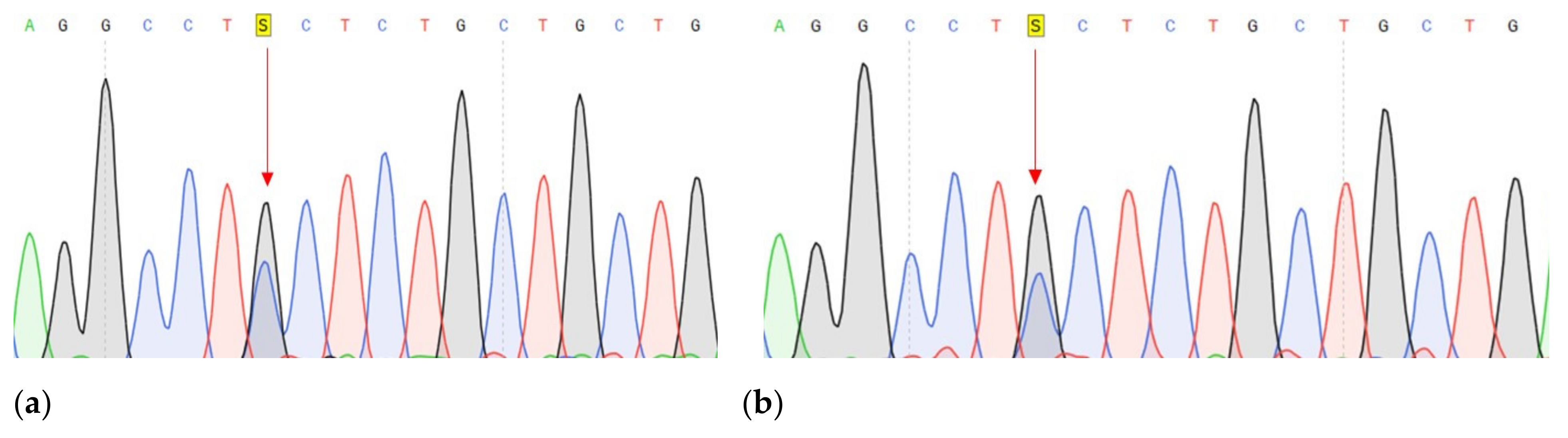
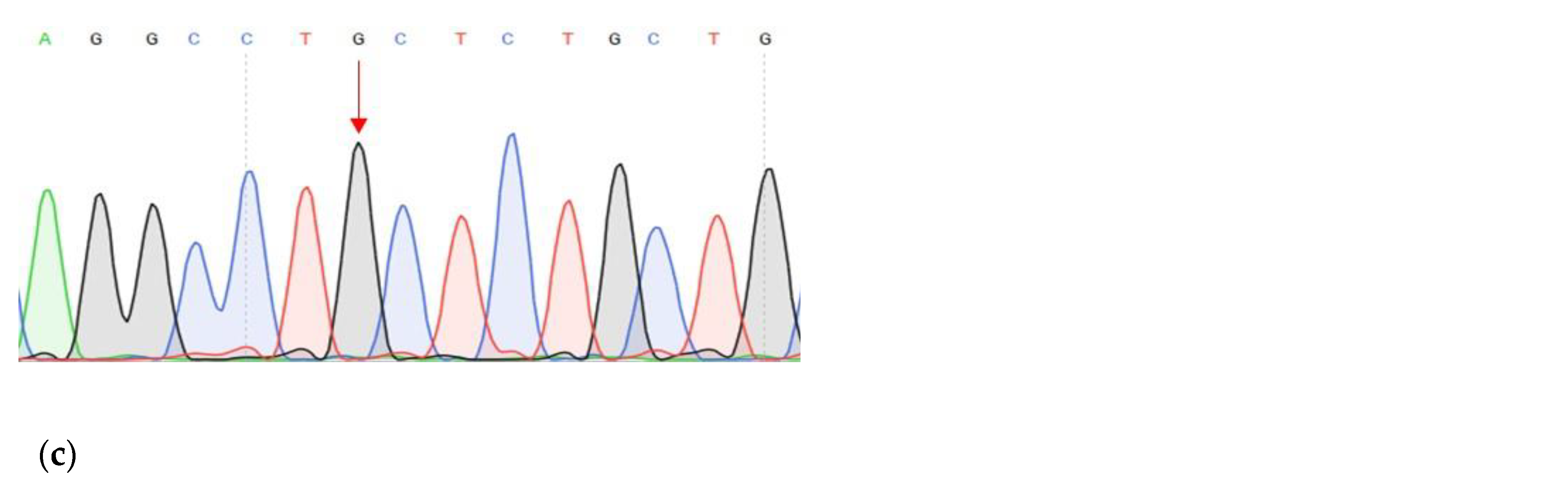
| DNA change | c.1320+1G>A | c.27_28insCCCAGCCCCAGCTACCA | c.G1874C |
| Protein change | - | p.A9fs | p.C625S |
| Frequency by 1000 Genomes | - | - | 0.001 |
| dbSNP | - | - | rs139235751 |
| PolyPhen-2 1 | - | - | D (0.986) |
| LTR 2 | - | - | D |
| Mutation Taster 3 | D | - | D |
| Mutation Assessor 4 | - | - | M |
| FATHMM 5 | D | ||
| CADD 6 | 34 | 23.5 | 26.4 |
| Sift 7 | - | - | D (0.006) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nurgalieva, A.; Galliamova, L.; Ekomasova, N.; Yankina, M.; Sakaeva, D.; Valiev, R.; Prokofyeva, D.; Dzhaubermezov, M.; Fedorova, Y.; Khusnutdinov, S.; et al. Whole Exome Sequencing Study Suggests an Impact of FANCA, CDH1 and VEGFA Genes on Diffuse Gastric Cancer Development. Genes 2023, 14, 280. https://doi.org/10.3390/genes14020280
Nurgalieva A, Galliamova L, Ekomasova N, Yankina M, Sakaeva D, Valiev R, Prokofyeva D, Dzhaubermezov M, Fedorova Y, Khusnutdinov S, et al. Whole Exome Sequencing Study Suggests an Impact of FANCA, CDH1 and VEGFA Genes on Diffuse Gastric Cancer Development. Genes. 2023; 14(2):280. https://doi.org/10.3390/genes14020280
Chicago/Turabian StyleNurgalieva, Alfiia, Lilia Galliamova, Natalia Ekomasova, Maria Yankina, Dina Sakaeva, Ruslan Valiev, Darya Prokofyeva, Murat Dzhaubermezov, Yuliya Fedorova, Shamil Khusnutdinov, and et al. 2023. "Whole Exome Sequencing Study Suggests an Impact of FANCA, CDH1 and VEGFA Genes on Diffuse Gastric Cancer Development" Genes 14, no. 2: 280. https://doi.org/10.3390/genes14020280