
Inter-Specific Genetic Exchange Despite Strong Divergence in Deep-Sea Hydrothermal Vent Gastropods of the Genus Alviniconcha

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
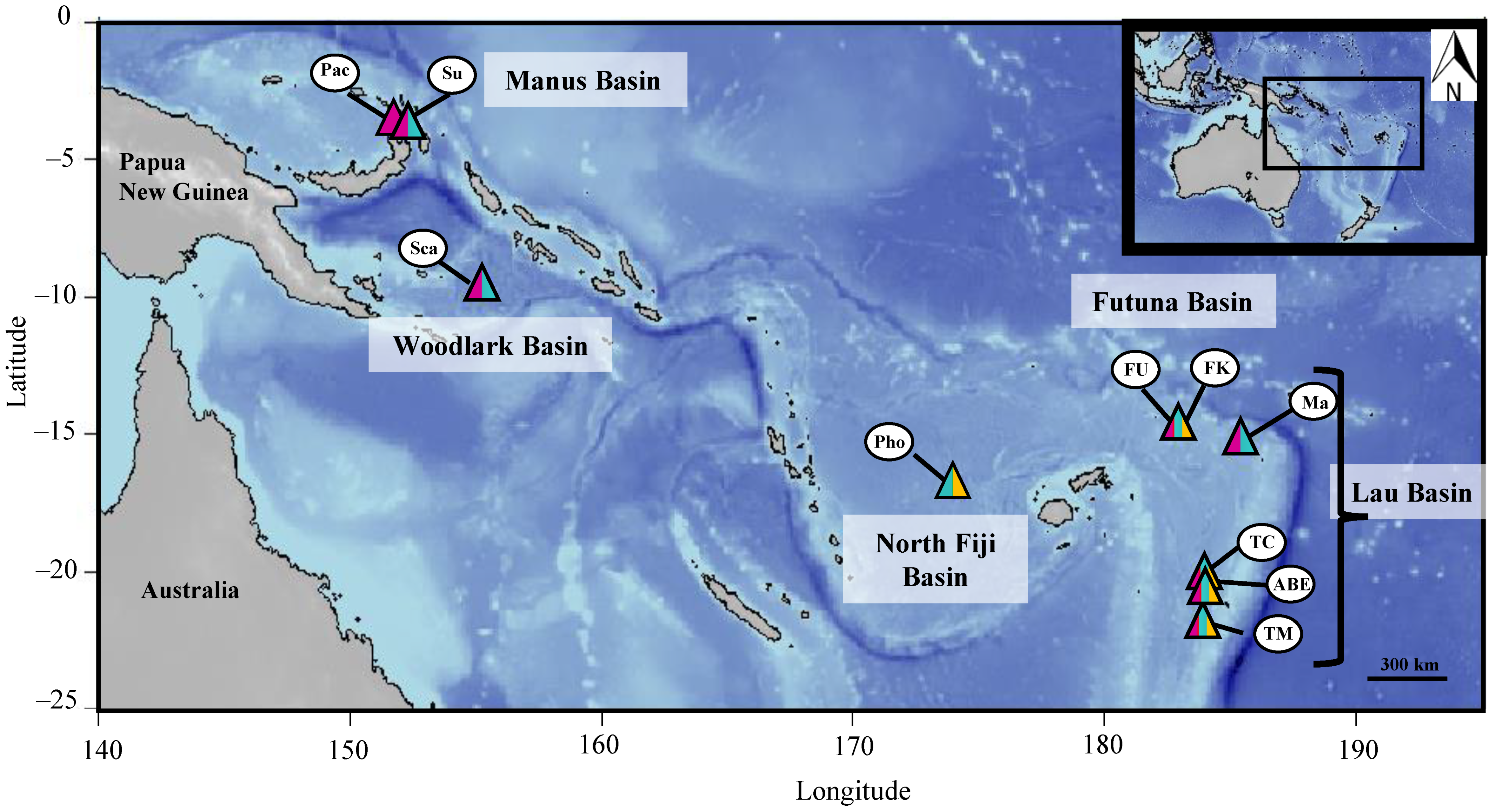
2.1. Sampling
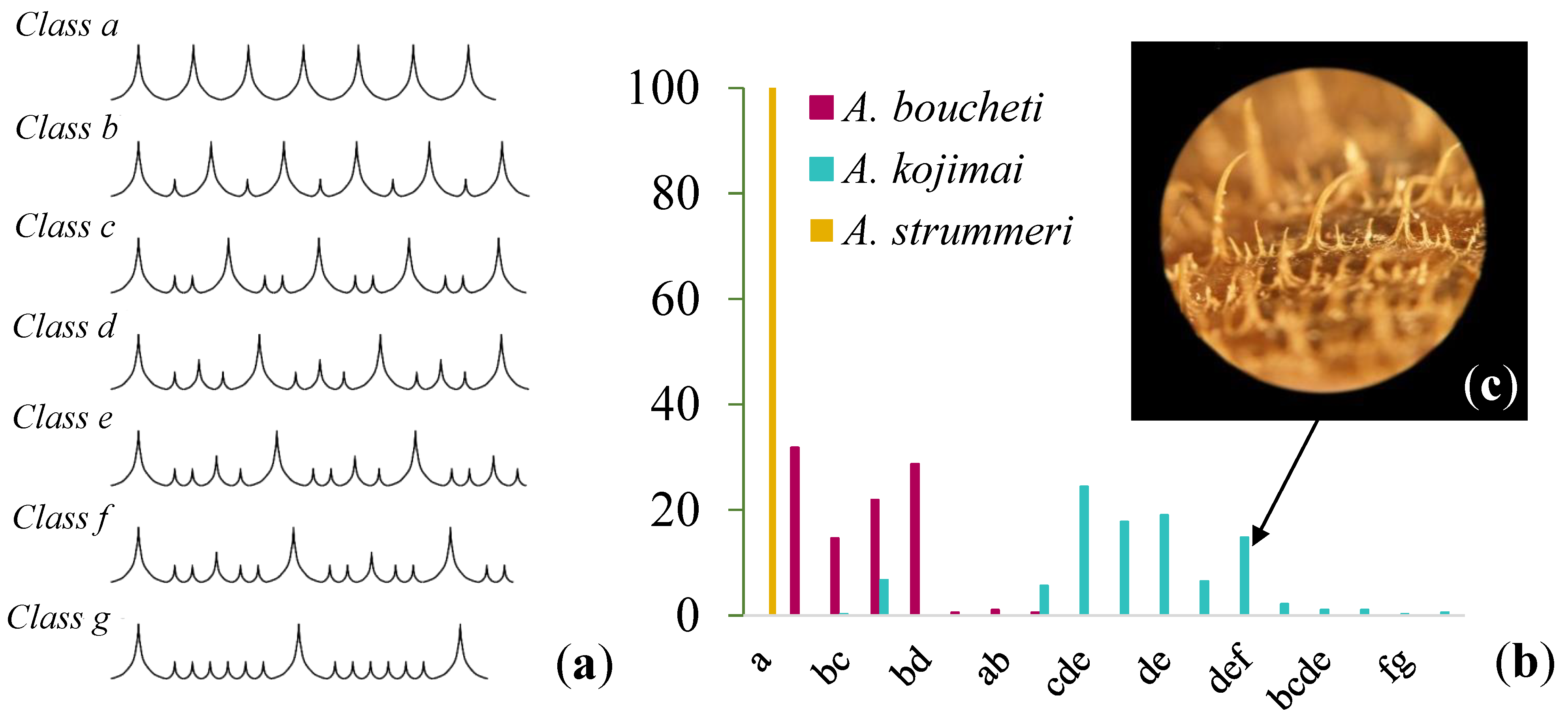
2.2. Morphological Analysis
2.3. Mitochondrial Cox1 Sequence Analysis
2.4. Nuclear Genome Analysis
2.4.1. ddRAD Library Preparation
2.4.2. Bioinformatic Filtering of Illumina Reads
2.4.3. Population Structure, Divergence, and Admixture
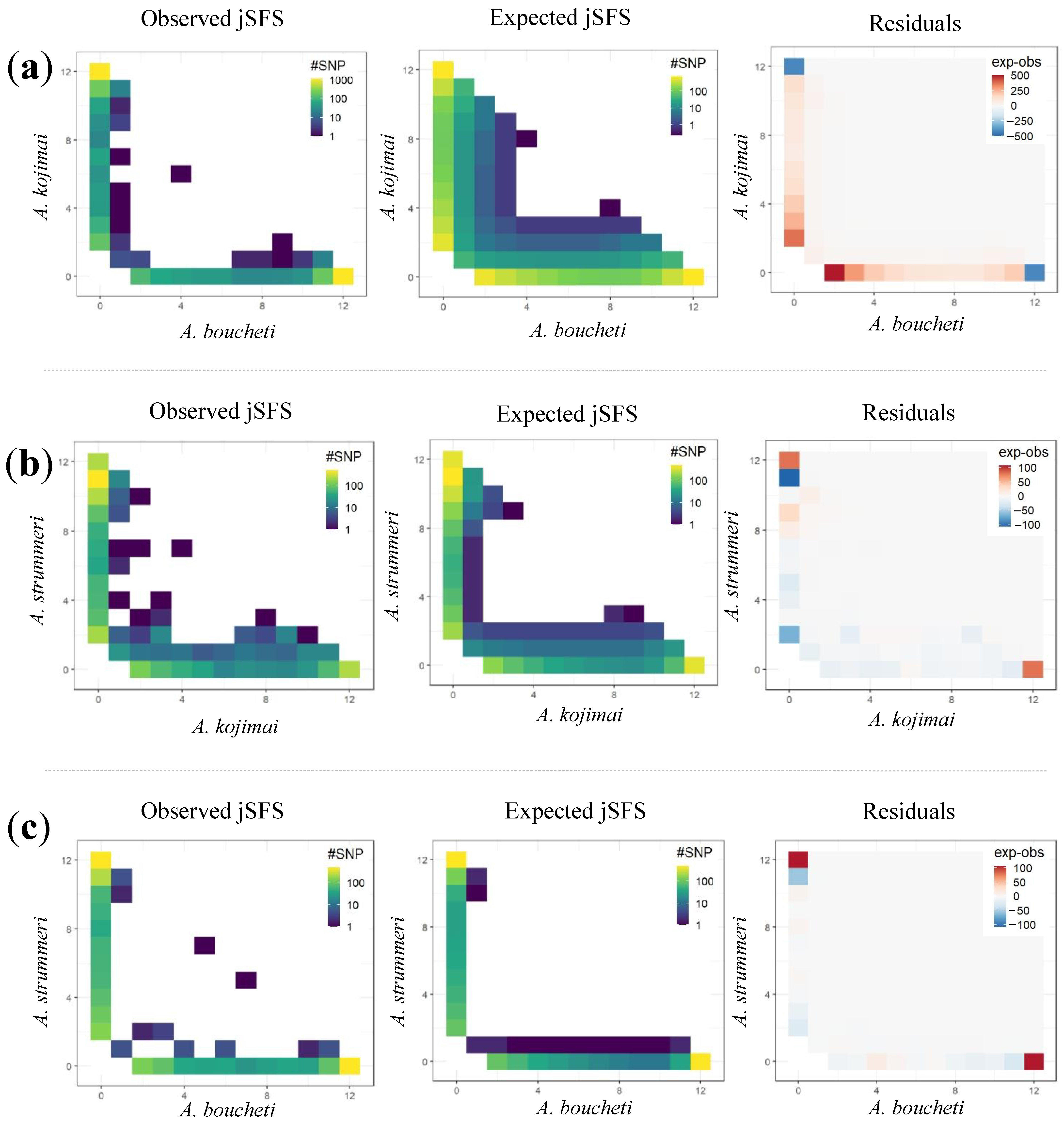
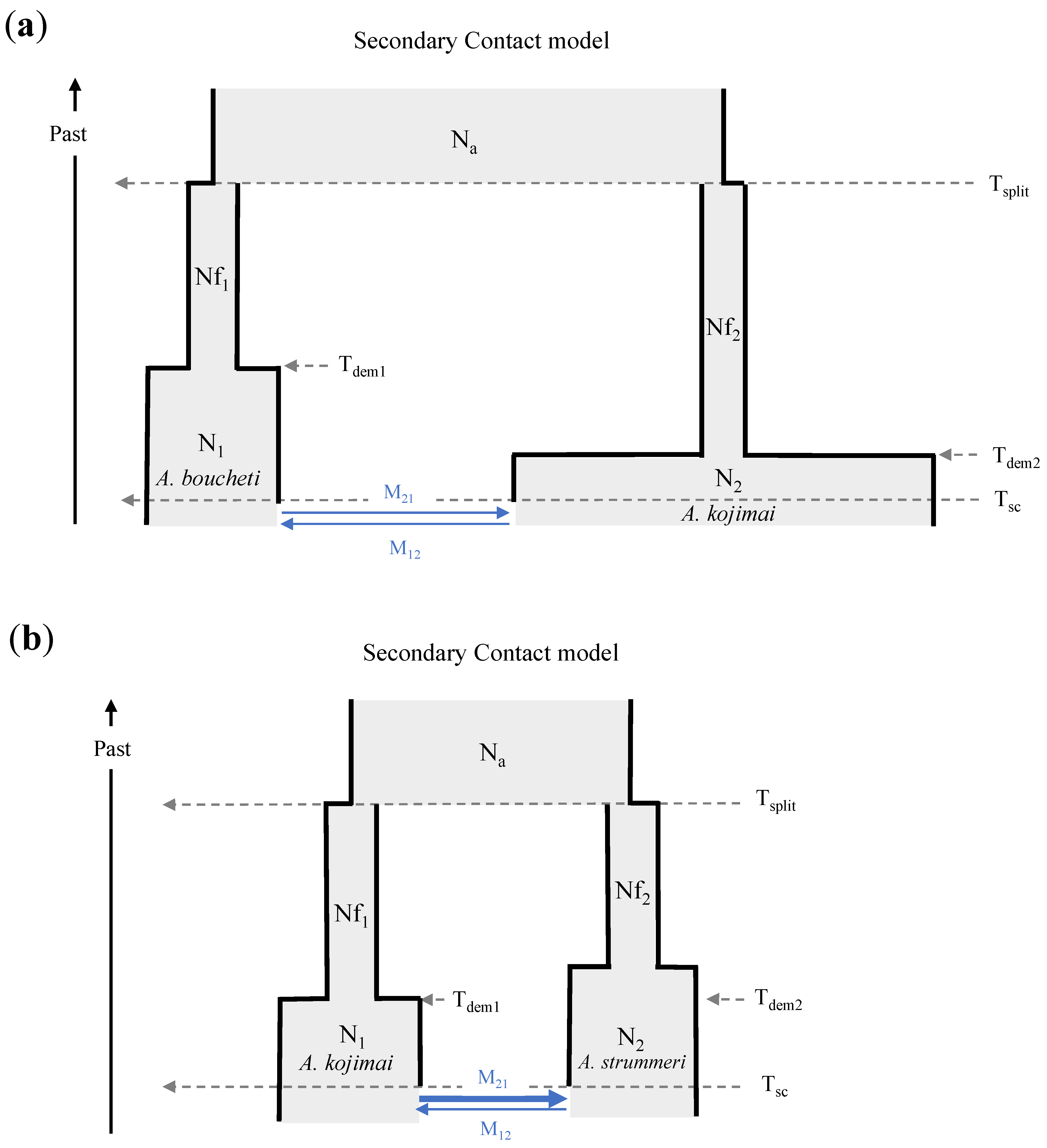
2.4.4. Demographic Inference
2.5. Transcriptome Analysis
3. Results
3.1. Species Distribution
3.2. Morphology
3.3. Mitochondrial Cox1 Gene Analysis
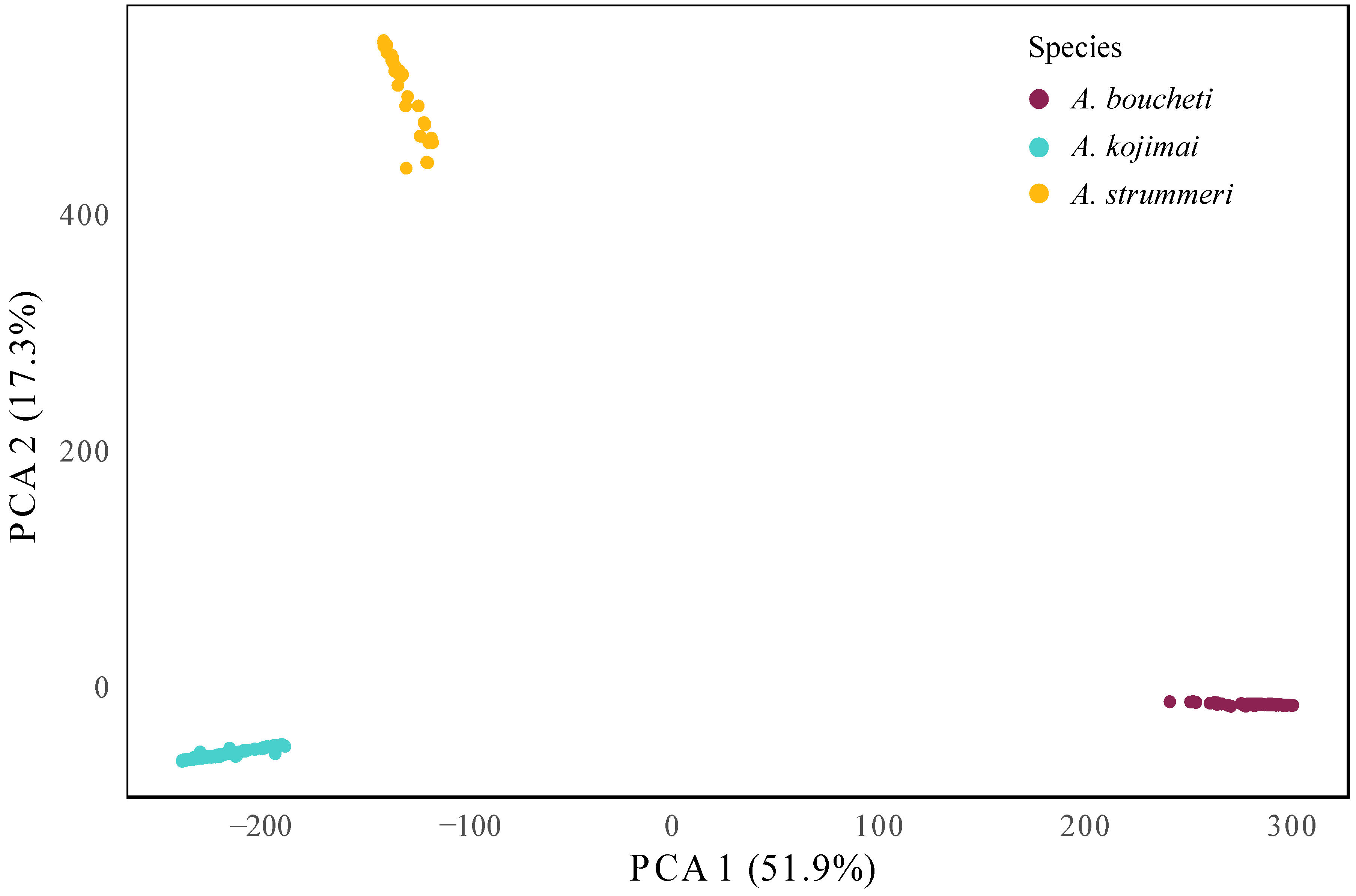
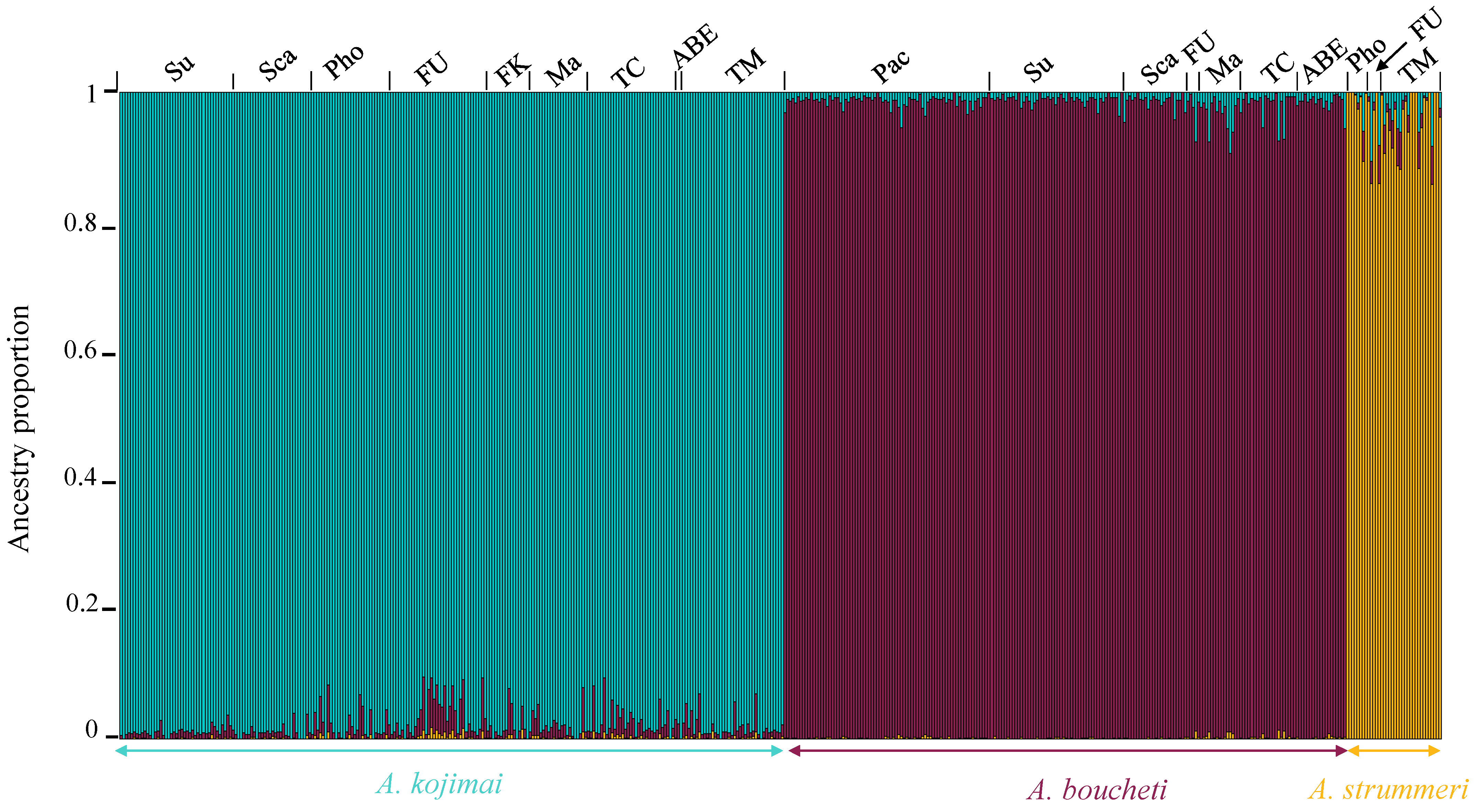
3.4. ddRAD-seq Analysis along the Nuclear Genome
3.5. Divergences in Coding Sequences
4. Discussion
4.1. Hydrothermal Vents in the Western Pacific Are Home to Three Sympatric Alviniconcha Species
4.2. A Long History of Divergence in Allopatry
4.3. Historical Scenarios of Divergence
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lonsdale, P. Clustering of suspension-feeding macrobenthos near abyssal hydrothermal vents at oceanic spreading centers. Deep Sea Res. 1977, 24, 857–863. [Google Scholar] [CrossRef]
- Tunnicliffe, V. The biology of hydrothermal vents: Ecology and evolution. Ocean. Mar. Biol. Annu. Rev. 1991, 29, 319–407. [Google Scholar]
- Moalic, Y.; Desbruyères, D.; Duarte, C.M.; Rozenfeld, A.F.; Bachraty, C.; Arnaud-Haond, S. Biogeography Revisited with Network Theory: Retracing the History of Hydrothermal Vent Communities. Syst. Biol. 2012, 61, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matabos, M.; Jollivet, D. Revisiting the Lepetodrilus elevatus species complex (Vetigastropoda: Lepetodrilidae), using samples from the Galápagos and Guaymas hydrothermal vent systems. J. Molluscan Stud. 2019, 85, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Vrijenhoek, R.C. Gene Flow and Genetic Diversity in Naturally Fragmented Metapopulations of Deep-Sea Hydrothermal Vent Animals. J. Hered. 1997, 88, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Jollivet, D.; Chevaldonné, P.; Planque, B. Hydrothermal-Vent Alvinellid Polychaete Dispersal in the Eastern Pacific. 2. a Metapopulation Model Based on Habitat Shifts. Evolution 1999, 53, 1128–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bris, N.; Sarradin, P.-M.; Caprais, J.-C. Contrasted sulphide chemistries in the environment of 13° N EPR vent fauna. Deep Sea Res. Part Oceanogr. Res. Pap. 2003, 50, 737–747. [Google Scholar] [CrossRef]
- Fontanillas, E.; Galzitskaya, O.V.; Lecompte, O.; Lobanov, M.Y.; Tanguy, A.; Mary, J.; Girguis, P.R.; Hourdez, S.; Jollivet, D. Proteome Evolution of Deep-Sea Hydrothermal Vent Alvinellid Polychaetes Supports the Ancestry of Thermophily and Subsequent Adaptation to Cold in Some Lineages. Genome Biol. Evol. 2017, 9, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Reeves, E.P.; Seewald, J.S.; Saccocia, P.; Bach, W.; Craddock, P.R.; Shanks, W.C.; Sylva, S.P.; Walsh, E.; Pichler, T.; Rosner, M. Geochemistry of hydrothermal fluids from the PACMANUS, Northeast Pual and Vienna Woods hydrothermal fields, Manus Basin, Papua New Guinea. Geochim. Cosmochim. Acta 2011, 75, 1088–1123. [Google Scholar] [CrossRef] [Green Version]
- Ondréas, H.; Cannat, M.; Fouquet, Y.; Normand, A. Geological context and vents morphology of the ultramafic-hosted Ashadze hydrothermal areas (Mid-Atlantic Ridge 13° N). Geochem. Geophys. Geosyst. 2012, 13, 11. [Google Scholar] [CrossRef]
- Podowski, E.L.; Moore, T.S.; Zelnio, K.A.; Luther, G.W.; Fisher, C.R. Distribution of diffuse flow megafauna in two sites on the Eastern Lau Spreading Center, Tonga. Deep Sea Res. Part Oceanogr. Res. Pap. 2009, 56, 2041–2056. [Google Scholar] [CrossRef]
- Podowski, E.L.; Ma, S.; Iii, G.W.L.; Wardrop, D.; Fisher, C.R. Biotic and abiotic factors affecting distributions of megafauna in diffuse flow on andesite and basalt along the Eastern Lau Spreading Center, Tonga. Mar. Ecol. Prog. Ser. 2011, 418, 25–45. [Google Scholar] [CrossRef]
- Johnson, S.B.; Warén, A.; Tunnicliffe, V.; Dover, C.V.; Wheat, C.G.; Schultz, T.F.; Vrijenhoek, R.C. Molecular taxonomy and naming of five cryptic species of Alviniconcha snails (Gastropoda: Abyssochrysoidea) from hydrothermal vents. Syst. Biodivers. 2015, 13, 278–295. [Google Scholar] [CrossRef]
- Breusing, C.; Johnson, S.B.; Tunnicliffe, V.; Clague, D.A.; Vrijenhoek, R.C.; Beinart, R.A. Allopatric and Sympatric Drivers of Speciation in Alviniconcha Hydrothermal Vent Snails. Mol. Biol. Evol. 2020, 37, 3469–3484. [Google Scholar] [CrossRef]
- Wu, C.-I. The genic view of the process of speciation. J. Evol. Biol. 2001, 14, 851–865. [Google Scholar] [CrossRef]
- Coyne, J.A.; Orr, H.A. Speciation, Volume 37 Sunderland; Sinauer Associates: Sunderland, MA, USA, 2004. [Google Scholar]
- Havird, J.C.; Sloan, D.B. The Roles of Mutation, Selection, and Expression in Determining Relative Rates of Evolution in Mitochondrial versus Nuclear Genomes. Mol. Biol. Evol. 2016, 33, 3042–3053. [Google Scholar] [CrossRef]
- Decker, S.K.; Ammerman, L.K. Phylogeographic analysis reveals mito-nuclear discordance in Dasypterus intermedius. J. Mammal. 2020, 101, 1400–1409. [Google Scholar] [CrossRef]
- Toews, D.P.L.; Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 2012, 21, 3907–3930. [Google Scholar] [CrossRef]
- Lima, T.G.; Burton, R.S.; Willett, C.S. Genomic scans reveal multiple mito-nuclear incompatibilities in population crosses of the copepod Tigriopus californicus. Evolution 2019, 73, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Pante, E.; Becquet, V.; Viricel, A.; Garcia, P. Investigation of the molecular signatures of selection on ATP synthase genes in the marine bivalve Limecola balthica. Aquat. Living Resour. 2019, 32, 3. [Google Scholar] [CrossRef]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J.W.; Baird, S.J.E.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C.A.; Buggs, R.; et al. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelkens, R.; Seehausen, O. Genetic Distance between Species Predicts Novel Trait Expression in Their Hybrids. Evolution 2009, 63, 884–897. [Google Scholar] [CrossRef] [Green Version]
- Roux, C.; Fraïsse, C.; Romiguier, J.; Anciaux, Y.; Galtier, N.; Bierne, N. Shedding Light on the Grey Zone of Speciation along a Continuum of Genomic Divergence. PLoS Biol. 2016, 14, e2000234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.T.; Jollivet, D.; Gentil, F.; Thiébaut, E.; Viard, F. Sharp genetic break between Atlantic and English Channel populations of the polychaete Pectinaria koreni, along the North coast of France. Heredity 2005, 94, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muths, D.; Jollivet, D.; Gentil, F.; Davoult, D. Large-scale genetic patchiness among NE Atlantic populations of the brittle star Ophiothrix fragilis. Aquat. Biol. 2009, 5, 117–132. [Google Scholar] [CrossRef]
- Roux, C.; Tsagkogeorga, G.; Bierne, N.; Galtier, N. Crossing the Species Barrier: Genomic Hotspots of Introgression between Two Highly Divergent Ciona intestinalis Species. Mol. Biol. Evol. 2013, 30, 1574–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laming, S.R.; Hourdez, S.; Cambon-Bonavita, M.-A.; Pradillon, F. Classical and computed tomographic anatomical analyses in a not-so-cryptic Alviniconcha species complex from hydrothermal vents in the SW Pacific. Front. Zool. 2020, 17, 12. [Google Scholar] [CrossRef]
- Denis, F.; Jollivet, D.; Moraga, D. Genetic separation of two allopatric populations of hydrothermal snails Alviniconcha spp. (Gastropoda) from two South Western Pacific back-arc basins. Biochem. Syst. Ecol. 1993, 21, 431–440. [Google Scholar] [CrossRef]
- Kojima, S.; Segawa, R.; Fijiwara, Y.; Fujikura, K.; Ohta, S.; Hashimoto, J. Phylogeny of Hydrothermal-Vent–Endemic Gastropods Alviniconcha spp. from the Western Pacific Revealed by Mitochondrial DNA Sequences. Biol. Bull. 2001, 200, 298–304. [Google Scholar] [CrossRef]
- Pante, E.; Simon-Bouhet, B. Marmap: A Package for Importing, Plotting and Analyzing Bathymetric and Topographic Data in R. PLoS ONE 2013, 8, e73051. [Google Scholar] [CrossRef]
- Chiu, Y.-W.; Chen, H.-C.; Lee, S.-C.; Chen, C.A. Morphometric Analysis of Shell and Operculum Variations in the Viviparid Snail, Cipangopaludina chinensis (Mollusca: Gastropoda), in Taiwan. Zool. Stud. 2002, 41, 321–331. [Google Scholar]
- Thioulouse, J.; Dray, S.; Dufour, A.-B.; Siberchicot, A.; Jombart, T.; Pavoine, S. Multivariate Analysis of Ecological Data with Ade4; Springer: New York, NY, USA, 2018; ISBN 978-1-4939-8848-8. [Google Scholar]
- Ripley, B.; Venables, B.; Bates, D.M.; Hornik, K.; Gebhardt, A.; Firth, D.; Ripley, M.B. Package ‘mass’. Cran R 2013, 538, 113–120. [Google Scholar]
- Doyle, J.J.; Dickson, E.E. Preservation of Plant Samples for Dna Restriction Endonuclease Analysis. Taxon 1987, 36, 715–722. [Google Scholar] [CrossRef]
- Jolly, M.; Viard, F.; Weinmayr, G.; Gentil, F.; Thiébaut, E.; Jollivet, D. Does the genetic structure of Pectinaria koreni (Polychaeta: Pectinariidae) conform to a source—Sink metapopulation model at the scale of the Baie de Seine? Helgol. Mar. Res. 2003, 56, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Kojima, S.; Sasaki, T.; Suzuki, M.; Utsumi, T.; Watanabe, H.; Urakawa, H.; Tsuchida, S.; Nunoura, T.; Hirayama, H.; et al. Host-Symbiont Relationships in Hydrothermal Vent Gastropods of the Genus Alviniconcha from the Southwest Pacific. Appl. Environ. Microbiol. 2006, 72, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for de Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Brelsford, A.; Dufresnes, C.; Perrin, N. High-density sex-specific linkage maps of a European tree frog (Hyla arborea) identify the sex chromosome without information on offspring sex. Heredity 2016, 116, 177–181. [Google Scholar] [CrossRef]
- Daguin-Thiébaut, C. Construction of Individual ddRAD Libraries V.1. 2021. Available online: https://www.protocols.io/view/construction-of-individual-ddrad-libraries-q26g7bwo9lwz/v1 (accessed on 10 February 2022).
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and Genotyping Loci de Novo from Short-Read Sequences. GenesGenomesGenetics 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Frichot, E.; Mathieu, F.; Trouillon, T.; Bouchard, G.; François, O. Fast and Efficient Estimation of Individual Ancestry Coefficients. Genetics 2014, 196, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraïsse, C.; Popovic, I.; Mazoyer, C.; Spataro, B.; Delmotte, S.; Romiguier, J.; Loire, É.; Simon, A.; Galtier, N.; Duret, L.; et al. DILS: Demographic inferences with linked selection by using ABC. Mol. Ecol. Resour. 2021, 21, 2629–2644. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Belshaw, R.; Katzourakis, A. BlastAlign: A program that uses blast to align problematic nucleotide sequences. Bioinformatics 2005, 21, 122–123. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z.; dos Reis, M. Statistical Properties of the Branch-Site Test of Positive Selection. Mol. Biol. Evol. 2011, 28, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinart, R.A.; Sanders, J.G.; Faure, B.; Sylva, S.P.; Lee, R.W.; Becker, E.L.; Gartman, A.; Luther, G.W.; Seewald, J.S.; Fisher, C.R.; et al. Evidence for the role of endosymbionts in regional-scale habitat partitioning by hydrothermal vent symbioses. Proc. Natl. Acad. Sci. USA 2012, 109, E3241–E3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wares, J.P.; Cunningham, C.W. Phylogeography and Historical Ecology of the North Atlantic Intertidal. Evolution 2001, 55, 2455–2469. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Evolution of the mutation rate. Trends Genet. 2010, 26, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrijenhoek, R.C. Cryptic species, phenotypic plasticity, and complex life histories: Assessing deep-sea faunal diversity with molecular markers. Deep Sea Res. Part II Top. Stud. Oceanogr. 2009, 56, 1713–1723. [Google Scholar] [CrossRef]
- Matabos, M.; Plouviez, S.; Hourdez, S.; Desbruyères, D.; Legendre, P.; Warén, A.; Jollivet, D.; Thiébaut, E. Faunal changes and geographic crypticism indicate the occurrence of a biogeographic transition zone along the southern East Pacific Rise. J. Biogeogr. 2011, 38, 575–594. [Google Scholar] [CrossRef]
- Warèn, A.; Bouchet, P. New records, species, genera, and a new family of gastropods from hydrothermal vents and hydrocarbon seeps. Zool. Scr. 1993, 22, 1–90. [Google Scholar] [CrossRef]
- Boulart, C.; Rouxel, O.; Scalabrin, C.; Le Meur, P.; Pelleter, E.; Poitrimol, C.; Thiébaut, E.; Matabos, M.; Castel, J.; Tran Lu, Y.A.; et al. Active hydrothermal vents in the Woodlark Basin may act as dispersing centres for hydrothermal fauna. Commun. Earth Environ. 2022, 3, 64. [Google Scholar] [CrossRef]
- Beinart, R.A.; Nyholm, S.V.; Dubilier, N.; Girguis, P.R. Intracellular Oceanospirillales inhabit the gills of the hydrothermal vent snail Alviniconcha with chemosynthetic, γ-Proteobacterial symbionts. Environ. Microbiol. Rep. 2014, 6, 656–664. [Google Scholar] [CrossRef]
- Duda, T.F. Patterns of variation of mutation rates of mitochondrial and nuclear genes of gastropods. BMC Ecol. Evol. 2021, 21, 13. [Google Scholar] [CrossRef]
- Allio, R.; Donega, S.; Galtier, N.; Nabholz, B. Large Variation in the Ratio of Mitochondrial to Nuclear Mutation Rate across Animals: Implications for Genetic Diversity and the Use of Mitochondrial DNA as a Molecular Marker. Mol. Biol. Evol. 2017, 34, 2762–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayral, P.; Melo-Ferreira, J.; Glémin, S.; Bierne, N.; Carneiro, M.; Nabholz, B.; Lourenco, J.M.; Alves, P.C.; Ballenghien, M.; Faivre, N.; et al. Reference-Free Population Genomics from Next-Generation Transcriptome Data and the Vertebrate–Invertebrate Gap. PLoS Genet. 2013, 9, e1003457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbiza, L.; Dopazo, J.; Dopazo, H. Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome. PLoS Comput. Biol. 2006, 2, e38. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Ina, Y. Variation in synonymous substitution rates among mammalian genes and the correlation between synonymous and nonsynonymous divergences. J. Mol. Evol. 1995, 41, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Scally, A.; Durbin, R. Revising the human mutation rate: Implications for understanding human evolution. Nat. Rev. Genet. 2012, 13, 745–753. [Google Scholar] [CrossRef]
- Tine, M.; Kuhl, H.; Gagnaire, P.-A.; Louro, B.; Desmarais, E.; Martins, R.S.T.; Hecht, J.; Knaust, F.; Belkhir, K.; Klages, S.; et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat. Commun. 2014, 5, 5770. [Google Scholar] [CrossRef] [Green Version]
- Tyler, P.A.; Young, C.M. Reproduction and dispersal at vents and cold seeps. J. Mar. Biol. Assoc. UK 1999, 79, 193–208. [Google Scholar] [CrossRef]
- Chevaldonné, P.; Jollivet, D.; Desbruyères, D.; Lutz, R.; Vrijenhoek, R. Sister-species of eastern Pacific hydrothermal vent worms (Ampharetidae, Alvinellidae, Vestimentifera) provide new mitochondrial COI clock calibration. Cah. Biol. Mar. 2002, 43, 367–370. [Google Scholar]
- Johnson, S.B.; Young, C.R.; Jones, W.J.; Warén, A.; Vrijenhoek, R.C. Migration, Isolation, and Speciation of Hydrothermal Vent Limpets (Gastropoda; Lepetodrilidae) across the Blanco Transform Fault. Biol. Bull. 2006, 210, 140–157. [Google Scholar] [CrossRef]
- Schellart, W.P.; Lister, G.S.; Toy, V.G. A Late Cretaceous and Cenozoic reconstruction of the Southwest Pacific region: Tectonics controlled by subduction and slab rollback processes. Earth-Sci. Rev. 2006, 76, 191–233. [Google Scholar] [CrossRef]
- Hall, R. Cenozoic geological and plate tectonic evolution of SE Asia and the SW Pacific: Computer-based reconstructions, model and animations. J. Asian Earth Sci. 2002, 20, 353–431. [Google Scholar] [CrossRef]
- Taylor, B.; Goodliffe, A.; Martinez, F.; Hey, R. Continental rifting and initial sea-floor spreading in the Woodlark basin. Nature 1995, 374, 534–537. [Google Scholar] [CrossRef]
- Auzende, J.-M.; Ishibashi, J.; Beaudoin, Y.; Charlou, J.-L.; Delteil, J.; Donval, J.-P.; Fouquet, Y.; Gouillou, J.-P.; Ildefonse, B.; Kimura, H.; et al. Les extrémités orientale et occidentale du bassin de Manus, Papouasie-Nouvelle-Guinée, explorées par submersible: La campagne Manaute. Comptes Rendus Académie Sci. Ser. IIA Earth Planet. Sci. 2000, 331, 119–126. [Google Scholar] [CrossRef]
- Mitarai, S.; Watanabe, H.; Nakajima, Y.; Shchepetkin, A.F.; McWilliams, J.C. Quantifying dispersal from hydrothermal vent fields in the western Pacific Ocean. Proc. Natl. Acad. Sci. USA 2016, 113, 2976–2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breusing, C.; Johnson, S.B.; Mitarai, S.; Beinart, R.A.; Tunnicliffe, V. Differential patterns of connectivity in Western Pacific hydrothermal vent metapopulations: A comparison of biophysical and genetic models. Evol. Appl. 2021, 1–14. [Google Scholar] [CrossRef]
- Bouchemousse, S.; Liautard-Haag, C.; Bierne, N.; Viard, F. Distinguishing contemporary hybridization from past introgression with postgenomic ancestry-informative SNPs in strongly differentiated Ciona species. Mol. Ecol. 2016, 25, 5527–5542. [Google Scholar] [CrossRef] [Green Version]
- Pinceel, J.; Jordaens, K.; Backeljau, T. Extreme mtDNA divergences in a terrestrial slug (Gastropoda, Pulmonata, Arionidae): Accelerated evolution, allopatric divergence and secondary contact. J. Evol. Biol. 2005, 18, 1264–1280. [Google Scholar] [CrossRef]
- Hamilton, Z.R.; Johnson, M.S. Hybridization between genetically and morphologically divergent forms of Rhagada (Gastropoda: Camaenidae) snails at a zone of secondary contact. Biol. J. Linn. Soc. 2015, 114, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Muths, D.; Davoult, D.; Gentil, F.; Jollivet, D. Incomplete cryptic speciation between intertidal and subtidal morphs of Acrocnida brachiata (Echinodermata: Ophiuroidea) in the Northeast Atlantic. Mol. Ecol. 2006, 15, 3303–3318. [Google Scholar] [CrossRef]
- Hauser, T.P. Frost sensitivity of hybrids between wild and cultivated carrots. Conserv. Genet. 2002, 3, 73–76. [Google Scholar] [CrossRef]
- Brannock, P.M.; Hilbish, T.J. Hybridization results in high levels of sterility and restricted introgression between invasive and endemic marine blue mussels. Mar. Ecol. Prog. Ser. 2010, 406, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Maheshwari, S.; Barbash, D.A. The Genetics of Hybrid Incompatibilities. Annu. Rev. Genet. 2011, 45, 331–355. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | K | H | Hd | π | |
|---|---|---|---|---|---|
| Mitochondrial (Cox1; 599 bp; 722 ind) | |||||
| A. kojimai | 454 | 89 | 102 | 0.8 | 0.003 |
| A. boucheti | 243 | 67 | 62 | 0.91 | 0.004 |
| A. strummeri | 25 | 31 | 19 | 0.96 | 0.008 |
| Transcriptomes (RNAseq; 1,186,131 bp; 7 ind) | |||||
| A. kojimai | 2 | 10,672 | 2 | 1 | 0.004 |
| A. boucheti | 3 | 10,851 | 3 | 1 | 0.003 |
| A. strummeri | 2 | 12,010 | 2 | 1 | 0.005 |
| Genome (ddRAD-seq; 498 ind) | |||||
| A. kojimai | 250 | 21,397 | 0.0013 | ||
| A. boucheti | 212 | 40,879 | 0.0014 | ||
| A. strummeri | 36 | 25,801 | 0.0014 |
| dA | dXY | FST | dN/dS | dN | dS | |
|---|---|---|---|---|---|---|
| Mitochondrial (Cox1; 599 bp; 722 ind) | ||||||
| A. kojimai/A. boucheti | 0.123 | 0.126 | 0.974 | 0.006 | 0.005 | 0.784 |
| A. kojimai/A. strummeri | 0.086 | 0.091 | 0.961 | 0.015 | 0.007 | 0.468 |
| A. boucheti/A. strummeri | 0.118 | 0.124 | 0.967 | 0.003 | 0.002 | 0.793 |
| Transcriptomes (RNAseq; 1,186,131 bp; 7 ind) | ||||||
| A. kojimai/A. boucheti | 0.028 | 0.031 | 0.133 | 0.013 | 0.097 | |
| A. kojimai/A. strummeri | 0.016 | 0.020 | 0.124 | 0.008 | 0.062 | |
| A. boucheti/A. strummeri | 0.027 | 0.031 | 0.134 | 0.013 | 0.096 | |
| Genome (ddRAD-seq; 60,084 SNPs; 498 ind) | ||||||
| A. kojimai/A. boucheti | 0.031 | 0.031 | 0.922 | |||
| A. kojimai/A. strummeri | 0.018 | 0.018 | 0.842 | |||
| A. boucheti/A. strummeri | 0.031 | 0.031 | 0.917 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castel, J.; Hourdez, S.; Pradillon, F.; Daguin-Thiébaut, C.; Ballenghien, M.; Ruault, S.; Corre, E.; Tran Lu Y, A.; Mary, J.; Gagnaire, P.-A.; et al. Inter-Specific Genetic Exchange Despite Strong Divergence in Deep-Sea Hydrothermal Vent Gastropods of the Genus Alviniconcha. Genes 2022, 13, 985. https://doi.org/10.3390/genes13060985
Castel J, Hourdez S, Pradillon F, Daguin-Thiébaut C, Ballenghien M, Ruault S, Corre E, Tran Lu Y A, Mary J, Gagnaire P-A, et al. Inter-Specific Genetic Exchange Despite Strong Divergence in Deep-Sea Hydrothermal Vent Gastropods of the Genus Alviniconcha. Genes. 2022; 13(6):985. https://doi.org/10.3390/genes13060985
Chicago/Turabian StyleCastel, Jade, Stéphane Hourdez, Florence Pradillon, Claire Daguin-Thiébaut, Marion Ballenghien, Stéphanie Ruault, Erwan Corre, Adrien Tran Lu Y, Jean Mary, Pierre-Alexandre Gagnaire, and et al. 2022. "Inter-Specific Genetic Exchange Despite Strong Divergence in Deep-Sea Hydrothermal Vent Gastropods of the Genus Alviniconcha" Genes 13, no. 6: 985. https://doi.org/10.3390/genes13060985