
GATA-1 Defects in Diamond–Blackfan Anemia: Phenotypic Characterization Points to a Specific Subset of Disease

 , , , and
, , , and
Abstract
:1. Introduction
2. Methods
2.1. Patients and Genetics
2.2. Literature Search
2.3. Bone Marrow Analysis
2.4. Ethics
3. Results
3.1. Hematological Characteristics of Two Novel DBA-Like Patients with GATA1 Defects
3.2. Comparative Analysis with Previously Reported Cases Illustrates Distinct DBA-Like Disease Characteristics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Diamond, L.; Blackfan, K. Hypoplastic anemia. Am. J. Dis. Child 1938, 56, 464–467. [Google Scholar]
- Lipton, J.M.; Ellis, S.R. Diamond-Blackfan Anemia: Diagnosis, Treatment, and Molecular Pathogenesis. Hematol. Clin. N. Am. 2009, 23, 261–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachos, A.; Ball, S.; Dahl, N.; Alter, B.P.; Sheth, S.; Ramenghi, U.; Meerpohl, J.; Karlsson, S.; Liu, J.M.; Leblanc, T.; et al. Diagnosing and treating Diamond Blackfan anaemia: Results of an international clinical consensus conference. Br. J. Haematol. 2008, 142, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.-N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef] [Green Version]
- Lezzerini, M.; Penzo, M.; O’Donohue, M.-F.; Vieira, C.M.D.S.; Saby, M.; Elfrink, H.L.; Diets, I.J.; Hesse, A.-M.; Couté, Y.; Gastou, M.; et al. Ribosomal protein gene RPL9 variants can differentially impair ribosome function and cellular metabolism. Nucleic Acids Res. 2019, 48, 770–787. [Google Scholar] [CrossRef] [Green Version]
- Choesmel, V.; Bacqueville, D.; Rouquette, J.; Noaillac-Depeyre, J.; Fribourg, S.; Crétien, A.; Leblanc, T.; Tchernia, G.; Da Costa, L.; Gleizes, P.-E. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood 2007, 109, 1275–1283. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, S.; Thomas, G. The Role of p53 in Ribosomopathies. Semin. Hematol. 2011, 48, 97–105. [Google Scholar] [CrossRef]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef] [Green Version]
- Horos, R.; Von Lindern, M. Molecular mechanisms of pathology and treatment in Diamond Blackfan Anaemia. Br. J. Haematol. 2012, 159, 514–527. [Google Scholar] [CrossRef]
- Yang, Z.; Keel, S.B.; Shimamura, A.; Liu, L.; Gerds, A.T.; Li, H.Y.; Wood, B.L.; Scott, B.L.; Abkowitz, J.L. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 2016, 8, 338ra67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moniz, H.; DBA Group of Société d’Hématologie et d’Immunologie Pédiatrique (SHIP); Gastou, M.; Leblanc, T.; Hurtaud, C.; Crétien, A.; Lécluse, Y.; Raslova, H.; Larghero, J.; Croisille, L.; et al. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012, 3, e356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rio, S.; Gastou, M.; Karboul, N.; Derman, R.; Suriyun, T.; Manceau, H.; Leblanc, T.; El Benna, J.; Schmitt, C.; Azouzi, S.; et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 2019, 133, 1358–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doty, R.; Yan, X.; Lausted, C.; Munday, A.D.; Yang, Z.; Yi, D.; Jabbari, N.; Liu, L.; Keel, S.B.; Tian, Q.; et al. Single-cell analyses demonstrate that a heme–GATA1 feedback loop regulates red cell differentiation. Blood 2019, 133, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Le Goff, S.; Boussaid, I.; Floquet, C.; Raimbault, A.; Hatin, I.; Andrieu-Soler, C.; Salma, M.; LeDuc, M.; Gautier, E.-F.; Guyot, B.; et al. p53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood 2020, 137, 89–102. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.-A.; Beggs, A.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Investig. 2012, 122, 2439–2443. [Google Scholar] [CrossRef] [Green Version]
- Klar, J.; Khalfallah, A.; Arzoo, P.S.; Gazda, H.T.; Dahl, N. RecurrentGATA1mutations in Diamond-Blackfan anaemia. Br. J. Haematol. 2014, 166, 949–951. [Google Scholar] [CrossRef]
- Parrella, S.; Aspesi, A.; Quarello, P.; Garelli, E.; Pavesi, E.; Carando, A.; Nardi, M.; Ellis, S.R.; Ramenghi, U.; Dianzani, I. Loss of GATA-1 full length as a cause of Diamond-Blackfan anemia phenotype. Pediatr. Blood Cancer 2014, 61, 1319–1321. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, L.S.; Gazda, H.T.; Eng, J.C.; Eichhorn, S.W.; Thiru, P.; Ghazvinian, R.; George, T.; Gotlib, J.R.; Beggs, A.H.; Sieff, C.A.; et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 2014, 20, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103.e19. [Google Scholar] [CrossRef] [Green Version]
- Boussaid, I.; Le Goff, S.; Floquet, C.; Gautier, E.-F.; Raimbault, A.; Viailly, P.-J.; Al Dulaimi, D.; Burroni, B.; Dusanter-Fourt, I.; Hatin, I.; et al. Integrated analyses of translatome and proteome identify the rules of translation selectivity in RPS14-deficient cells. Haematologica 2021, 106, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, L.; Caballero, N.; Fernández-Calleja, L.; Karkoulia, E.; Strouboulis, J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life 2019, 72, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Byrska-Bishop, M.; VanDorn, D.; Campbell, A.E.; Betensky, M.; Arca, P.R.; Yao, Y.; Gadue, P.; Costa, F.F.; Nemiroff, R.L.; Blobel, G.A.; et al. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. J. Clin. Investig. 2015, 125, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Chlon, T.M.; McNulty, M.; Goldenson, B.; Rosinski, A.; Crispino, J.D. Global transcriptome and chromatin occupancy analysis reveal the short isoform of GATA1 is deficient for erythroid specification and gene expression. Haematologica 2015, 100, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, K.E.; Crispino, J.; Poncz, M.; White, J.G.; Orkin, S.H.; Maris, J.M.; Weiss, M. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet. 2000, 24, 266–270. [Google Scholar] [CrossRef]
- Russo, R.; Andolfo, I.; Gambale, A.; De Rosa, G.; Manna, F.; Arillo, A.; Wandroo, F.; Bisconte, M.G.; Iolascon, A. GATA1 erythroid-specific regulation of SEC23B expression and its implication in the pathogenesis of congenital dyserythropoietic anemia type II. Haematologica 2017, 102, e371–e374. [Google Scholar] [CrossRef] [Green Version]
- Bouchghoul, H.; Quelin, C.; Loget, P.; Encha-Razavi, F.; Senat, M.-V.; Maheut, L.; Galimand, J.; Collardeau-Frachon, S.; Da Costa, L.; Martinovic, J. Fetal cerebral hemorrhage due to X-linkedGATA1gene mutation. Prenat. Diagn. 2018, 38, 772–778. [Google Scholar] [CrossRef]
- Mehaffey, M.G.; Newton, A.L.; Gandhi, M.J.; Crossley, M.; Drachman, J.G. X-linked thrombocytopenia caused by a novel mutation ofGATA-1. Blood 2001, 98, 2681–2688. [Google Scholar] [CrossRef] [Green Version]
- Freson, K.; Wijgaerts, A.; Van Geet, C. GATA1 gene variants associated with thrombocytopenia and anemia. Platelets 2017, 28, 731–734. [Google Scholar] [CrossRef]
- Freson, K.; Matthijs, G.; Thys, C.; Mariën, P.; Hoylaerts, M.F.; Vermylen, J.; Van Geet, C. Different substitutions at residue D218 of the X-linked transcription factor GATA1 lead to altered clinical severity of macrothrombocytopenia and anemia and are associated with variable skewed X inactivation. Hum. Mol. Genet. 2002, 11, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Niakan, K.; Matsushita, M.; Stamatoyannopoulos, G.; Orkin, S.H.; Raskind, W.H. X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood 2002, 100, 2040–2045. [Google Scholar] [CrossRef]
- Hollanda, L.M.; Lima, C.S.P.; Cunha, A.F.; Albuquerque, D.M.; Vassallo, J.; Ozelo, M.C.; Joazeiro, P.P.; Saad, S.T.O.; Costa, F.F. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat. Genet. 2006, 38, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.D.; Steensma, D.P.; Pulsipher, M.A.; Spangrude, G.J.; Kushner, J.P. Congenital erythropoietic porphyria due to a mutation in GATA1: The first trans-acting mutation causative for a human porphyria. Blood 2007, 109, 2618–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundschau, G.; Gurbuxani, S.; Gamis, A.S.; Greene, M.E.; Arceci, R.J.; Crispino, J.D. Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 2003, 101, 4298–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechsler, J.; Greene, M.E.; McDevitt, M.A.; Anastasi, J.; Karp, J.E.; Le Beau, M.M.; Crispino, J. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. 2002, 32, 148–152. [Google Scholar] [CrossRef]
- Yoshida, K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; Park, M.-J.; Terui, K.; Suzuki, H.; et al. The landscape of somatic mutations in Down syndrome–related myeloid disorders. Nat. Genet. 2013, 45, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.E.; Mundschau, G.; Wechsler, J.; McDevitt, M.; Gamis, A.; Karp, J.; Gurbuxani, S.; Arceci, R.; Crispino, J.D. Mutations in GATA1 in both transient myeloproliferative disorder and acute megakaryoblastic leukemia of Down syndrome. Blood Cells Mol. Dis. 2003, 31, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.M.; Leblanc, T.M.; Mohandas, N. Diamond-Blackfan anemia. Blood 2020, 136, 1262–1273. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B. Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Dis. Mark. 2015, 2015, 635670. [Google Scholar] [CrossRef] [Green Version]
- Ling, T.; Crispino, J.D. GATA1 mutations in red cell disorders. IUBMB Life 2019, 72, 106–118. [Google Scholar] [CrossRef]
- Crispino, J.D.; Horwitz, M.S. GATA factor mutations in hematologic disease. Blood 2017, 129, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, L.; Tsukamoto, S.; Suzuki, M.; Yamamoto-Mukai, H.; Yamamoto, M.; Philipsen, S.; Ohneda, K. Ablation of Gata in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood 2008, 111, 4375–4385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Onodera, K.; Motohashi, H.; Suwabe, N.; Hayashi, N.; Yanai, N.; Nabesima, Y.; Yamamoto, M. Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J. Biol. Chem. 1997, 272, 12611–12615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.T.; Khandros, E.; Bailey, L.C.; Nichols, K.E.; Vakoc, C.R.; Yao, Y.; Huang, Z.; Crispino, J.D.; Hardison, R.C.; Blobel, G.A.; et al. Graded repression of PU.1/Sfpi1 gene transcription by GATA factors regulates hematopoietic cell fate. Blood 2009, 114, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieminen, T.T.; O’Donohue, M.-F.; Wu, Y.; Lohi, H.; Scherer, S.; Paterson, A.D.; Ellonen, P.; Abdel-Rahman, W.M.; Valo, S.; Mecklin, J.-P.; et al. Germline Mutation of RPS20, Encoding a Ribosomal Protein, Causes Predisposition to Hereditary Nonpolyposis Colorectal Carcinoma without DNA Mismatch Repair Deficiency. Gastroenterology 2014, 147, 595–598.e5. [Google Scholar] [CrossRef] [Green Version]
- Sulima, S.O.; Hofman, I.J.F.; De Keersmaecker, K.; Dinman, J.D. How Ribosomes Translate Cancer. Cancer Discov. 2017, 7, 1069–1087. [Google Scholar] [CrossRef] [Green Version]
- Fancello, L.; Kampen, K.R.; Hofman, I.J.; Verbeeck, J.; De Keersmaecker, K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget 2017, 8, 14462–14478. [Google Scholar] [CrossRef] [Green Version]
- Iskander, D.; Roberts, I.; Rees, C.; Szydlo, R.; Alikian, M.; Neale, M.; Harrington, Y.; Kelleher, P.; Karadimitris, A.; De La Fuente, J. Impaired cellular and humoral immunity is a feature of Diamond-Blackfan anaemia; experience of 107 unselected cases in the United Kingdom. Br. J. Haematol. 2019, 186, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Gianferante, D.M.; Wlodarski, M.W.; Atsidaftos, E.; Da Costa, L.; Delaporta, P.; Farrar, J.E.; Goldman, F.D.; Hussain, M.; Kattamis, A.; Leblanc, T.; et al. Genotype-phenotype association and variant characterization in Diamond-Blackfan anemia caused by pathogenic variants in RPL35A. Haematologica 2020, 106, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Fargo, J.H.; Kratz, C.P.; Giri, N.; Savage, S.A.; Wong, C.; Backer, K.; Alter, B.P.; Glader, B. Erythrocyte adenosine deaminase: Diagnostic value for Diamond-Blackfan anaemia. Br. J. Haematol. 2013, 160, 547–554. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
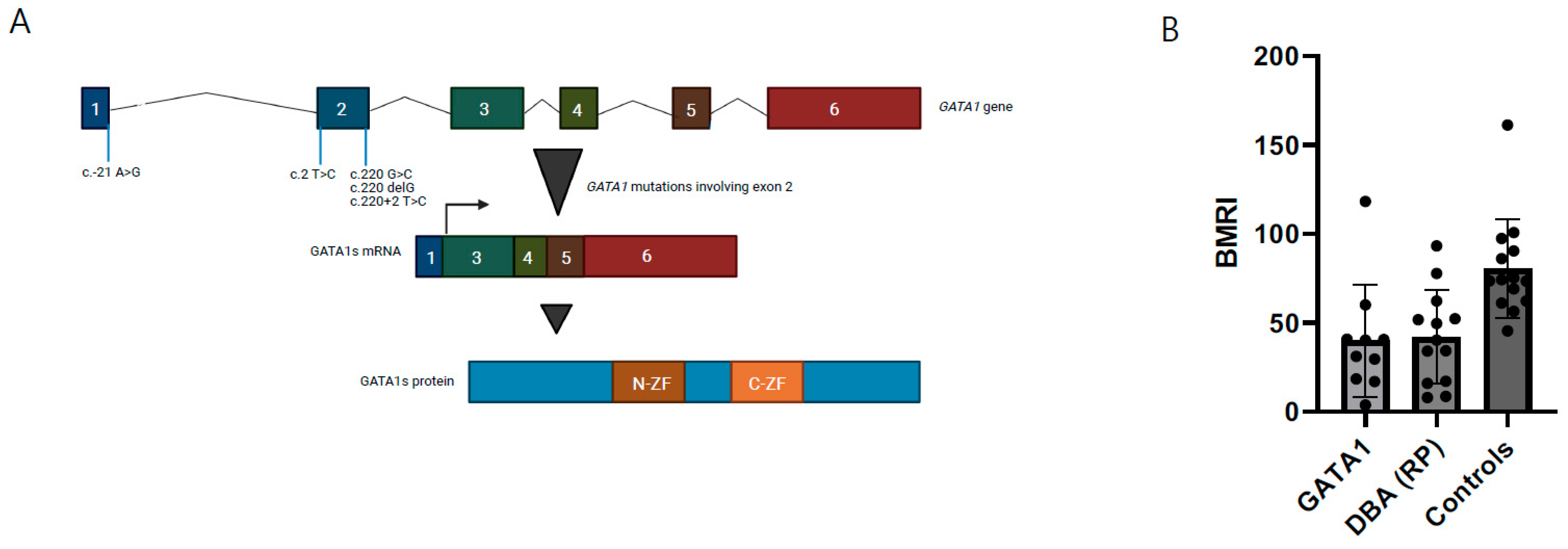{kind=link}
| Index# | Reported | Molecular Defect | Type | Age Diagnosis | Hgb (g/dL) | HbF (%) | MCV (fL) | Retics (×109/L) | WBC (×109/L) | ANC (×109/L) | PLts (×109/L) | eADA | Steroid Responsive | Current Treatment | Bone Marrow |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I-I | Hollanda et al. 2006 | c.220G>C | missense | 40 years | 10.9 | NA | 100.6 | NA | 5.9 | 2.8 | 352 | NA | NA | none | Moderate hypercellularity in 3 lineages. Moderate reduction in relationship between G/E. Retardation of maturation of the granulocytic and erythroblast series. Moderate number of micromegakaryocytes. Neutrophils with pseudo Pelger–Hüet anomaly. |
| I-II | c.220G>C | missense | 24 years | 7.7 | NA | 99.0 | 56 | 3.7 | 1.2 | 161 | NA | NA | deceased (severe pneumonia) | Hypocellularity with normal number of megakaryocytes and frequent micromegakaryocytes. | |
| I-III | c.220G>C | missense | 35 years | 11.8 | NA | 100.8 | 139 | 5.2 | 2.0 | 190 | NA | NA | none | NA | |
| I-IV | c.220G>C | missense | 12 years | 5.2 | ↑ | 94 | 108 | 2 | 0.6 | 135 | NA | NA | remission after allogeneic BMT | Moderate hypocellularity with multinucleation and nuclear karyorrhexis in erythroblasts. Neutrophils with pseudo Pelger–Hüet anomaly and moderate number of micromegakaryocytes. No ringed sideroblasts were seen. | |
| I-V | c.220G>C | missense | 20 years | 3.8 | ↑ | 101 | 108 | 1.6 | 0.5 | 144 | NA | NA | remission after allogeneic BMT | Moderate hypocellularity with multinucleation and nuclear karyorrhexis in erythroblasts. Neutrophils with pseudo Pelger–Hüet anomaly and moderate number of micromegakaryocytes. No ringed sideroblasts were seen. | |
| I-VI | c.220G>C | missense | 2 months | 6.1 | NA | 93 | NA | 7.5 | NP | 345 | NA | NA | regular transfusions | Normocellularity with moderate reduction in the relationship between G/E. Maturation preserved and moderate number of megakaryocytes, with micromegakaryocytes. | |
| I-VII | c.220G>C | missense | 4 years | 5.3 | ↑ | 88.1 | 44.2 | 2.2 | 0.6 | 294 | NA | NA | regular transfusions | Moderate hypocellularity with multinucleation and nuclear karyorrhexis in erythroblasts. Neutrophils with pseudo Pelger–Hüet anomaly and moderate number of micromegakaryocytes. No ringed sideroblasts were seen. | |
| I-VIII | c.220G>C | missense | 17 years | 9.6 | ↑ | 102.8 | 58.1 | 3.7 | 1 | 400 | NA | NA | deceased (‘unrelated cause’) | Moderate hypocellularity with multinucleation and nuclear karyorrhexis in erythroblasts. Neutrophils with pseudo Pelger–Hüet anomaly and moderate number of micromegakaryocytes. No ringed sideroblasts were seen. | |
| II-I | Sankaran et al. 2012 | c.220G>C | missense | birth | (lab 23y) 8.4 | ↑ | NA | 21 | 3.1 | 0.83 | 71 | normal | YES (temporarily) | regular transfusions | Erythroid hypoplasia without abnormalities of the other hematopoietic lineages. |
| II-II | c.220G>C | missense | 1.6 months | (lab 19y) 8.3 | NA | 100 | 68 | 4.5 | 2.1 | 201 | NA | YES (temporarily) | regular transfusions | Erythroid hypoplasia without abnormalities of the other hematopoietic lineages. | |
| II-III | c.220delG | frameshift | 6 weeks | 3.5 | ↑ * | not reported | unknown | 6.8 | 1.6 (?) | 362 (?) | normal | YES | glucocorticoids | Not documented. | |
| III-I | Ludwig et al. 2014 | c.2T>C | not reported | 9.7 | NA | 101.8 | 73.2 | 6.2 | 3.9 | 239 | NP | NA | regular transfusions | Not documented. | |
| IV-I | Parrella et al. 2014 | c.2T>C | 9 months | 5.5 | NA | 93 | 40 | normal range | normal range | normal range | ↑ | partial response | remission after allogeneic BMT | Selective deficiency in erythroid precursors without abnormalities in the other hematopoietic lineages --> 4 yrs: severe hypocellular bone marrow with a 45XY, −7 clone (65%) and a further 50XXY, +3, +8, +21 clone (MDS). | |
| V-I | Klar et al. 2014 | c.220G>C | missense | <3 months | 5.0–9.0 | ↑ | 104–108 | “low” | normal range | normal range | normal range | ↑ | YES (temporarily) | regular transfusions | Erythroid hypoplasia with otherwise normal cellularity. |
| V-II | c.220G>C | missense | <3 months | 5.0–9.0 | ↑ | 104–108 | “low” | normal range | normal range | normal range | ↑ | YES (temporarily) | regular transfusions | Erythroid hypoplasia with otherwise normal cellularity. | |
| V-III | c.220G>C | missense | <3 months | 5.0–9.0 | ↑ | 104–108 | “low” | normal range | normal range | normal range | unknown | NA | none | Erythroid hypoplasia with otherwise normal cellularity. | |
| VI-I | this paper, patient 1 | c.220+2T>C | missense | 7 months | 9.5 | ↑ | 100 | 75.4 | 5.5 | 4.1 | 525 | normal | YES | glucocorticoids | Mildly decreased erthropoiesis, dyserythropoiesis, increased megakaryopoiesis. |
| VII-I | this paper, patient 2 | c.2T>C | missense | 5 years | 8.2 | ↑ | 97 | NA | 4.8 | 3.1 | 652 | NA | YES,(partial) | glucocorticoids | Normal cellularity and erythroid activity with megaloblastic changes, no significant dysplastic features. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Dooijeweert, B.; Kia, S.K.; Dahl, N.; Fenneteau, O.; Leguit, R.; Nieuwenhuis, E.; van Solinge, W.; van Wijk, R.; Da Costa, L.; Bartels, M. GATA-1 Defects in Diamond–Blackfan Anemia: Phenotypic Characterization Points to a Specific Subset of Disease. Genes 2022, 13, 447. https://doi.org/10.3390/genes13030447
van Dooijeweert B, Kia SK, Dahl N, Fenneteau O, Leguit R, Nieuwenhuis E, van Solinge W, van Wijk R, Da Costa L, Bartels M. GATA-1 Defects in Diamond–Blackfan Anemia: Phenotypic Characterization Points to a Specific Subset of Disease. Genes. 2022; 13(3):447. https://doi.org/10.3390/genes13030447
Chicago/Turabian Stylevan Dooijeweert, Birgit, Sima Kheradmand Kia, Niklas Dahl, Odile Fenneteau, Roos Leguit, Edward Nieuwenhuis, Wouter van Solinge, Richard van Wijk, Lydie Da Costa, and Marije Bartels. 2022. "GATA-1 Defects in Diamond–Blackfan Anemia: Phenotypic Characterization Points to a Specific Subset of Disease" Genes 13, no. 3: 447. https://doi.org/10.3390/genes13030447