
In Silico Analysis Identified Putative Pathogenic Missense nsSNPs in Human SLITRK1 Gene

 , ,
, ,  ,
,  , ,
, ,
Abstract
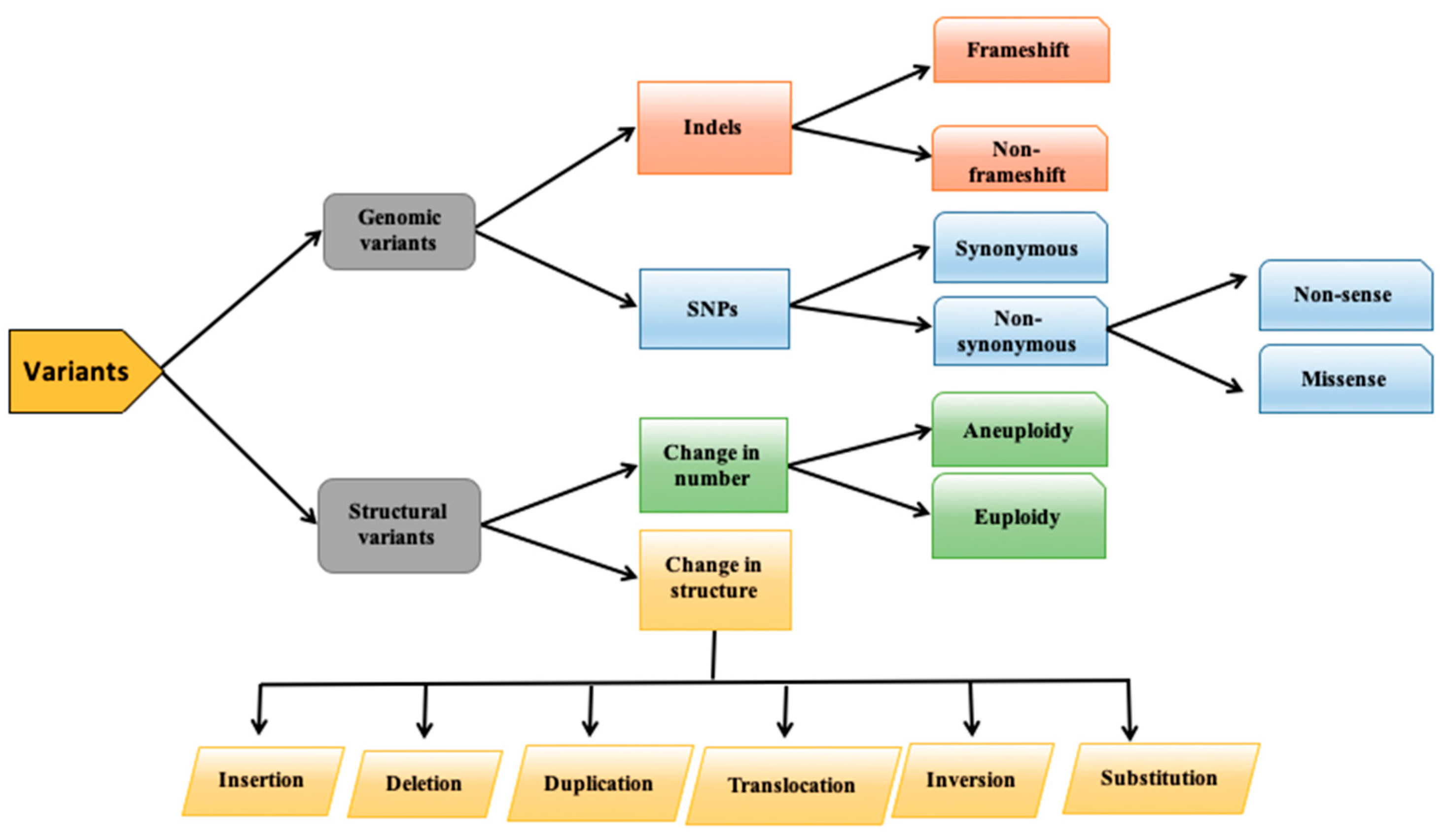
:1. Introduction
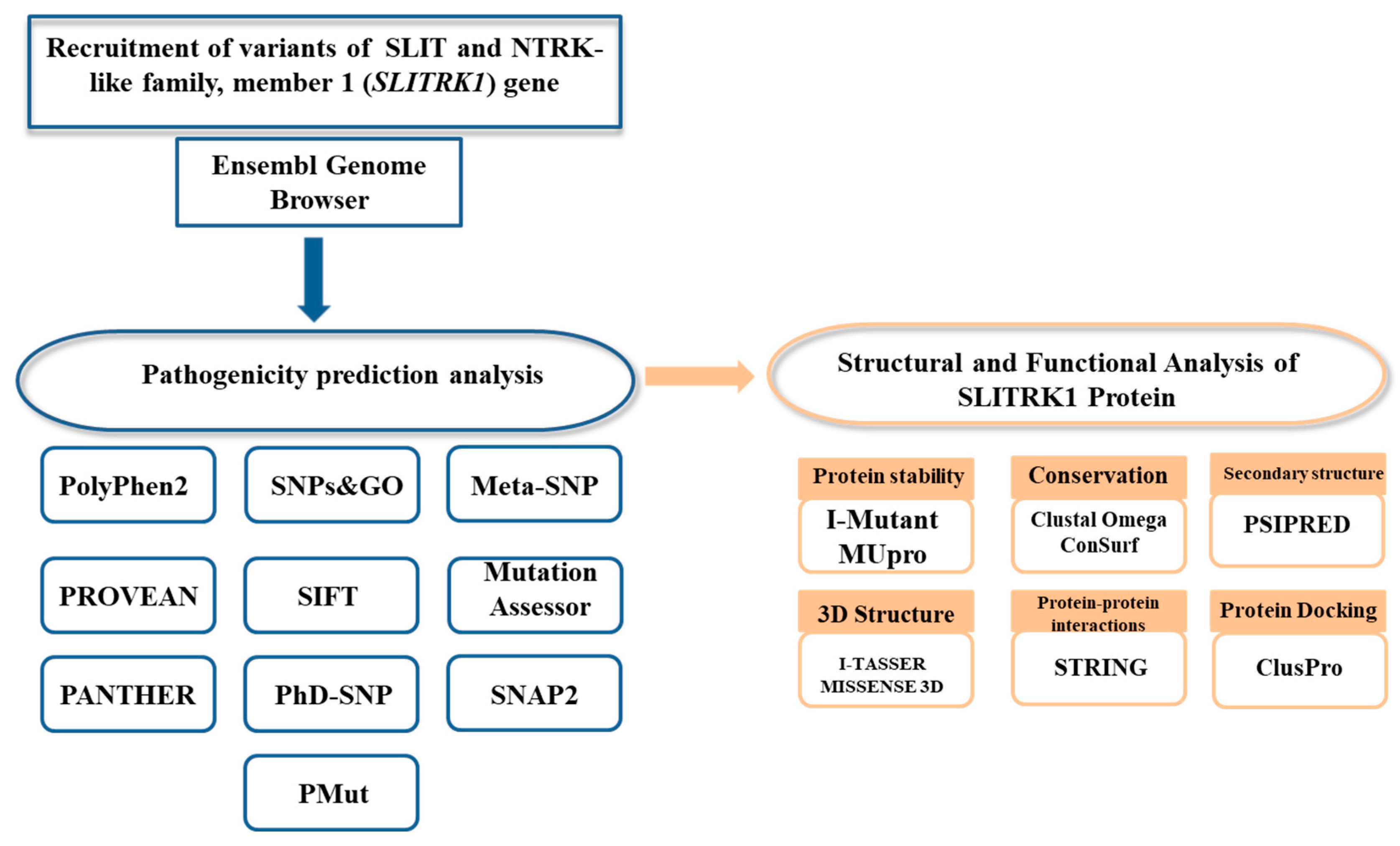
2. Materials and Methods
2.1. Variant Recruitment
2.2. Predicting Pathogenicity of Missense nsSNPs
2.3. Variant Frequency
2.4. Secondary Structure Prediction
2.5. Protein Stability Analysis
2.6. Conservation Analysis
2.7. Protein 3D Structure Prediction
2.8. Protein–Protein Interactions
2.9. Protein-Protein Docking

3. Results
3.1. Variant Recruitment
3.2. Pathogenicity Prediction of Variants
3.3. Variant Frequency
3.4. Secondary Structure Prediction
3.5. Protein Stability Analysis
3.6. Amino Acid Conservation
3.6.1. Clustal Omega
3.6.2. ConSurf
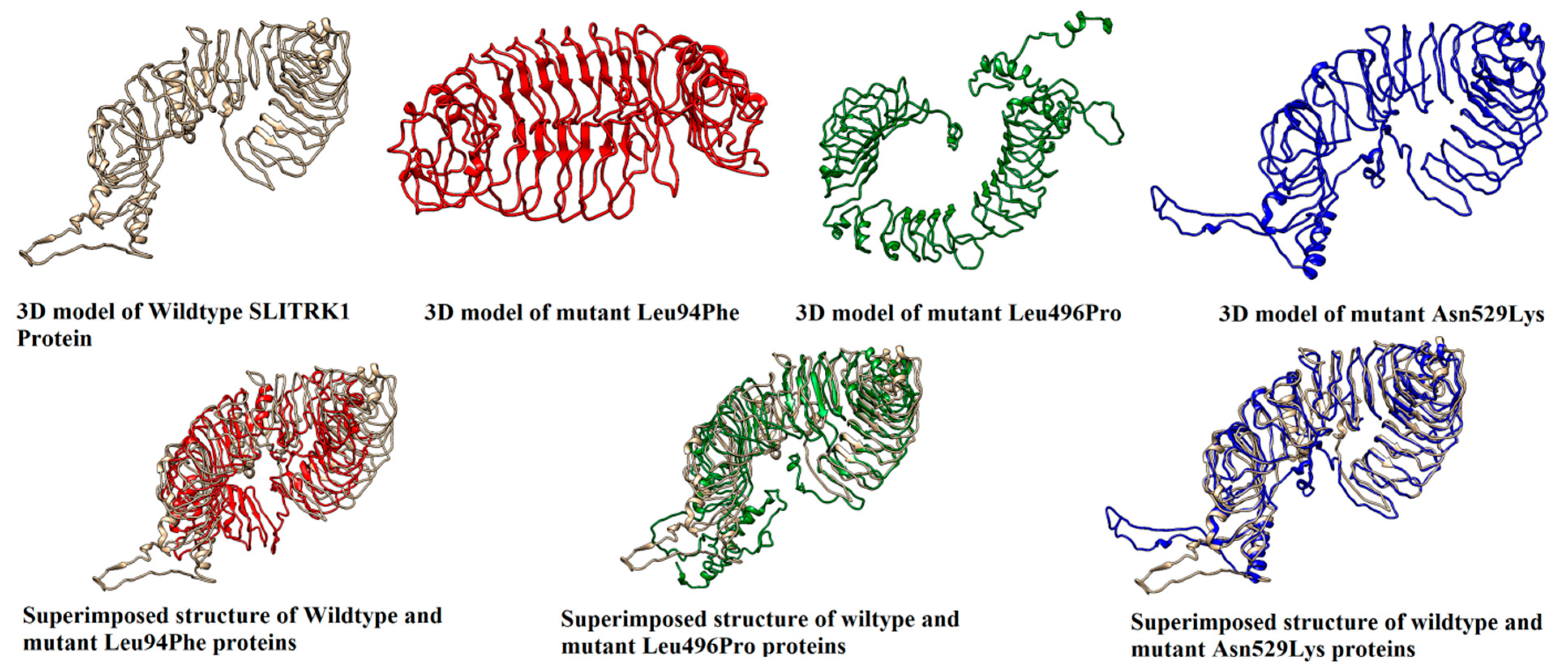
3.7. 3D Structure Predictions
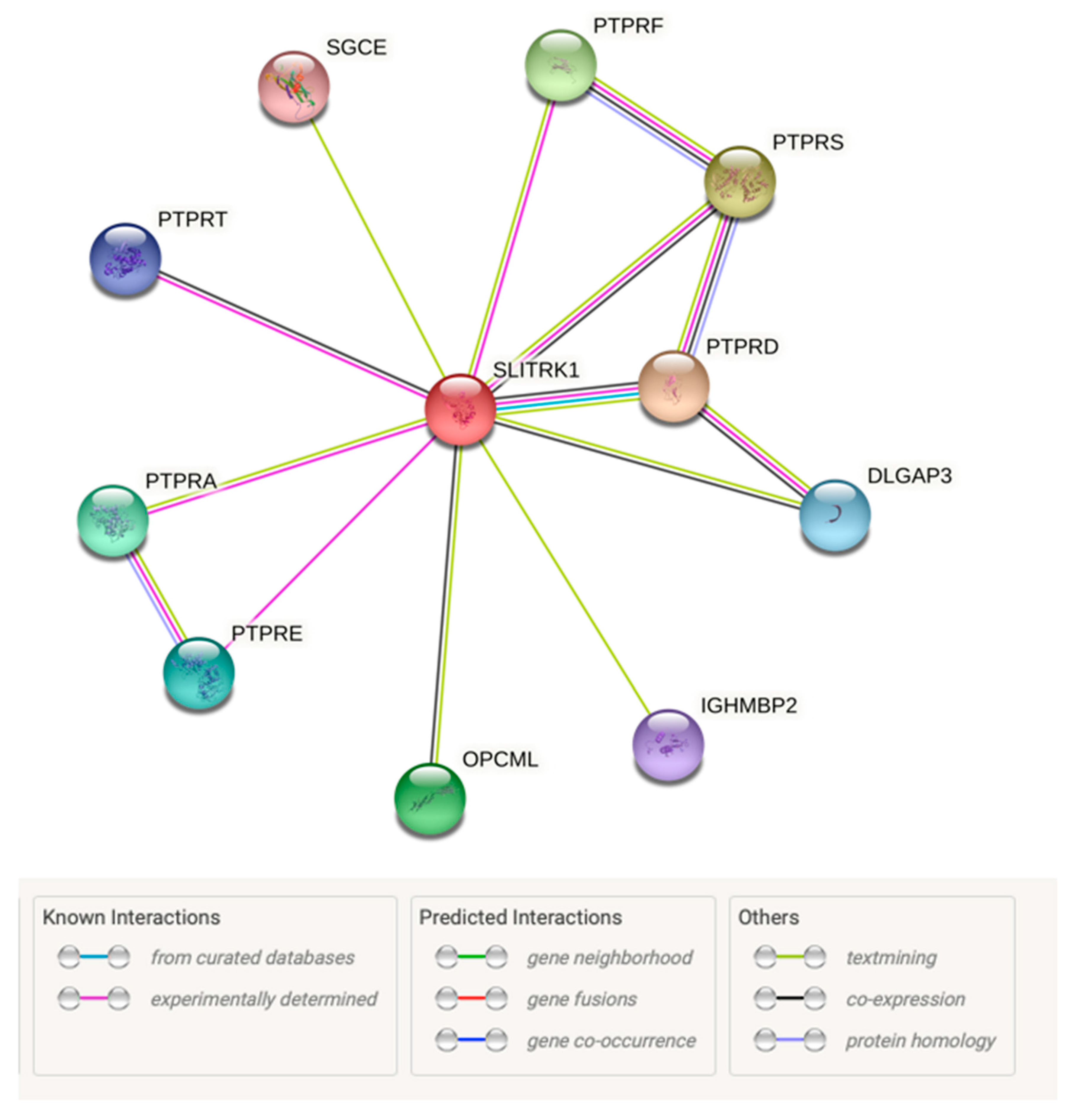
3.8. Protein–Protein Interactions
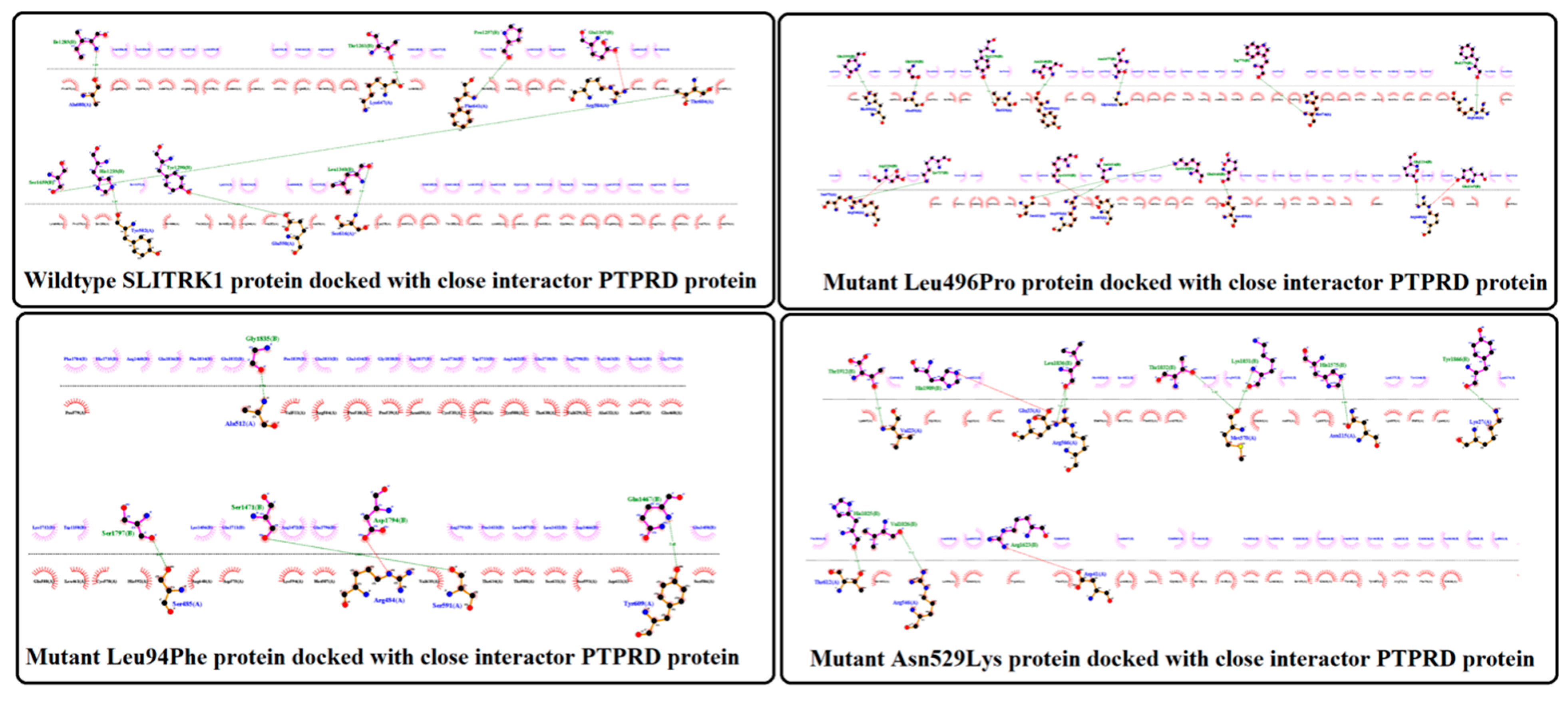
3.9. Protein–Protein Docking
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lantican, D.V.; Cortaga, C.Q.; Manohar, A.N.C.; dela Cueva, F.M.; Sison, M.L.J. Resistance gene analogs of mango: Insights on molecular defenses and evolutionary dynamics. Philipp. J. Sci. 2020, 149, 915–934. [Google Scholar]
- Hasan, N.; Choudhary, S.; Naaz, N.; Sharma, N.; Laskar, R.A. Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes. J. Genet. Eng. Biotechnol. 2021, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Cortaga, C.Q.; Latina, R.A.; Habunal, R.R.; Lantican, D.V. Identification and characterization of genome-wide resistance gene analogs (RGAs) of durian (Durio zibethinus L.). J. Genet. Eng. Biotechnol. 2022, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, J.M.; Mills, R.E.; Pittard, W.S.; Devine, S.E. Small insertions and deletions (INDELs) in human genomes. Hum. Mol. Genet. 2010, 19, R131–R136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, R.E.; Luttig, C.T.; Larkins, C.E.; Beauchamp, A.; Tsui, C.; Pittard, W.S.; Devine, S.E. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006, 16, 1182–1190. [Google Scholar] [CrossRef] [Green Version]
- Rozman, V.; Kunej, T. Harnessing omics big data in nine vertebrate species by genome-wide prioritization of sequence variants with the highest predicted deleterious effect on protein function. Omics A J. Integr. Biol. 2018, 22, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S. The new genomics: Global views of biology. Science 1996, 274, 536–539. [Google Scholar] [CrossRef] [Green Version]
- Doniger, S.W.; Kim, H.S.; Swain, D.; Corcuera, D.; Williams, M.; Yang, S.P.; Fay, J.C. A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet. 2008, 4, e1000183. [Google Scholar] [CrossRef] [Green Version]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins Struct. Funct. Bioinform. 2010, 78, 365–380. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Von Mering, C.; et al. STRING v9. 1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Marks, L.; May, G.H.; Wilson, J.B. The genetic basis of disease. Essays Biochem. 2018, 62, 643–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Gough, J.; Rost, B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet. 2006, 2, e29. [Google Scholar] [CrossRef] [Green Version]
- Ahonen, T.; Saltevo, J.; Laakso, M.; Kautiainen, H.; Kumpusa-lo, E.; Vanhala, M. Gender differences relating to metabolic syndrome and proinflammation in Finnish subjects with elevated blood pressure. Mediators Inflamm. 2009, 2009, 959281. [Google Scholar] [CrossRef] [Green Version]
- Degner, J.F.; Pai, A.A.; Pique-Regi, R.; Veyrieras, J.B.; Gaffney, D.J.; Pickrell, J.K.; De Leon, S.; Michelini, K.; Lewellen, N.; Crawford, G.E. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 2012, 482, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Trynka, G.; Sandor, C.; Han, B.; Xu, H.; Stranger, B.E.; Liu, X.S.; Raychaudhuri, S. Chromatin marks identify critic-al cell types for fine mapping complex trait variants. Nat. Genet. 2013, 45, 124–130. [Google Scholar] [CrossRef]
- Kapoor, A.; Sekar, R.B.; Hansen, N.F.; Fox-Talbot, K.; Morley, M.; Pihur, V.; Chatterjee, S.; Brandimarto, J.; Moravec, C.S.; Pulit, S.L. An enhancer polymorphism at the car- diomyocyte intercalated disc protein NOS1AP locus is a major regulator of the QT interval. Am. J. Hum. Genet. 2014, 94, 854–869. [Google Scholar] [CrossRef] [Green Version]
- Pai, A.A.; Pritchard, J.K.; Gilad, Y. The genetic and mechanistic basis for variation in gene regulation. PLoS Genet. 2015, 11, e1004857. [Google Scholar] [CrossRef] [Green Version]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarti, A. Single base differences between human genomes underlie differences in susceptibility to, or protection from, a host of diseases. Hence the great potential of such information in medicine. Nature 2001, 409, 822–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genom. Hum. Genet. 2006, 7, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Castejon, M.; Marin, F.; Soler-Rivas, C.; Reglero, G.; Visioli, F.; Rodriguez-Casado, A. Functional non-synonymous polymorphisms prediction methods: Current approaches and future developments. Curr. Med. Chem. 2011, 18, 5095–5103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, R.J.; Munroe, P.B.; Caulfield, M.J.; Saqi, M.A. Predicting deleterious nsSNPs: An analysis of sequence and structural attributes. BMC Bioinform. 2006, 7, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Purohit, R. Computational screening and molecular dynamics simulation of disease associated nsSNPs in CENP-E. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2012, 738, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, V.; Sethumadhavan, R. Drug resistance mechanism of PncA in Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2014, 32, 209–221. [Google Scholar] [CrossRef]
- Rajendran, V.; Gopalakrishnan, C.; Sethumadhavan, R. Pathological role of a point mutation (T315I) in BCR-ABL1 protein—A computational insight. J. Cell. Biochem. 2018, 119, 918–925. [Google Scholar] [CrossRef]
- Aruga, J.; Yokota, N.; Mikoshiba, K. Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003, 315, 87–94. [Google Scholar] [CrossRef]
- Takahashi, H.; Katayama, K.I.; Sohya, K.; Miyamoto, H.; Prasad, T.; Matsumoto, Y.; Ota, M.; Yasuda, H.; Tsumoto, T.; Aruga, J.; et al. Selective control of inhibitory synapse development by Slitrk3-PTPδ trans-synaptic interaction. Nat. Neurosci. 2012, 15, 389–398. [Google Scholar] [CrossRef]
- Um, J.W.; Kim, K.H.; Park, B.S.; Choi, Y.; Kim, D.; Kim, C.Y.; Kim, S.J.; Kim, M.; Ko, J.S.; Lee, S.G.; et al. Structural basis for LAR-RPTP/Slitrk complex-mediated synaptic adhesion. Nat. Commun. 2014, 5, 5423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.A.; Jeon, S.; Um, J.W.; Ko, J. Emergent synapse organizers: LAR-RPTPs and their companions. Int. Rev. Cell Mol. Biol. 2016, 324, 39–65. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Nakayama, M.; Nakajima, D.; Kikuno, R.; Ohara, O. Prediction of the coding sequences of unidentified human genes. XX. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2001, 8, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Abelson, J.F.; Kwan, K.Y.; O’Roak, B.J.; Baek, D.Y.; Stillman, A.A.; Morgan, T.M.; Mathews, C.A.; Pauls, D.L.; Rašin, M.R.; Gunel, M.; et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science 2005, 310, 317–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, P.J.; Dietrich, A.; Edwards, M.J.; Elamin, I.; Martino, D. Environmental factors in ourette syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1040–1049. [Google Scholar] [CrossRef]
- Mathews, C.A.; Scharf, J.M.; Miller, L.L.; Macdonald-Wallis, C.; Lawlor, D.A.; Ben-Shlomo, Y. Association between pre-and perinatal exposures and Tourette syndrome or chronic tic disorder in the ALSPAC cohort. Br. J. Psychiatry 2014, 204, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalanithi, P.S.; Zheng, W.; Kataoka, Y.; DiFiglia, M.; Grantz, H.; Saper, C.B.; Schwartz, M.L.; Leckman, J.F.; Vaccarino, F.M. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 13307–13312. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.S.; Thomas, P.; Kane, M.J.; Scahill, L.; Zhang, H.; Bronen, R.; King, R.A.; Leckman, J.F.; Staib, L. Basal ganglia volumes in patients with Gilles de la Tourette syndrome. Arch. Gen. Psychiatry 2003, 60, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Müller-Vahl, K.R.; Meyer, G.J.; Knapp, W.H.; Emrich, H.M.; Gielow, P.; Brücke, T.; Berding, G. Serotonin transporter binding in Tourette Syndrome. Neurosci. Lett. 2005, 385, 120–125. [Google Scholar] [CrossRef]
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 72–95. [Google Scholar] [CrossRef] [Green Version]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins Struct. Funct. Bioinform. 2011, 79, 2444–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | Chr:bp | Alleles | AA | AA Coord | Polyphen2 | SNPs&Go | MetaSNP | Provean | SIFT | Mutation Assessor | Panther | PHD SNP | SNAP2 | PMut | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pred | Prob | Pred | Prob | Pred | Score | Pred | Score | Pred | Score | F I | FI Score | Pred | Preservation Time | Pred | Score | Pred | Score | Pred | Score | |||||
| 1 | 13:83879772 | G/A | Pro/Leu | 579 | ProD | 0.997 | D | 0.736 | D | 0.657 | D | −8.14 | D | 0 | M | 2.995 | ProD | 750 | D | 8 | E | 5 | D | 0.6827 |
| 2 | 13:83879921 | G/T | Asn/Lys | 529 | ProD | 1 | D | 0.853 | D | 0.81 | D | −5.77 | D | 0 | M | 3.065 | ProD | 456 | D | 8 | E | 82 | D | 0.7961 |
| 3 | 13:83879932 | G/A | Leu/Phe | 526 | ProD | 0.997 | D | 0.673 | D | 0.603 | D | −3.85 | D | 0 | M | 2.74 | ProD | 750 | D | 7 | E | 71 | D | 0.7852 |
| 4 | 13:83879997 | T/A | Asn/Ile | 504 | ProD | 0.999 | D | 0.907 | D | 0.825 | D | −8.14 | D | 0 | H | 4.565 | ProD | 750 | D | 8 | E | 85 | D | 0.8016 |
| 5 | 13:83880021 | A/G | Leu/Pro | 496 | ProD | 1 | D | 0.834 | D | 0.786 | D | −6.15 | D | 0 | H | 4.75 | ProD | 456 | D | 6 | E | 90 | D | 0.8058 |
| 6 | 13:83880024 | G/C | Ser/Trp | 495 | ProD | 0.985 | D | 0.616 | D | 0.72 | D | −4.2 | D | 0.01 | H | 4.165 | ProD | 456 | D | 1 | E | 65 | D | 0.7634 |
| 7 | 13:83880354 | T/C | Asn/Ser | 385 | ProD | 0.999 | D | 0.812 | D | 0.721 | D | −4.37 | D | 0 | M | 2.995 | PosD | 361 | D | 8 | E | 72 | D | 0.5334 |
| 8 | 13:83880432 | C/T | Cys/Tyr | 359 | ProD | 0.999 | D | 0.922 | D | 0.816 | D | −9.47 | D | 0 | M | 2.62 | PosD | 361 | D | 4 | E | 82 | D | 0.8359 |
| 9 | 13:83880435 | T/A | Asn/Ile | 358 | PosD | 0.775 | D | 0.636 | D | 0.761 | D | −6.24 | D | 0 | M | 2.3 | PosD | 361 | D | 3 | E | 58 | D | 0.7522 |
| 10 | 13:83880469 | A/C | Cys/Gly | 347 | ProD | 1 | D | 0.757 | D | 0.733 | D | −9.53 | D | 0 | M | 2.62 | ProD | 750 | D | 6 | E | 86 | D | 0.7549 |
| 11 | 13:83880930 | G/A | Pro/Leu | 193 | ProD | 0.949 | D | 0.767 | D | 0.676 | D | −6.7 | D | 0 | M | 2.93 | ProD | 750 | D | 8 | E | 17 | D | 0.6558 |
| 12 | 13:83881017 | T/C | Asn/Ser | 164 | ProD | 1 | D | 0.833 | D | 0.766 | D | −4.75 | D | 0 | M | 3.375 | ProD | 750 | D | 8 | E | 66 | D | 0.7106 |
| 13 | 13:83881162 | T/A | Asn/Tyr | 116 | ProD | 1 | D | 0.907 | D | 0.865 | D | −7.59 | D | 0 | H | 4.72 | ProD | 750 | D | 7 | E | 83 | D | 0.7989 |
| 14 | 13:83881226 | C/A | Leu/Phe | 94 | ProD | 0.98 | D | 0.744 | D | 0.532 | D | −2.96 | D | 0.01 | M | 3.18 | ProD | 456 | D | 7 | E | 46 | D | 0.8303 |
| 15 | 13:83881276 | A/G | Phe/Leu | 78 | PosD | 0.831 | D | 0.741 | D | 0.511 | D | −4.8 | D | 0.02 | M | 2.035 | ProD | 750 | D | 8 | E | 52 | D | 0.5225 |
| 16 | 13:83881305 | T/C | Asn/Ser | 68 | ProD | 0.985 | D | 0.753 | D | 0.69 | D | −4.72 | D | 0 | H | 3.555 | ProD | 750 | D | 1 | E | 64 | D | 0.8179 |
| Variant No. | rs ID | AA | AA Coord | I-Mutant | MuPro | ||
|---|---|---|---|---|---|---|---|
| Stability | RI | Stability | Score | ||||
| I | rs1048143268 | Asn/Lys | 529 | Decrease | 4 | Decrease | −0.86 |
| II | rs1219903976 | Leu/Phe | 526 | Decrease | 8 | Decrease | −0.99 |
| III | rs1226852299 | Leu/Pro | 496 | Decrease | 6 | Decrease | −0.992 |
| IV | rs1472728808 | Asn/Ser | 385 | Decrease | 5 | Decrease | −0.971 |
| V | rs1277399796 | Cys/Gly | 347 | Decrease | 7 | Decrease | −0.994 |
| VI | rs1429907885 | Pro/Leu | 193 | Decrease | 6 | Decrease | −0.778 |
| VII | rs774612607 | Asn/Ser | 164 | Decrease | 5 | Decrease | −0.999 |
| VIII | rs1410244448 | Leu/Phe | 94 | Decrease | 8 | Decrease | −0.999 |
| IX | rs954218528 | Phe/Leu | 78 | Decrease | 7 | Decrease | −0.661 |
| X | rs1228122404 | Asn/Ser | 68 | Decrease | 2 | Decrease | −0.751 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.Z.; Farid, A.; Ahmad, S.; Muzammal, M.; Mohaini, M.A.; Alsalman, A.J.; Al Hawaj, M.A.; Alhashem, Y.N.; Alsaleh, A.A.; Almusalami, E.M.; et al. In Silico Analysis Identified Putative Pathogenic Missense nsSNPs in Human SLITRK1 Gene. Genes 2022, 13, 672. https://doi.org/10.3390/genes13040672
Ali MZ, Farid A, Ahmad S, Muzammal M, Mohaini MA, Alsalman AJ, Al Hawaj MA, Alhashem YN, Alsaleh AA, Almusalami EM, et al. In Silico Analysis Identified Putative Pathogenic Missense nsSNPs in Human SLITRK1 Gene. Genes. 2022; 13(4):672. https://doi.org/10.3390/genes13040672
Chicago/Turabian StyleAli, Muhammad Zeeshan, Arshad Farid, Safeer Ahmad, Muhammad Muzammal, Mohammed Al Mohaini, Abdulkhaliq J. Alsalman, Maitham A. Al Hawaj, Yousef N. Alhashem, Abdulmonem A. Alsaleh, Eman M. Almusalami, and et al. 2022. "In Silico Analysis Identified Putative Pathogenic Missense nsSNPs in Human SLITRK1 Gene" Genes 13, no. 4: 672. https://doi.org/10.3390/genes13040672