
The Complete Chloroplast Genome of Endangered Species Stemona parviflora: Insight into the Phylogenetic Relationship and Conservation Implications



1
College of Life Science and Technology, Xinjiang University, Urumqi 830046, China
2
Laboratory of Adaptation and Evolution of Plateau Biota, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China
*
Author to whom correspondence should be addressed.
Genes 2022, 13(8), 1361; https://doi.org/10.3390/genes13081361
Submission received: 7 July 2022
/
Revised: 24 July 2022
/
Accepted: 27 July 2022
/
Published: 29 July 2022
(This article belongs to the Section Plant Genetics and Genomics)
Abstract
:Stemona parviflora is an endangered species, narrowly endemic to Hainan and Southwest Guangdong. The taxonomic classification of S. parviflora remains controversial. Moreover, studying endangered species is helpful for current management and conservation. In this study, the first complete chloroplast genome of S. parviflora was assembled and compared with other Stemona species. The chloroplast genome size of S. parviflora was 154,552 bp, consisting of 87 protein-coding genes, 38 tRNA genes, 8 rRNA genes, and one pseudogene. The ψycf1 gene was lost in the cp genome of S. sessilifolia, but it was detected in four other species of Stemona. The inverted repeats (IR) regions have a relatively lower length variation compared with the large single copy (LSC) and small single copy (SSC) regions. Long repeat sequences and simple sequence repeat (SSR) were detected, and most SSR were distributed in the LSC region. Codon usage bias analyses revealed that the RSCU value of the genus Stemona has almost no difference. As with most angiosperm chloroplast genomes, protein-coding regions were more conservative than the inter-gene spacer. Seven genes (atpI, ccsA, cemA, matK, ndhA, petA, and rpoC1) were detected under positive selection in different Stemona species, which may result from adaptive evolution to different habitats. Phylogenetic analyses show the Stemona cluster in two main groups; S. parviflora were closest to S. tuberosa. A highly suitable region of S. parviflora was simulated by Maxent in this study; it is worth noting that the whole territory of Taiwan has changed to a low fitness area and below in the 2050 s, which may not be suitable for the introduction and cultivation of S. parviflora. In addition, limited by the dispersal capacity of S. parviflora, it is necessary to carry out artificial grafts to expand the survival areas of S. parviflora. Our results provide valuable information on characteristics of the chloroplast genome, phylogenetic relationships, and potential distribution range of the endangered species S. parviflora.
1. Introduction
S. parviflora, belonging to the genus Stemona, is narrowly endemic to Hainan and Southwest Guangdong. It is born in valleys, streams, and stone cracks at an altitude of 700 m [1]. Stemona mainly relies on ants to spread seeds. Due to the limited diffusion ability of ants, the vast majority of Stemonaceae species cannot cross the barrier of the ocean and become a narrow endemic group [2,3]. Amplified Fragment Length Polymorphism (AFLP) analyses demonstrated that S. parviflora has high genetic diversity and low genetic differences. In addition to the insect vector, the water vector and wind vector may also appear in S. parviflora at the same time [4]. S. parviflora can be used as a medicine; its effective component is an alkaloid, which has an antitussive effect. Phytochemical studies have shown that 17 alkaloids and 22 non-alkaloids have been extracted from parts of S. parviflora [5,6]. Given its great economic value and the fact the limited population of S. psrviflora are threatened by habitat fragmentation by human activity, the S. parviflora was considered an endangered species (EN) in the Threatened Species List of China’s Higher Plants [7].
Taxonomic classification has remained controversial in S. parviflora due to the use of the different molecular markers. S. parviflora were separated from S. japonica, S. sessilifolia, and S. tuberosa and were more closely related to S. phyllantha, S. aphylla, Stemona sp. based on trnH-psbA cpDNA markers [8]. The phylogenetic analyses were based on five cpDNA (atpI-atpH, psbB-psbH, rpl4-rpl36, trnC-ycf6, trnL-trnF), implying that S. parviflora were sister to S. javanica with an extremely low bootstrap value of 52% [9]. Recent studies based on three cpDNA markers (matK, rbcL, psbK-psbI) [3] suggest that S. parviflora was sister to S. tuberosa.
Climatic oscillation plays a crucial role in the pattern of a species distribution area on historical, current, and future scenarios. Comparative history and the current potential distribution range can help to us comprehend the historical dynamic processes of the population [10]. Understanding future invasion ranges is crucial for nature conservation and management. Maximum entropy (MaxEnt) has been widely applied to predict the potential distribution area of many taxa, especially in economic crops and endangered species [11]. For example, MaxEnt modeling has excellently predicted the potential distribution range of Mentha pulegium, which shows a northeastward to center-eastward shift of suitable regions, and the high suitable areas would decrease in 2050 s and 2070 s compared with the current distribution, thus providing useful information for current management [12]. In addition, MaxEnt was more suitable for predicting the distribution area of a small population under current and future circumstances [13]. The distribution pattern of S. parviflora is relatively unknown. Identifying potential distribution areas of S. parviflora will be meaningful for current management.
The structure of the chloroplast genome is relatively conservative in land plants, with a large single copy (LSC), a small single copy (SSC), and two inverted repeats (IR) in most angiosperm plants [14]. However, chloroplast genome variation occurs in different plant taxa, even at an intraspecific level, e.g., the length of gene expanded to the boundary of IR/SSC and IR/LSC region vary in different degrees among genera, interspecies, and interspecific levels [15]. Gene loss and pseudogenization occur in many angiosperms. For example, the gene infA was lost from some genus of Aroideae and Lemnoideae [16]. The chloroplast genome is widely used in angiosperm phylogenetic analysis, species definition, and the inference of species’ divergence time and geographical origin due to its sequence conservation and parthenogenetic characteristics [17,18,19].
Studies on endangered species are significant for us to understand the population’s evolutionary history. In this study, the first complete chloroplast genome of the endangered species S. parviflora was assembled and compared with other Stemona species. Our main goals were: (1) to characterize and compare the chloroplast genome of genus Stemona; (2) to assess the taxonomic position of S. parviflora; and (3) to simulate the potential distribution range of S. parviflora and provide protective measures.
2. Materials and Methods
2.1. Sample Collection, DNA Sequencing, and Chloroplast Genome Assembly
The leaf materials of S. parviflora were collected from Bopian Village, Qiongshan District, Hainan Provence (19°55′ N, 110°20′ E). The silica gel-dried leaves of S. parviflora were sent to Genepioneer Biotechnologies (Nanjing, China) for sequencing. A tissue material of 30 mg was added to liquid nitrogen, and then DNA was extracted using the DP305 kit. The samples were sequenced on the NovaSeq 6000 sequencing platform, and the length of sequencing was 150 BP. High-quality clean data were obtained by removing low-quality sequences. The S. parviflora cp genome was assembled using the GetOrganelle v1.7.5.0 pipeline with the Stemona japonica cp genome as the reference [20]. The bandage was used to confirm that the assembly graph was a circle [21].
2.2. Chloroplast Genome Annoation
The annotation of S. parviflora cp genome was completed using the online program GeSeq https://chlorobox.mpimp-golm.mpg.de/geseq.html (accessed on 16 August 2021). Sequin software modifies the GenBank file of S. parviflora. The online tools at https://chlorobox.mpimp-golm.mpg.de/OGDraw.html (accessed on 16 August 2021) were used to visualize the structure map of the chloroplast genome. The cp genome of S. parviflora was submitted by the Banklt platform under the accession number MZ151339.
2.3. Condon Usage Bais and Repeat Sequence Analyses
The codon preferences of protein sequences encoded by five species of Stemona were counted by MEGA X software (https://www.megasoftware.net/dload_win_gui, accessed on 16 August 2021) [22]. The long repeat sequences were predicted by REPuter [23], with the minimum repeat size set at 30 and a Hamming distance of 3. MIcroSAtellite identification tool (MISA) software was used to check simple sequence repeats (SSRs), with 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, and pentanucleotide sequences [24].
2.4. Comparative Analysis of the Chloroplast Genomes
The IR/SSC and IR/LSC junctions of S. japonica, S. tuberosa, S. parviflora, S. mairei, and S. sessilifolia were compared using the online tool Irscope (https://irscope.shinyapps.io/irapp/ (accessed on 12 June 2022)). The online tool mVISTA (https://genome.lbl.gov/vista/mvista/submit.shtml (accessed on 12 June 2022)) was used to compare the complete chloroplast genomes of the five species, with the annotation of S. parviflora as the reference.
2.5. Ka/Ks Value Analyses
To understand the natural selection pressure in the evolution of the genus Stemona, homologous protein sequences between S. parviflora and other Stemona species were obtained using BLASTN. Then, the shared protein-coding genes were aligned using MAFFT version 7 [25]. The non-synonymous (Ka) and synonymous (Ks) ratios (Ka/Ks) were calculated using KaKs_Calculator version 2 [26].
2.6. Phylogenetic Analysis
To explore the phylogenetic relationships of S. japonica, S. tuberosa, S. parviflora, S. mairei, and S. sessilifolia, 80 protein-coding genes were extracted, maffted, and concatenated by PhyloSuite [27]. Bayesian Inference (BI) analyses were conducted using MrBayes version 3.2.6 in PhyloSuite, which is under the GTR + F + G4 model (two parallel runs, 1,000,000 generations) from ModelFinder.
2.7. Ecological Niche Modeling
The longitude and latitude information of S. parviflora was collected from published research papers and the National Specimen Information Infrastructure (http://www.nsii.org.cn/2017/home.php (accessed on 23 June 2022)) (Table S1). After coordinate point redundancy analysis, 19 effective distribution information points were reserved for subsequent analysis. Nineteen environmental and climatic factors were downloaded from WorldClim https://worldclim.org/data/index.html (accessed on 23 June 2022), and ENMTools version 1.4.0 software was used for the correlation analysis of environmental factors [28]. A pairwise correlation comparison between current climate factors and future climate factors was carried out, respectively, and the climate factors with a correlation coefficient greater than 0.8 (R2 ≥ |0.8|) were removed. Eight current climate factors have been retained, including mean diurnal range/°C (Bio2), Isothermality (Bio3), temperature (standard deviation) seasonality (Bio4), minimum temperature of coldest month/°C (Bio6), precipitation of wettest month/mm (Bio13), precipitation of driest month/mm (Bio14), precipitation of warmest quarter/mm (Bio18), and precipitation of coldest quarter/mm (Bio19). Nine future climate factors were retained for predicting the future potential distribution range, including mean diurnal range/°C (Bio2), minimum temperature of coldest month/°C (Bio6), annual temperature range/°C (Bio7), mean temperature of wettest quarter/°C (Bio8), annual precipitation/mm (Bio12), precipitation of driest month/mm (Bio14), precipitation seasonality (Coefficient of Variation)/mm (Bio15), precipitation of warmest quarter/mm (Bio18), and precipitation of coldest quarter/mm (Bio19) (Table S2). Based on the selected climate factors, MaxEnt version 3.4.4 was used to simulate the current and future (2050 s) potential distribution areas of the S. parviflora [29]. Of the distribution point data, 80% was used for the training set and 20% was used for random detection. The operation was repeated 10 times, and the number of iterations was 5000.
3. Results
3.1. General Features of the Chloroplast Genome
The chloroplast genome of S. parviflora had a conventional quadripartite structure characteristic of most land plants, with an LSC (82,410 bp), SSC (17,966 bp), and two IR regions (27,088 bp) (Figure 1). The length of the chloroplast genome of Stemona was about 15.4 KB, of which the longest size of the plastid genome was S. parviflora (154,552 bp). S. sessilifolia had the shortest chloroplast genome in size (154,037 bp). Among the five species of Stemona, the length difference of the LSC region was 461 bp, the length difference of the SSC region was 77 bp, and the length difference of IR regions was 22 bp. The IR regions had a relatively lower length variation compared with the LSC and SSC regions. The LSC region had a high variation in the length of the plastid genome. The GC content of S. sessilifolia and S. japonica was 38.0%; however, the GC content of S. parviflora and S. tuberosa accounted for 37.9%. S. mairei had the lowest GC content at 37.9%. The cp genome of S. japonica, S. tuberosa, S. parviflora, and S. mairei had 131 genes, including 87 protein-coding genes, 38 tRNA genes, 8 rRNA genes, and one pseudogene. One pseudogene was lost in the cp genome of S. sessilifolia (Table 1).
3.2. Long Repeat Sequence and SSR Analyses
In total, 62, 16, 3, 4, 5, and 6 SSRs represented by mono-, di-, tri-, tetra-, penta-, and hexanucleotides repeats were found in S. parviflora, respectively. Of the SSRs, 98, 106, 107, 96, and 111 were detected in the S. parviflora, S. sessilifloia, S. mairei, S. tuberose, and S. japonica chloroplast genome (Figure 2A). The LSC region harbored the largest number of SSRs, followed by the SSC region. The IR regions (IRa and IRb) had a relatively lower number of SSRs compared with the LSC and SSC regions, as the majority of land plants’ mononucleotides’ repeats represent the most abundant of SSRs in the plastid genome (Figure 2B).
A total of 49 long repeats were identified in the cp genome of S. parviflora, including 24 palindromic, 14 forward, 4 complement, and 7 reverse repeats. The size of the long repeats in S. parviflora ranged from 30 to 56 bp (Figure 2C). The number of four types of long repeats was the same compared with other congeneric species. However, there was a significantly different number of palindromic, forward, complement, and reverse repeats, respectively. For example, no complement repeats were detected in the cp genome of S. mairei; the rest of the four species harbored four, one, six, and seven complement repeats, respectively. Moreover, most long repeats fell into 30–35 bp (Figure 2D).
3.3. Comparative Analysis of Chloroplast Genome of Stemona
The complete chloroplast genome sequence of S. parviflora was compared with congeneric species (S. japonica, S. mairei, S. sessilifolia, and S. tuberosa) using the online genome alignment tool mVISTA (Figure 3). Large sequence variations were detected between S. parviflora and the other congeneric species. These variations were mainly distributed in the LSC and SSC regions. In the protein-coding gene regions, chloroplast genome sequence variations mainly appeared in rps16, trnQ-UUG, psbI, trnS-UGA, ycf3, trnS-GGA, trnL-UAA, psbJ, clpP, petD, rpl16, rpl22, and ycf1. The most divergent regions in the intergenic spacer regions were found in trnK-UUU_rps6, rps16_trnQ-UUG, trnS-GCU_trnG-UUC, atpF_atpH, atpH_atpI, rpoB_trnC-GCA, petN_psbM, psbM_trnD-GCU, trnT-UGU_trnL-UAA, ndhC_trnV-UAC, atpB_rbcL, rbcL_accD, accD_psaI, cemA_psbJ, psbE_petL, rpl20_rps12, ndhI_ndhJ, trnL-UAG_rpl32, and rpl32_ndhF. Like most other land plans, the IR regions were more conservative than the LSC and SSC regions.
3.4. IR Expansion and Contraction
The content and the number of genes in the boundary of IR/LSC and IR/SSC regions were compared among the five species of the genus Stemona (Figure 4). Except for S. sessilifolia, the rest of the four species contained the same content and the same number of genes in the IR/LSC and IR/SSC boundary, including rps3, rpl22, trnN, ycf1, ndhF, ψycf,1, and psbA. The gene rps3 is completely located in the LSC region with 6 bp away from the IRb region. The rpl22 and trnN genes are distributed in the LSC region. The gene ycf1 in S. sessilifolia was located in the SSC region with 23 bp away from IRb region; however, ycf1 in the rest of the four species all spanned the IRb/SSC boundary with 1206 bp extending to the IRb region. Furthermore, one pseudogene, ψycf1, was lost in the chloroplast genome of S. sessiliflora.
3.5. Codon Usage Bais of Stenoma Species
The average number of codons in Stemona was 22795. Stemona japonica had the largest number of codons at 22,808. The RSCU values of S. japonica, S. parviflora, S. mairei, and S. sessilifolia, all with 20 codons, were greater than 1, whereas the S. tuberosa with 29 codons was greater than 1. The A/U contents at the third codon position were the most preferred (Table S3). There was almost no difference in the RSCU value of Stemona species (Figure 5).
3.6. Selective Pressure Analyses
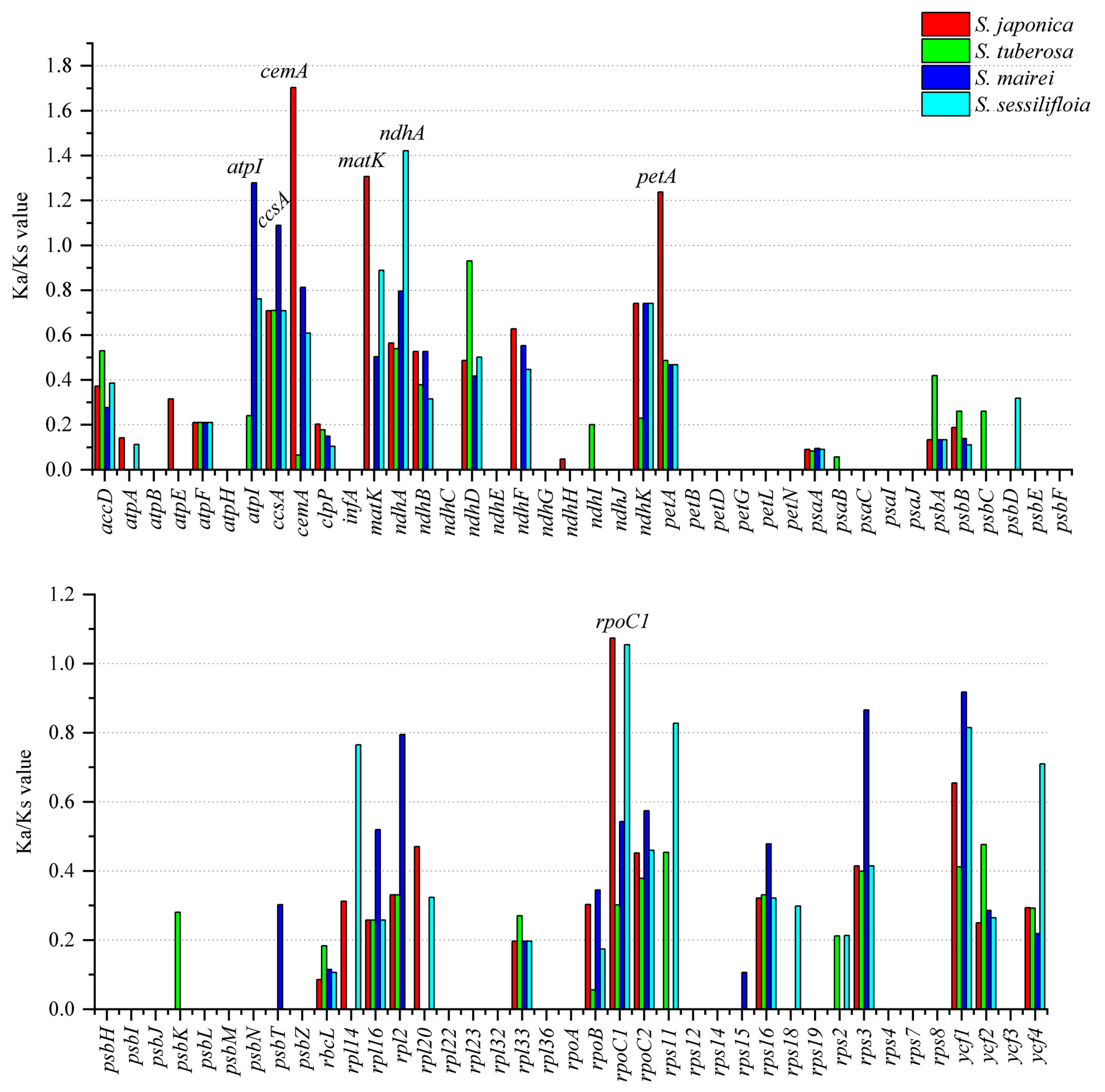
The non-synonymous (Ka) and synonymous (Ks) ratio (Ka/Ks) was calculated among the protein-coding genes of S. parviflora and four Stemona species (S. japonica, S. tuberosa, S. mairei, and S. sessilifloia) (Figure 6). The majority of protein-coding genes with Ka/Ks were valued less than one, indicating that these genes are under the purifying selection effect. The Ks/Ks values of atpI, ccsA, cemA, matK, ndhA, petA, and rpoC1 were greater than one, indicating that these genes are under positive selection.
3.7. Phylogenetic Relationships between S. parviflora and Other Stemona Species
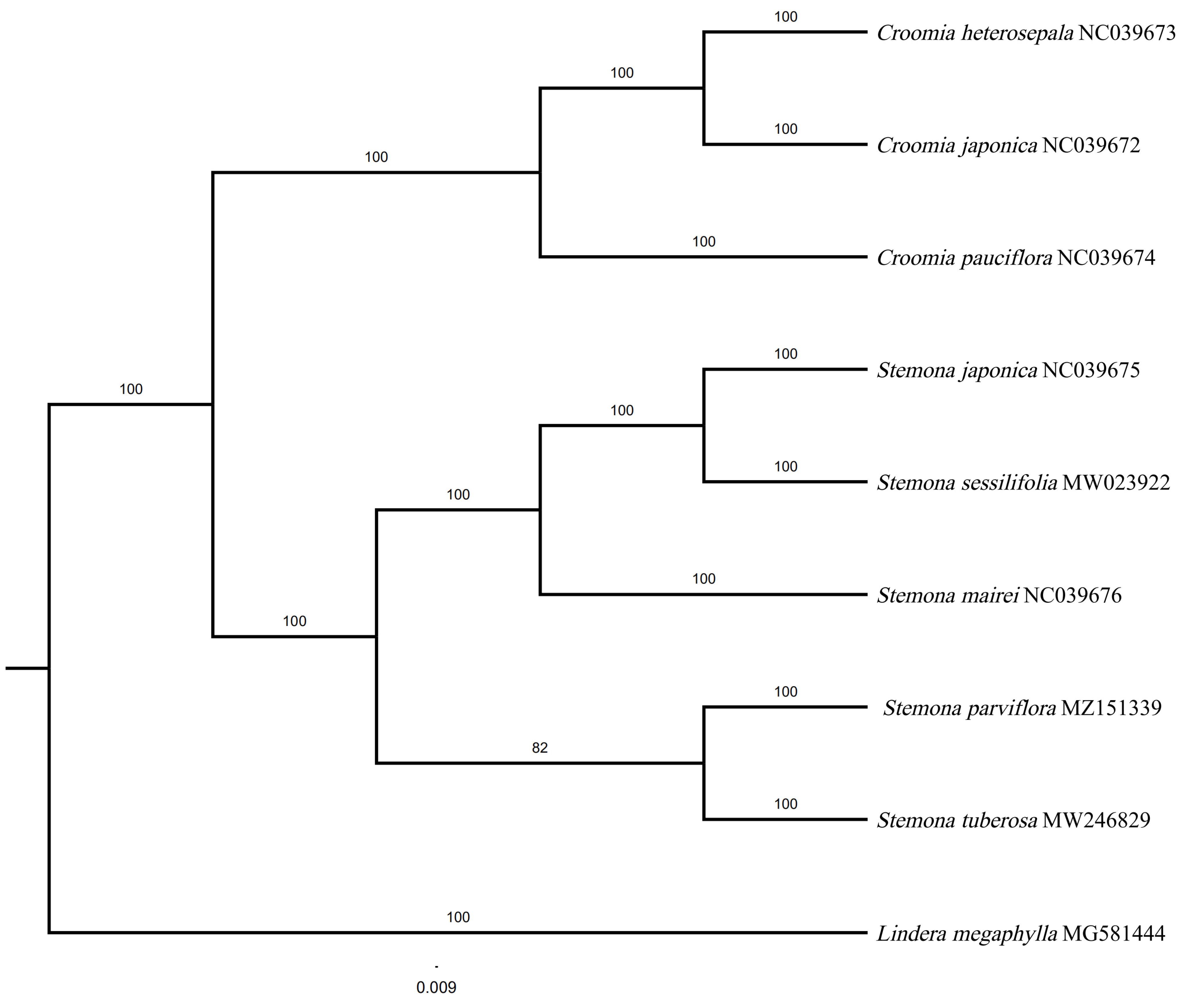
A BI tree was constructed based on 79 protein-coding genes using PhyloSuite (Figure 7). The tree received high bootstrap values in most nodes. In our phylogenetic tree, the monophyly of Stemona and Croomia received a high Bayesian posterior probability (100%). Stemona was sistered to Croomia with strong support (100%). The genus Croomia comprises three species worldwide (C. heterosepala, C. japonica, and C. pauciflora), of which only one species is distributed in China (C. japonica). C. japonica was closest to C. heterosepala. Five species in Stemona were mainly divided into two clades in the phylogenetic tree. S. japonica, S. sessilifolia, and S. mairei clustered to one clade. S. parviflora were sister to S. tuberosa, implying the close relationship between the two species.
3.8. Niche Analyses
The AUC values of the MaxEnt results were all greater than 0.95, indicating that the model is very reliable. The results of MaxEnt show that the southwest of Guangdong and Guangxi, the southwest of Taiwan, and the vast majority of Hainan are highly suitable areas for S. parviflora, of which Hainan is the main suitable area. The future simulation results show that Hainan is still the main and highest fitness area of the S. parviflora, the southeast of Sichuan has changed from a medium fitness area to a high fitness area, the whole territory of Taiwan has changed to a low fitness area and below, and the southeast of Guangdong and its coastal areas are still high fitness areas of the S. parviflora. It is worth noting that the junction of Yunnan Province, Sichuan Province, and Chongqing is also highly suitable for the survival of S. parviflora (Figure 8).
4. Discussion
The structure of cp genomes was conservative in the genus Stemona, as well as in most land plants. The average size of cp genomes in Stemona was larger than its sister genus Croomia [30]. As most reported in other land plants, the LSC region has a relatively higher length variation compared with the IR and SSC regions, suggesting the IR and SSC regions were more conservative [31]. The GC content of the chloroplast genome ranged from 24.78% to 57.664%; the average of this occupied 36.82 % [32]. The GC content varied between different species in Stemona. For example, the GC content of S. sessilifolia and S. japonica was 38.0%; however, S. parviflora, S. mairei, and S. tuberosa had a GC content that accounted for 37.9%. The differentiation of GC content in the same genus was also reported in other studies [33]. Pseudogenization was commonly in the evolution of chloroplast genomes, such as the accD, ccsA, ycf1, ycf2, ycf15, ycf86, rps19, and psbB pseudogenes [32,34,35,36]. One pseudogene, ψycf1, was lost in the cp genome of S. sessilifolia, but was detected in four other species of Stemona. The pseudogenization of the ycf1 gene may be interpreted as wastage in the course of species evolution, or as the fact that the species does not specifically need the gene. Moreover, the pseudogenization of ycf1 was also detected in the genera Paeonia and Aconitum [37,38,39].
In the course of evolution, species will form a set of adaptive codon usage patterns. The codon usage bias is similar, indicating that species have similar living environmental or close genetic relationships [40,41]. Codon usage bias analyses have been widely used to investigate the evolution and phylogeny of land plants [42]. The value of relative synonymous codon usage (RSCU) was greater than one, indicating that there was codon usage bias. There were 29 to 30 codons greater than one among the five plastid genomes. A higher A/U content at the third codon position was detected with high RSCU values. These were different from other species that enriched AT at the third codon position [43]. The RSCU value of the genus Stemona, with almost no differences, was consistent with sect. Moutan species [37].
SSR has been used for population genetic and phylogenetic analyses between intraspecific and interspecific levels [44]. A total of 98, 106, 107, 96, and 111 SSRs were detected in S. parviflora, S. sessilifloia, S. mairei, S. tuberose, and the S. japonica chloroplast genome, respectively. Mononucleotides and dinucleotides were the most abundant repeat type in the plastid genome, as has been reported in other species [45]. In addition, we found that most of the SSR distributed in the LSC and SSC regions are consistent with the plastid genome of Dracunculus clade (Aroideae, Araceae) [46]. Long repeats sequences were also analyzed in this study. A total of 49 long repeats of four types were detected in the plastid genome of S. parviflora. As reported in other species, palindromic repeats were the most abundant repeat type [47].
The length of the plastid genome and genes in the IR/SSC and IR/LSC regions may change during the contraction and expansion of the IR region [48,49]. Prior research has demonstrated that the length of gene extent to IR/SSC and IR/LSC is associated with systematic analyses [50]. The content and length of the gene in the IR/LSC and IR/SSC boundary regions vary from congeneric species, as has been reported in the genus Pedicularis [36]. The genes rps3 and trnN were more conservative compared with other genes in the genus Stemona. The genes rpl22, ycf1, and psbA from different species vary in gene length and distance from the IR/LSC and IR/SSC boundary regions. The genes ndhF and ψycf1 had the same pattern in the rest of the four species, except for S. sessilifloi.
As reported in other angiosperm species, the LSC region was the most divergent, and IR regions were the most conservative [51]. The inter-gene spacer was more divergent compared with protein-coding sequences [52]. Twelve divergence regions were found in protein-coding sequences, including rps16, trnQ-UUG, psbI, trnS-UGA, ycf3, trnS-GGA, trnL-UAA, psbJ, clpP, petD, rpl16, rpl22, and ycf1. The ycf1 gene had been used as DNA barcode for phylogenetic analyses [53]; moreover, significant variations were found in ycf1 from many taxa, such as the Quercus, Vicatia, and Curcuma chloroplast genome [54,55]. In addition, we found the protein-coding gene petD was the most divergent compared among the five species of Stemona. S. tuberosa had little difference compared with S. parvifloa in the gene petD, which may be due to the close phylogenetic relationship between the two species.
The value of Ka/Ks was related to gene adaptive evolution, such as positive selection and purification selection effect [56]. The genes that were under positive selection might result from natural selection and adaptation to the living environment [57]. Seven genes (atpI, ccsA, cemA, matK, ndhA, petA, and rpoC1) were under positive selection in genus Stemona. The positive selection gene matK has been widely reported in other land plants, such as Paphiopedilum [58], Chrysosplenium [59], and tribe Selineae species [60]. The genes atpI, ndhA, and petA were related to photosynthesis; prior research has implied that light plays an important role in gene mutation rates, and these positive selection genes might be associated with sunlight conditions [59]. Furthermore, except for rpoC1, the rest of the six positive selection genes only appeared in one species, indicating that genes in different species of Stemona may have undergone different degrees of selective evolutionary pressure. There was no gene under positive selection between S. parviflora and S. tuberosa, which may result from the overlapping habitats of these two species. Thus, environmental selection pressure between the two species was not obvious.
In our phylogenetic tree, the monophyly of Stenoma and Croomia was well-supported. Our results also support Stemona as a sister to the genus Croomia, which is consistent with prior research [30,61]. Two main branches were shown in the phylogenetic tree and had a Bayesian posterior probability of 100%. Clade 1 contains S. japonica, S. sessilifolia, and S. mairei. S. japonica were closest to S. sessilifolia. In clade 2, S. parviflora and S. tubersa were clustered in a single branch, implying the close relationship between the two species. The S. parviflora and S. tubeorsa clustered in one branch, consisting of phylogenetic analyses that were based on matK, rbcL, and psbK-psbI cpDNA markets [3].
Species are fundamental to biodiversity [62]. Predicting the potential distribution range in currently and in the future will be significant for the protection and management of endangered species [63]. A population genetic study that investigated S. parviflora was last performed five years ago; besides, AFLP molecular markers are insufficient to represent the genetic diversity of species and are rarely used in current research [4]. Genome-wide SNP data should reassess the population diversity of S. parviflora. Some protective measures have been proposed by previous research, such as seed germination, tissue culture, introduction and conservation, and pollination by the insect protection [4]. The highly suitable region of S. parviflora was not provided in the previous study, which makes it difficult for current introduction and cultivation measures. Fragmentation of the distribution range leads to S. parviflora being more vulnerable to threats. In addition, the actual distribution area is narrower than the simulation results. Limited by seed dispersal mechanisms (mainly by ants) [2], the spread distance of S. parviflora was is narrow. Although the niche simulation results show that most areas in Southwest Guangdong and Hainan are highly suitable for the survival of S. parviflora, due to the limited dispersal capacity of S. parviflora, it is necessary to carry out artificial grafts to expand the survival areas of S. parviflora. Furthermore, in the medium- and above-suitable regions revealed in this study, the whole territory of Taiwan has changed to a low fitness area and below in the 2050 s, which may not be suitable for the introduction and cultivation of S. parviflora. The suitability of the junction between Yunnan Province, Sichuan Province, and Chongqing for the survival of S. parviflora needs more transplant and cultivation measures to be verified. Overall, our results provide multiple highly suitable areas for S. parviflora, which may play a crucial role in current and future conservation measures.
5. Conclusions
In this study, the chloroplast genome of the endangered species S. parviflora was assembled and compared with four other Stemona species. The structure of the chloroplast genome was extremely conservative in the genus Stemona. Long repeat and SSR sequences were detected in this study, which may be used for population genetic analyses. We also compared the IR/LSC and IR/SSC boundary region, code usage bias, and the whole plastid genome. Seven genes (atpI, ccsA, cemA, matK, ndhA, petA, and rpoC1) were detected under positive selection. Phylogenetic analyses implied that S. parviflora was closest to S. tubersa. Niche simulation results showed that the southeast of Guangdong, the majority of Hainan, and Southwest Taiwan were highly suitable for S. parviflora survival in the current period. The changes in highly suitable areas in the future (2050s) are also discussed in this study.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13081361/s1, Table S1 Effective latitude and longitude coordinates of S. parviflora used in this study, Table S2 Correlation of 19 climatic factors, Table S3 Relative synonymous codon usage (RSCU) of 79 protein-coding genes of Stemona.
Author Contributions
Q.L. conceived of the project, designed the research, and conducted sequencing; R.W. and Q.L. wrote the paper; R.W. analyzed the data; Q.L. conducted the format review and modification. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The chloroplast genome sequences have been deposited in GenBank under the accession number: MZ151339. Row data are available at SRA under the accession number: PRJNA850147.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hu, C.; Kelso, S. Flora of China. In Flora of China; Science Press: Beijing, China, 1996; pp. 99–108. [Google Scholar]
- Lengyel, S.; Gove, A.D.; Latimer, A.M.; Majer, J.D.; Dunn, R.R. Convergent evolution of seed dispersal by ants, and phylogeny and biogeography in flowering plants: A global survey. Perspect. Plant Ecol. Evol. Syst. 2010, 12, 43–55. [Google Scholar] [CrossRef]
- Chen, Y.S.; Zeng, C.X.; Muellner-Riehl, A.N.; Wang, Z.H.; Sun, L.; Schinnerl, J.; Kongkiatpaiboon, S.; Kadota, Y.; Cai, X.H.; Chen, G. Invertebrate-mediated dispersal plays an important role in shaping the current distribution of a herbaceous monocot. J. Biogeogr. 2021, 48, 1101–1111. [Google Scholar] [CrossRef]
- Zhang, X.; Ge, J.; Yang, J.; Dunn, B.; Chen, G. Genetic diversity of Stemona parviflora: A threatened myrmecochorous medicinal plant in China. Biochem. Syst. Ecol. 2017, 71, 193–199. [Google Scholar] [CrossRef]
- Huang, S.Z.; Kong, F.D.; Ma, Q.Y.; Guo, Z.K.; Zhou, L.M.; Wang, Q.; Dai, H.F.; Zhao, Y.X. Nematicidal stemona alkaloids from stemona parviflora. J. Nat. Prod. 2016, 79, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Z.; Kong, F.D.; Chen, G.; Cai, X.H.; Zhou, L.M.; Ma, Q.Y.; Wang, Q.; Mei, W.L.; Dai, H.F.; Zhao, Y.X. A phytochemical investigation of Stemona parviflora roots reveals several compounds with nematocidal activity. Phytochemistry 2019, 159, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Yang, Y.; Dong, S.; He, Q.; Jia, Y.; Zhao, L.; Yu, S.; Liu, H.; Liu, B.; Yan, Y.; et al. Threatened species list of China’s higher plants. Biodivers. Sci. 2017, 25, 696–744. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.L.; Zhu, S.; Chen, H.B.; Yang, D.H.; Cai, S.Q.; Komatsu, K. Molecular analysis of Stemona plants in China based on sequences of four chloroplast DNA regions. Biol. Pharm. Bull. 2009, 32, 1439–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.X.; Zhang, W.Q.; Fu, C.X.; Li, E.X. Species delimitation of Stemona (Stemonaceae) based on sequences of five plastid DNA regions. Phytotaxa 2018, 374, 291–296. [Google Scholar] [CrossRef]
- Dai, X.; Wu, W.; Ji, L.; Tian, S.; Yang, B.; Guan, B.; Wu, D. MaxEnt model-based prediction of potential distributions of Parnassia wightiana (Celastraceae) in China. Biodivers. Data J. 2022, 10, e81073. [Google Scholar] [CrossRef]
- Zhang, K.; Yao, L.; Meng, J.; Tao, J. Maxent modeling for predicting the potential geographical distribution of two Peony species under climate change. Sci. Total Environ. 2018, 634, 1326–1334. [Google Scholar] [CrossRef]
- Soilhi, Z.; Sayari, N.; Benalouache, N.; Mekki, M. Predicting current and future distributions of Mentha pulegium L. in Tunisia under climate change conditions, using the MaxEnt model. Ecol. Inform. 2022, 68, 101533. [Google Scholar] [CrossRef]
- Wisz, M.S.; Hijmans, R.J.; Li, J.; Peterson, A.T.; Graham, C.H.; Guisan, A.; Elith, J.; Dudík, M.; Ferrier, S.; Huettmann, F.; et al. Effects of sample size on the performance of species distribution models. Divers. Distrib. 2008, 14, 763–773. [Google Scholar] [CrossRef]
- Miao, H.; Bao, J.; Li, X.; Ding, Z.; Tian, X. Comparative analyses of chloroplast genomes in ‘Red Fuji’ apples: Low rate of chloroplast genome mutations. PeerJ 2022, 10, e12927. [Google Scholar] [CrossRef]
- Lian, C.; Yang, H.; Lan, J.; Zhang, X.; Zhang, F.; Yang, J.; Chen, S. Comparative analysis of chloroplast genomes reveals phylogenetic relationships and intraspecific variation in the medicinal plant Isodon rubescens. PLoS ONE 2022, 17, e0266546. [Google Scholar] [CrossRef]
- Choi, K.S.; Park, K.T.; Park, S.J. The chloroplast genome of Symplocarpus renifolius: A comparison of chloroplast genome structure in araceae. Genes 2017, 8, 324. [Google Scholar] [CrossRef] [Green Version]
- Namgung, J.; Do, H.D.K.; Kim, C.; Choi, H.J.; Kim, J.-H. Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of Allioideae (Amaryllidaceae). Sci. Rep. 2021, 11, 3262. [Google Scholar] [CrossRef]
- Zhang, X.J.; Liu, K.J.; Wang, Y.C.; He, J.; Wu, Y.M.; Zhang, Z.X. Complete chloroplast genomes of three Salix species: Genome structures and phylogenetic analysis. Forests 2021, 12, 1681. [Google Scholar] [CrossRef]
- Liu, S.; Xu, Q.; Liu, K.; Zhao, Y.; Chen, N. Chloroplast Genomes for Five Skeletonema Species: Comparative and Phylogenetic Analysis. Front. Plant Sci. 2021, 12, 774617. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit Incorporating γ-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Warren, D.L.; Glor, R.E.; Turelli, M. ENMTools: A toolbox for comparative studies of environmental niche models. Ecography 2010, 33, 607–611. [Google Scholar] [CrossRef]
- Phillips, S.B.; Aneja, V.P.; Kang, D.; Arya, S.P. Modelling and analysis of the atmospheric nitrogen deposition in North Carolina. Ecol. Modell. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Ye, W.; Lu, R.; Xu, W.; Qiu, Y. Phylogenomic and comparative analyses of complete plastomes of Croomia and Stemona (Stemonaceae). Int. J. Mol. Sci. 2018, 19, 2383. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.H.; Liu, W.W.; Duan, B.Z.; Zhang, H.Z.; Chen, X.B.; Wang, Y.; Xia, C. long The first complete chloroplast genome of Vicatia thibeticade Boiss.: Genome features, comparative analysis, and phylogenetic relationships. Physiol. Mol. Biol. Plants 2022, 28, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Long, H.; Zhang, L.; Liu, Z.; Cao, H.; Shi, M.; Tan, X. The complete chloroplast genome sequence of tung tree (Vernicia fordii): Organization and phylogenetic relationships with other angiosperms. Sci. Rep. 2017, 7, 1869. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Tang, D.; Wei, K.; Qin, F.; Li, L.; Lin, Y.; Zhu, Y.; Khan, A.; Kashif, M.H.; Miao, J. The complete chloroplast genome sequence of the medicinal plant Sophora tonkinensis. Sci. Rep. 2020, 10, 12473. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, K.; Wiland-Szymańska, J.; Buczkowska-Chmielewska, K.; Drapikowska, M.; Maślak, M.; Myszczyński, K.; Szczecińska, M.; Ślipiko, M.; Sawicki, J. The complete chloroplast genome of a rare orchid species Liparis loeselii (L.). Conserv. Genet. Resour. 2018, 10, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.P.; Huang, J.P.; Wu, C.S.; Hsu, C.Y.; Chaw, S.M. Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome Biol. Evol. 2010, 2, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, J.B.; Wang, H.; Song, Y.; Corlett, R.T.; Yao, X.; Li, D.Z.; Yu, W. Bin Plastid NDH Pseudogenization and Gene Loss in a Recently Derived Lineage from the Largest Hemiparasitic Plant Genus Pedicularis (Orobanchaceae). Plant Cell Physiol. 2021, 62, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Nie, L.; Xu, Z.; Li, P.; Wang, Y.; He, C.; Song, J.; Yao, H. Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Three Paeonia Section Moutan Species (Paeoniaceae). Front. Genet. 2020, 11, 980. [Google Scholar] [CrossRef]
- Dong, B.R.; Zhao, Z.L.; Ni, L.H.; Wu, J.R.; Danzhen, Z.G. Comparative analysis of complete chloroplast genome sequences within Gentianaceae and significance of identifying species. Chin. Tradit. Herb. Drugs 2020, 51, 1641–1649. [Google Scholar] [CrossRef]
- Li, Q.; Xia, M.; Yu, J.; Chen, S.; Zhang, F. Plastid genome insight to the taxonomic problem for Aconitum pendulum and A. flavum (Ranunculaceae). Biologia 2022, 77, 953–966. [Google Scholar] [CrossRef]
- Shen, Z.; Gan, Z.; Zhang, F.; Yi, X.; Zhang, J.; Wan, X. Analysis of codon usage patterns in citrus based on coding sequence data. BMC Genom. 2020, 21, 234. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, Q.; Wang, Y.; Zhao, J.; Qiao, L.; Wu, B.; Yan, S.; Zheng, J.; Zheng, X. Comparative Analysis of Genomic and Transcriptome Sequences Reveals Divergent Patterns of Codon Bias in Wheat and Its Ancestor Species. Front. Genet. 2021, 12, 732432. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, Q.; Wang, Y.; Li, M.; Wang, C.; Wang, Z.; Jiao, C.; Xu, C.; Wang, H.; Zhang, Z. Comparative Analysis of Codon Bias in the Chloroplast Genomes of Theaceae Species. Front. Genet. 2022, 13, 824610. [Google Scholar] [CrossRef]
- Guo, X.L.; Zheng, H.Y.; Price, M.; Zhou, S.D.; He, X.J. Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome. Plants 2020, 9, 965. [Google Scholar] [CrossRef]
- Chen, X.D.; Yang, J.; Guo, Y.F.; Zhao, Y.M.; Zhou, T.; Zhang, X.; Ju, M.M.; Li, Z.H.; Zhao, G.F. Spatial Genetic Structure and Demographic History of the Dominant Forest Oak Quercus fabri Hance in Subtropical China. Front. Plant Sci. 2021, 11, 583284. [Google Scholar] [CrossRef] [PubMed]
- Chincoya, D.A.; Sanchez-Flores, A.; Estrada, K.; Díaz-Velásquez, C.E.; González-Rodríguez, A.; Vaca-Paniagua, F.; Dávila, P.; Arias, S.; Solórzano, S. Identification of high molecular variation loci in complete chloroplast genomes of Mammillaria (Cactaceae, caryophyllales). Genes 2020, 11, 830. [Google Scholar] [CrossRef] [PubMed]
- Abdullah; Henriquez, C.L.; Mehmood, F.; Hayat, A.; Sammad, A.; Waseem, S.; Waheed, M.T.; Matthews, P.J.; Croat, T.B.; Poczai, P.; et al. Chloroplast genome evolution in the Dracunculus clade (Aroideae, Araceae). Genomics 2021, 113, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Khayi, S.; Gaboun, F.; Pirro, S.; Tatusova, T.; El Mousadik, A.; Ghazal, H.; Mentag, R. Complete chloroplast genome of Argania spinosa: Structural organization and phylogenetic relationships in sapotaceae. Plants 2020, 9, 1354. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xing, Y.; Xu, L.; Bao, G.; Zhan, Z.; Yang, Y.; Wang, J.; Li, S.; Zhang, D.; Kang, T. Comparative analysis of the complete chloroplast genome sequences of six species of Pulsatilla Miller, Ranunculaceae. Chin. Med. 2019, 14, 53. [Google Scholar] [CrossRef] [Green Version]
- Li, Q. The Complete Chloroplast Genomes of Primula obconica Provide Insight That Neither Species nor Natural Section Represent Monophyletic Taxa in Primula (Primulaceae). Genes 2022, 13, 567. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Guo, S.; Yin, Y.; Zhang, J.; Yin, X.; Liang, C.; Wang, Z.; Huang, B.; Liu, Y.; Xiao, S.; et al. Complete chloroplast genome sequence and phylogenetic analysis of Aster tataricus. Molecules 2018, 23, 2426. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.X.; Xian, Y.F.; Xiang, L.; Zhang, D.; Shi, Y.H.; Wu, M.L.; Dong, G.Q.; Ip, S.P.; Lin, Z.X.; Wu, L.; et al. Complete chloroplast genomes from Sanguisorba: Identity and variation among four species. Molecules 2018, 23, 2137. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Zhang, Y.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Yang, R. The Complete Chloroplast Genome Sequences of 14 Curcuma Species: Insights Into Genome Evolution and Phylogenetic Relationships Within Zingiberales. Front. Genet. 2020, 11, 802. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Gao, J.; Liao, P.C.; Li, J.Q.; Ma, W.B. Insights Into Comparative Analyses and Phylogenomic Implications of Acer (Sapindaceae) Inferred From Complete Chloroplast Genomes. Front. Genet. 2022, 12, 791628. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ye, H.; Zhang, N.; Ma, J.; Wang, J.; Hu, G.; Li, M.; Zhao, P. Comparative Analyses of Chloroplast Genomes Provide Comprehensive Insights into the Adaptive Evolution of Paphiopedilum (Orchidaceae). Horticulturae 2022, 8, 391. [Google Scholar] [CrossRef]
- Wu, Z.; Liao, R.; Yang, T.; Dong, X.; Lan, D.; Qin, R.; Liu, H. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genom. 2020, 21, 621. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Aou, X.; Tian, R.; Li, Z.; Peng, C.; He, X. Complete Chloroplast Genome of Cnidium monnieri (Apiaceae) and Comparisons with Other Tribe Selineae Species. Diversity 2022, 14, 323. [Google Scholar] [CrossRef]
- Mennes, C.B.; Smets, E.F.; Moses, S.N.; Merckx, V.S.F.T. New insights in the long-debated evolutionary history of Triuridaceae (Pandanales). Mol. Phylogenet. Evol. 2013, 69, 994–1004. [Google Scholar] [CrossRef]
- Hausdorf, B.; Hennig, C. Species delimitation using dominant and codominant multilocus markers. Syst. Biol. 2010, 59, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ren, Y.; Huang, Q.; Deng, X.; Chen, C.; Deng, H. Habitat suitability assessment of ebdangered plant Alsophila spinulosa in Chishui River area based on GIS and Maxent model. Acta Ecol. Sin. 2021, 41, 6123–6133. [Google Scholar] [CrossRef]
Figure 1.
Chloroplast genome map of Stemona parviflora.

Figure 2.
Distribution of repeats sequence in Stemona. (A) Number of SSR in the IR, SSC, and LSC regions; (B) Number of SSR in the five species of Stemona; (C) Number of four types of repeats in Stemona; (D) Length of four types of repeat sequences.
Figure 2.
Distribution of repeats sequence in Stemona. (A) Number of SSR in the IR, SSC, and LSC regions; (B) Number of SSR in the five species of Stemona; (C) Number of four types of repeats in Stemona; (D) Length of four types of repeat sequences.

Figure 3.
Comparison of Stemona chloroplast genomes using mVISTA, with S. parviflora chloroplast genome as reference.
Figure 3.
Comparison of Stemona chloroplast genomes using mVISTA, with S. parviflora chloroplast genome as reference.

Figure 4.
Comparisons of LSC, SSC, and IRs junctions among Stemona species.

Figure 5.
Codon distribution of protein-coding genes of the chloroplast genomes of Stemona species.

Figure 6.
Detection of positive selection sites of chloroplast genes in Stemona, with S. parviflora as a reference genome.
Figure 6.
Detection of positive selection sites of chloroplast genes in Stemona, with S. parviflora as a reference genome.

Figure 7.
Molecular phylogenetic tree of Stemona using Bayesian inference method.

Figure 8.
Potential distribution areas of S. parviflora in the current and future periods.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparative analysis of the complete chloroplast genome of genus Stemona.
| Species | Length | LSC | SSC | IR | GC | Number of Gene | tRNA | rRNA | Pesedogene |
| S. japonica | 154,224 bp | 82,117 bp | 17,943 bp | 27,082 bp | 38.0% | 134 | 38 | 8 | 1 |
| S. sessilifolia | 154,037 bp | 81,949 bp | 17,952 bp | 27,068 bp | 38.0% | 133 | 38 | 8 | 0 |
| S. parviflora | 154,552 bp | 82,410 bp | 17,966 bp | 27,088 bp | 37.9% | 134 | 38 | 8 | 1 |
| S. tuberosa | 154,374 bp | 82,305 bp | 17,929 bp | 27,070 bp | 37.9% | 134 | 38 | 8 | 1 |
| S. mairei | 154,307 bp | 82,254 bp | 17,889 bp | 27,082 bp | 38.0% | 134 | 38 | 8 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wei, R.; Li, Q. The Complete Chloroplast Genome of Endangered Species Stemona parviflora: Insight into the Phylogenetic Relationship and Conservation Implications. Genes 2022, 13, 1361. https://doi.org/10.3390/genes13081361
AMA Style
Wei R, Li Q. The Complete Chloroplast Genome of Endangered Species Stemona parviflora: Insight into the Phylogenetic Relationship and Conservation Implications. Genes. 2022; 13(8):1361. https://doi.org/10.3390/genes13081361
Chicago/Turabian StyleWei, Ran, and Qiang Li. 2022. "The Complete Chloroplast Genome of Endangered Species Stemona parviflora: Insight into the Phylogenetic Relationship and Conservation Implications" Genes 13, no. 8: 1361. https://doi.org/10.3390/genes13081361
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.