
Phenotypic Expression of Known and Novel Hemoglobin A2-Variants, Hemoglobin A2-Mae Phrik [Delta 52(D3) Asp > Gly, HBD:c.158A > G], Associated with Hemoglobin E [Beta 26(B8) Glu > Lys, HBB:c.79G > A] in Thailand

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
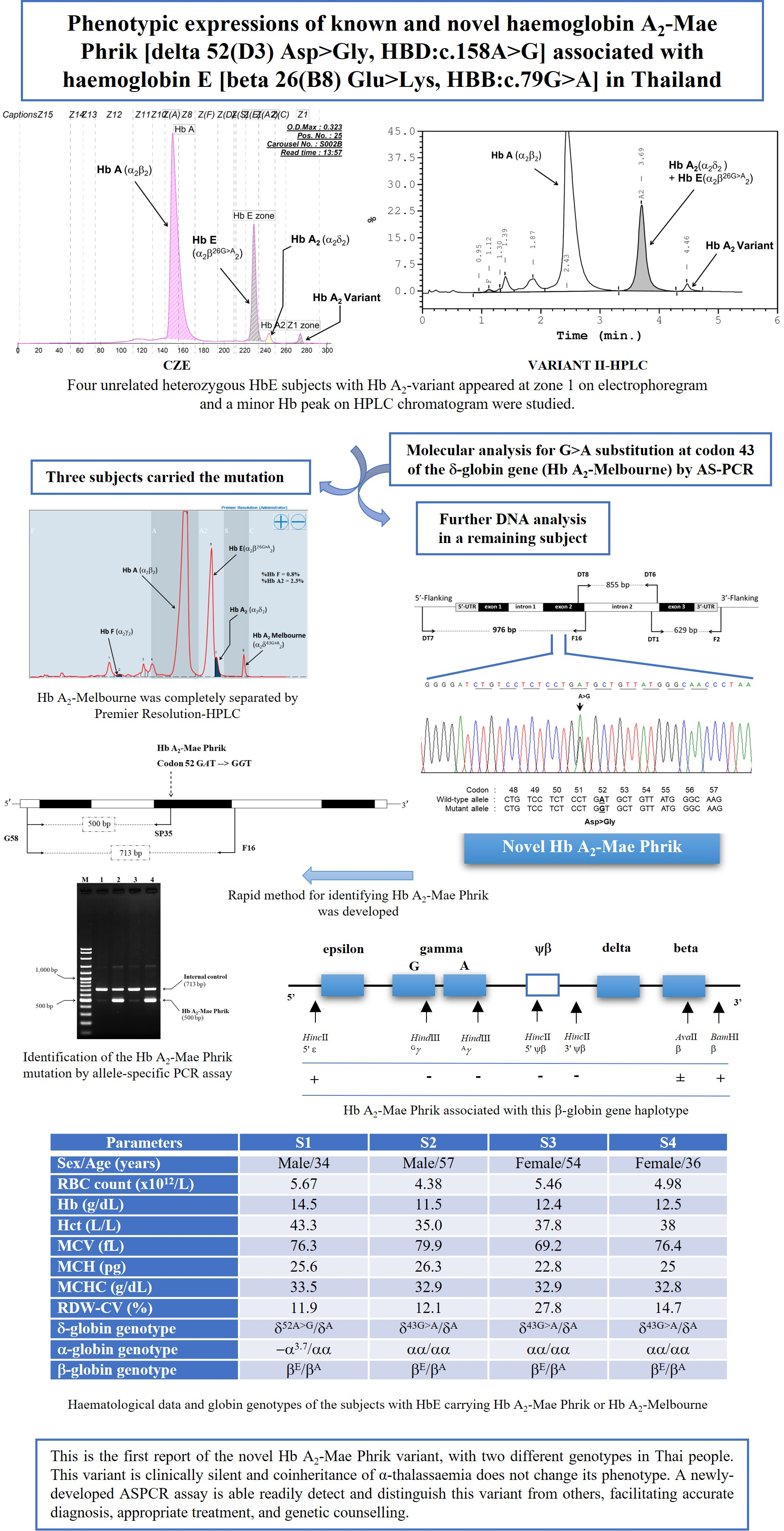
2.1. Subjects and Hematological Studies
2.2. DNA Analysis
2.3. β-Globin Gene Haplotype Analysis
2.4. Development of An Allele-Specific Polymerase Chain Reaction Assay (ASPCR) for Identification of the Hb A2-Mae Phrik Variant
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mosca, A.; Paleari, R. The role of haemoglobin A(2) testing in the diagnosis of thalassaemias and related haemoglobinopathies. J. Clin. Pathol. 2009, 62, 13–17. [Google Scholar] [CrossRef]
- Giambona, A.; Passarello, C.; Renda, D.; Maggio, A. The significance of the hemoglobin A(2) value in screening for hemoglobinopathies. Clin. Biochem. 2009, 42, 1786–1796. [Google Scholar] [CrossRef]
- HbVar, A Database of Human Hemoglobin Variants and Thalassemias. Available online: http://globin.cse.psu.edu/hbvar/menu.html (accessed on 17 April 2022).
- Waye, J.S.; Eng, B.; Hellens, L.; Hohenadel, B.A.; Nakamura, L.M.; Walker, L. Normal Hb A2 β-thalassemia trait: Frameshift mutation (HBB: c.187_251dup) in cis with the Hb A2’ δ-globin gene missense mutation (HBD: c.49G>C). Hemoglobin 2013, 37, 201–204. [Google Scholar] [CrossRef]
- Intasai, N.; Phasit, A.; Panyasai, S.; Pornprasert, S. A Case Report of Compound Heterozygosity for β0/β+-Thalassemia resulting from under diagnosed β-Thalassemia found in a Hb A’2 Sample. Hemoglobin 2019, 43, 63–65. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.; Mao, T.; Zhang, X.; Liang, L.; Meng, M. Detection of a Hb A2 -Melbourne (HBD: c.130G>A) combined with β-thalassemia in a Chinese individual. J. Clin. Lab. Anal. 2020, 34, e23401. [Google Scholar] [CrossRef]
- Kasmi, C.; Amri, Y.; Hadj-Fredj, S.; Oueslati, S.; Dabboussi, M.; Mahjoub, R.; Hammami, S.; Aljane, I.; Mami, F.B.; Jamoussi, H.; et al. Analysis of δ-globin gene alleles in Tunisians: Description of three new delta-thalassemia mutations. Mol. Biol. Rep. 2021, 48, 5923–5933. [Google Scholar] [CrossRef]
- Fucharoen, S.; Winichagoon, P. Haemoglobinopathies in southeast Asia. Indian J. Med. Res. 2011, 134, 498–506. [Google Scholar]
- Srivorakun, H.; Singha, K.; Fucharoen, G.; Sanchaisuriya, K.; Fucharoen, S. A large cohort of hemoglobin variants in Thailand: Molecular epidemiological study and diagnostic consideration. PLoS ONE 2014, 9, e108365. [Google Scholar] [CrossRef] [Green Version]
- Fucharoen, S.; Winichagoon, P. Hemoglobinopathies in Southeast Asia. Hemoglobin 1987, 11, 65–88. [Google Scholar] [CrossRef]
- Chaibunruang, A.; Fucharoen, G.; Fucharoen, S. First description of a Hb A2 variant in Thailand. Identification of Hb A2-Melbourne [δ43(CD2)Glu→Lys] in Thai individuals. Hemoglobin 2012, 36, 80–84. [Google Scholar] [CrossRef]
- Panyasai, S.; Fucharoen, G.; Fucharoen, S. Known and new hemoglobin A2 variants in Thailand and implication for β-thalassemia screening. Clin. Chim. Acta 2015, 438, 226–230. [Google Scholar] [CrossRef]
- Nuinoon, M.; Jeenduang, N.; Kesornsit, A.; Horpet, D.; Plyduang, T. Hematological and Molecular Characterization of a Novel Hb A2 Variant with Homozygous α-Thalassemia-2 in a Southern Thai Individual. Hemoglobin 2017, 41, 213–215. [Google Scholar] [CrossRef]
- Panyasai, S.; Pornprasert, S. Association of Hb A2 variants with several forms of α- and β-Thalassemia in Thailand. Hemoglobin 2020, 44, 179–183. [Google Scholar] [CrossRef]
- Prajantasen, T.; Prayalaw, P.; Panyasai, S.; Binlee, S.; Nongnuan, S. Development of a High Resolution Melting Curve Analysis for the Detection of Hemoglobin δ-Chain Variants in Thailand and Identification of Hb A2-Walsgrave [codon 52 (GAT>CAT), Asp→His; HBD:c.157G>C] in a Pregnant Woman from Southern Thailand. Genet. Test Mol. Biomark. 2021, 25, 426–433. [Google Scholar] [CrossRef]
- Singha, K.; Fucharoen, G.; Fucharoen, S. δ-Hemoglobinopathies in Thailand: Screening, molecular basis, genotype-phenotype interaction, and implication for prevention and control of thalassemia. Ann. Hematol. 2021, 100, 1953–1963. [Google Scholar] [CrossRef]
- Jomoui, W.; Panichchob, P.; Rujirachaivej, P.; Panyasai, S.; Tepakhan, W. Coinheritance of Hb A2-Melbourne (HBD: c.130G>A) and Hb E (HBB: c.79G>A) in Laos and Simultaneous High Resolution Melt Detection of Hb A2-Melbourne and Hb A2-Lampang (HBD: c.142G>A) in a Single Tube. Hemoglobin 2019, 43, 214–217. [Google Scholar] [CrossRef]
- Sea-ung, N.; Fucharoen, G.; Sanchaisuriya, K.; Fucharoen, S. Alpha 0-Thalassemia and related disorder in Northeast Thailand: A molecular and hematological characterization. Acta Haematol. 2007, 117, 78–82. [Google Scholar] [CrossRef]
- Fucharoen, S.; Fucharoen, G.; Sanchaisuriya, K.; Pengjam, Y. Molecular analysis of a thai beta-thalassaemia heterozygote with normal haemoglobin A2 level: Implication for population screening. Ann. Clin. Biochem. 2002, 39, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Fucharoen, S.; Sanchaisuriya, K.; Fucharoen, G.; Panyasai, S.; Devenish, R.; Luy, L. Interaction of hemoglobin E and several forms of alpha-thalassemia in Cambodian families. Haematologica 2003, 88, 1092–1098. [Google Scholar]
- Fucharoen, S.; Fucharoen, G.; Ratanasiri, T.; Jetsrisuparb, A.; Fukumaki, Y. A simple non-radioactive assay for hemoglobin E gene in prenatal diagnosis. Clin. Chim. Acta 1994, 229, 197–203. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgelish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Fucharoen, G.; Fucharoen, S.; Sanchaisuriya, K.; Sae-Ung, N.; Suyasunanond, U.; Sriwilai, P.; Chinorak, P. Frequency Distribution and Haplotypic Heterogeneity of beta(E)-Globin Gene among Eight Minority Groups of Northeast Thailand. Hum. Hered. 2002, 53, 18–22. [Google Scholar] [CrossRef]
- Storz, J.F. Hemoglobin: Insights into Protein Structure, Function, and Evolution; Oxford University Press: Glasgow, UK, 2018. [Google Scholar]
- Hariharan, P.; Colaco, S.; Colah, R.; Ghosh, K.; Nadkarni, A. Delta globin gene variations leading to reduction in HbA2 levels. Int. J. Lab. Hematol. 2016, 38, 610–615. [Google Scholar] [CrossRef]
- Sae-ung, N.; Srivorakun, H.; Fucharoen, G.; Yamsri, S.; Sanchaisuriya, K.; Fucharoen, S. Phenotypic expression of hemoglobins A₂, E and F in various hemoglobin E related disorders. Blood Cells Mol. Dis. 2012, 48, 11–16. [Google Scholar] [CrossRef]
- Khalil, M.S.; Marouf, S.; Element, D.; Timbs, A.; Gallienne, A.; Schuh, A.; Old, J.M.; Henderson, S. A study of δ-globin gene mutations in the UK population: Identification of three novel variants and development of a novel DNA test for Hb A’2. Hemoglobin 2014, 38, 201–206. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Family 1 | |||||
|---|---|---|---|---|---|---|
| Mother | Father | S1 | S2 | S3 | S4 | |
| Sex/Age (years) | 67 | 68 | Male/34 | Male/57 | Female/54 | Female/36 |
| RBC count (×1012/L) | 4.45 | 5.29 | 5.67 | 4.38 | 5.46 | 4.98 |
| Hb (g/dL) | 10.8 | 13.8 | 14.5 | 11.5 | 12.4 | 12.5 |
| Hct (L/L) | 33.9 | 43.4 | 43.3 | 35.0 | 37.8 | 38 |
| MCV (fL) | 76.1 | 82.0 | 76.3 | 79.9 | 69.2 | 76.4 |
| MCH (pg) | 24.3 | 26.1 | 25.6 | 26.3 | 22.8 | 25 |
| MCHC (g/dL) | 31.9 | 31.8 | 33.5 | 32.9 | 32.9 | 32.8 |
| RDW-CV (%) | 12.3 | 12.7 | 11.9 | 12.1 | 27.8 | 14.7 |
| CE-Hb Profile a | A2A with A2-variant | EA | EA with A2-variant | EA with A2-variant | EA with A2-variant | EA with A2-variant |
| Hb A (%) a | 97.9 | 73.5 | 75.5 | 69.5 | 75.6 | 72.3 |
| Hb A2 (%) a | 1.3 | 3.4 | 1.1 | 1.0 | 1.3 | 1.4 |
| Hb E (%) a | 0 | 23.1 | 22.0 | 24.6 | 21.5 | 24.6 |
| Hb F (%) a | 0 | 0 | 0 | 0 | 0 | 0 |
| Hb A2-variant (%) a | 0.8 | 0 | 1.4 | 1.3 | 1.6 | 1.7 |
| HPLC-Hb Profile b | A2A with A2-variant | EA | EA with A2-variant | EA with A2-variant | EA with A2-variant | EA with A2-variant |
| Hb A (%) b | 85.8 | 62.6 | 64.6 | 58.1 | 62.3 | 61.2 |
| Hb A2 + E (%) b | 2.0 | 26.8 | 24.2 | 22.2 | 26.2 | 26.9 |
| Hb F (%) b | 0.2 | 0.6 | 0.6 | 1.8 | 0.9 | 1.3 |
| Hb A2-variant (%) b | 0.9 | 0 | 1.4 | 1.3 | 1.4 | 1.7 |
| Iron profile | ||||||
| Ferritin (μg/L) | 120.5 | 33.9 | 74.2 | 893.1 | 34.2 | 82.7 |
| Serum iron (μg/dL) | 65.3 | 48.4 | 130.8 | 159.1 | 54.1 | 82.3 |
| TIBC (μg/dL) | 262.9 | 320.8 | 349.4 | 227.9 | 333.0 | 363.7 |
| %Tsat (%) | 24.8 | 15.1 | 37.4 | 67.8 | 16.3 | 22.6 |
| δ-globin genotype | δ52A > G/δA | δA/δA | δ52A > G/δA | δ43G > A/δA | δ43G > A/δA | δ43G > A/δA |
| α-globin genotype | αα/αα | −α3.7/αα | −α3.7/αα | αα/αα | αα/αα | αα/αα |
| β-globin genotype | βA/βA | βE/βA | βE/βA | βE/βA | βE/βA | βE/βA |
| β-Globin Haplotype (5′ > 3′) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Subjects | β-Genotype | HincII5’ ε | HindIIIGγ | HindIIIAγ | HincII5’ ψβ | HincII3’ ψβ | AvaIIβ | BamHI3’ β | Hb A2-Mae Phrik Linked Haplotype | |
| Proband | δ52A > G/δA | [+/−] | [+/−] | [−/−] | [+/−] | [+/−] | [+/−] | [+/+] | δ52A > G δA | [+ − − − − ± +] [− + − + + ± +] |
| Mother | δ52A > G/δA | [+/+] | [−/−] | [−/−] | [−/−] | [−/−] | [+/−] | [+/−] | δ52A > G δA | [+ − − − − ± +] [+ − − − − ± +] |
| Father | δA/δA | [+/−] | [+/−] | [−/−] | [+/−] | [+/−] | [+/−] | [+/−] | δA δA | [− + −+ + ± +] [+ − − − − ± −] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phasit, A.; Panyasai, S.; Mayoon, M.; Jettawan, N.; Satthakarn, S. Phenotypic Expression of Known and Novel Hemoglobin A2-Variants, Hemoglobin A2-Mae Phrik [Delta 52(D3) Asp > Gly, HBD:c.158A > G], Associated with Hemoglobin E [Beta 26(B8) Glu > Lys, HBB:c.79G > A] in Thailand. Genes 2022, 13, 959. https://doi.org/10.3390/genes13060959
Phasit A, Panyasai S, Mayoon M, Jettawan N, Satthakarn S. Phenotypic Expression of Known and Novel Hemoglobin A2-Variants, Hemoglobin A2-Mae Phrik [Delta 52(D3) Asp > Gly, HBD:c.158A > G], Associated with Hemoglobin E [Beta 26(B8) Glu > Lys, HBB:c.79G > A] in Thailand. Genes. 2022; 13(6):959. https://doi.org/10.3390/genes13060959
Chicago/Turabian StylePhasit, Amphai, Sitthichai Panyasai, Monthon Mayoon, Niphawan Jettawan, and Surada Satthakarn. 2022. "Phenotypic Expression of Known and Novel Hemoglobin A2-Variants, Hemoglobin A2-Mae Phrik [Delta 52(D3) Asp > Gly, HBD:c.158A > G], Associated with Hemoglobin E [Beta 26(B8) Glu > Lys, HBB:c.79G > A] in Thailand" Genes 13, no. 6: 959. https://doi.org/10.3390/genes13060959