
Genome-Wide RNA-Sequencing Reveals Massive Circular RNA Expression Changes of the Neurotransmission Genes in the Rat Brain after Ischemia–Reperfusion

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Transient Cerebral Ischemia Rat Model
2.3. Magnetic Resonance Imaging
2.4. RNA Isolation
2.5. RNA-Seq
2.6. RNA-Seq Data Analysis
2.7. Total RNA RNase R-Treatment
2.8. cDNA Synthesis
2.9. Reverse Transcription Polymerase Chain Reaction (RT–PCR)
2.10. Electrophoresis of PCR Products
2.11. Sequencing of PCR Products
2.12. Real-Time RT–PCR
2.13. Data Analysis of Real-time RT-PCR and Statistics
2.14. Functional Analysis
2.15. Construction of circ-miRNA-mRNA Network
2.16. Availability of Data and Material
3. Results
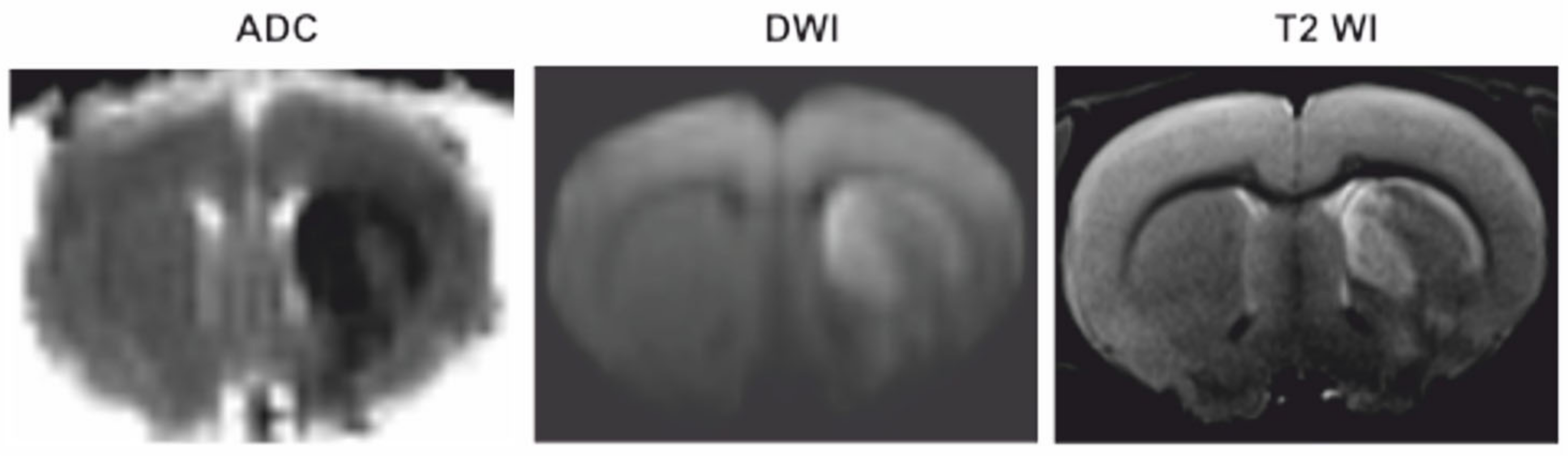
3.1. Magnetic Resonance Imaging (MRI)
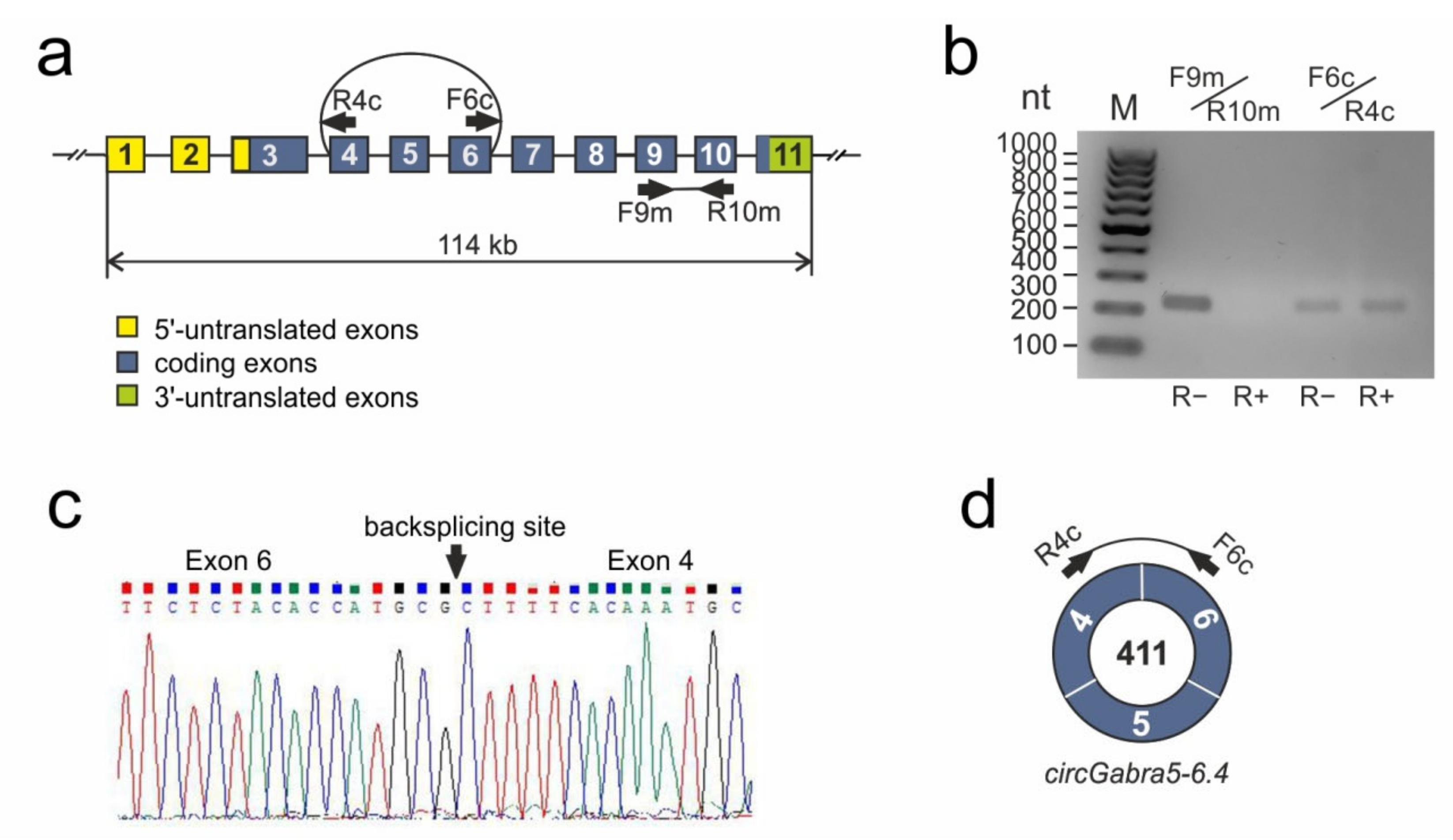
3.2. RNA-Seq Analysis of circRNA Diversity in tMCAO Rat Model Conditions
3.3. RNA-Seq Analysis of Differential Expression of circRNAs at 24 h after tMCAO
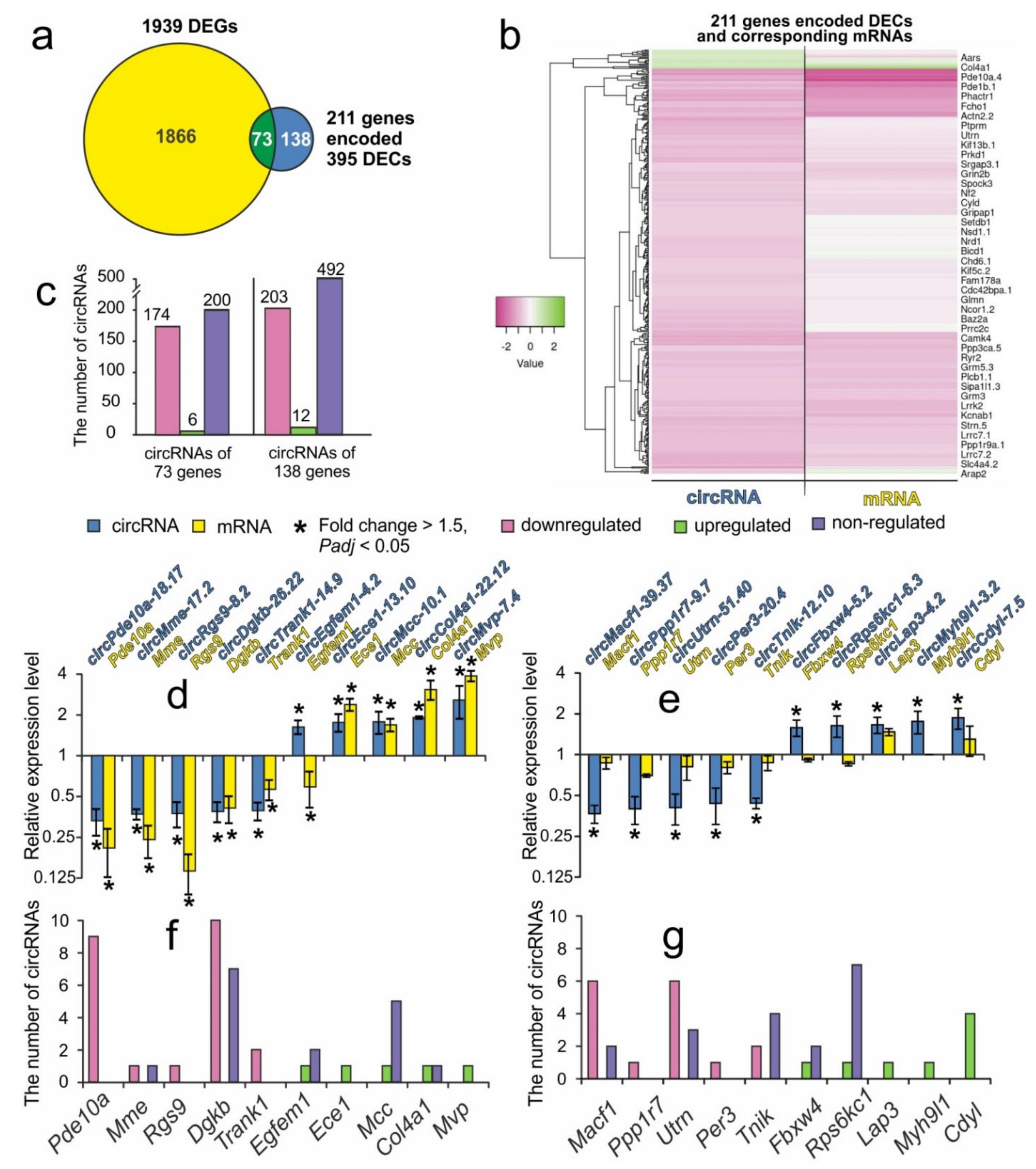
3.4. Comparative Analysis of Transcriptome Profile Changes of mRNAs and circRNAs at 24 h after tMCAO
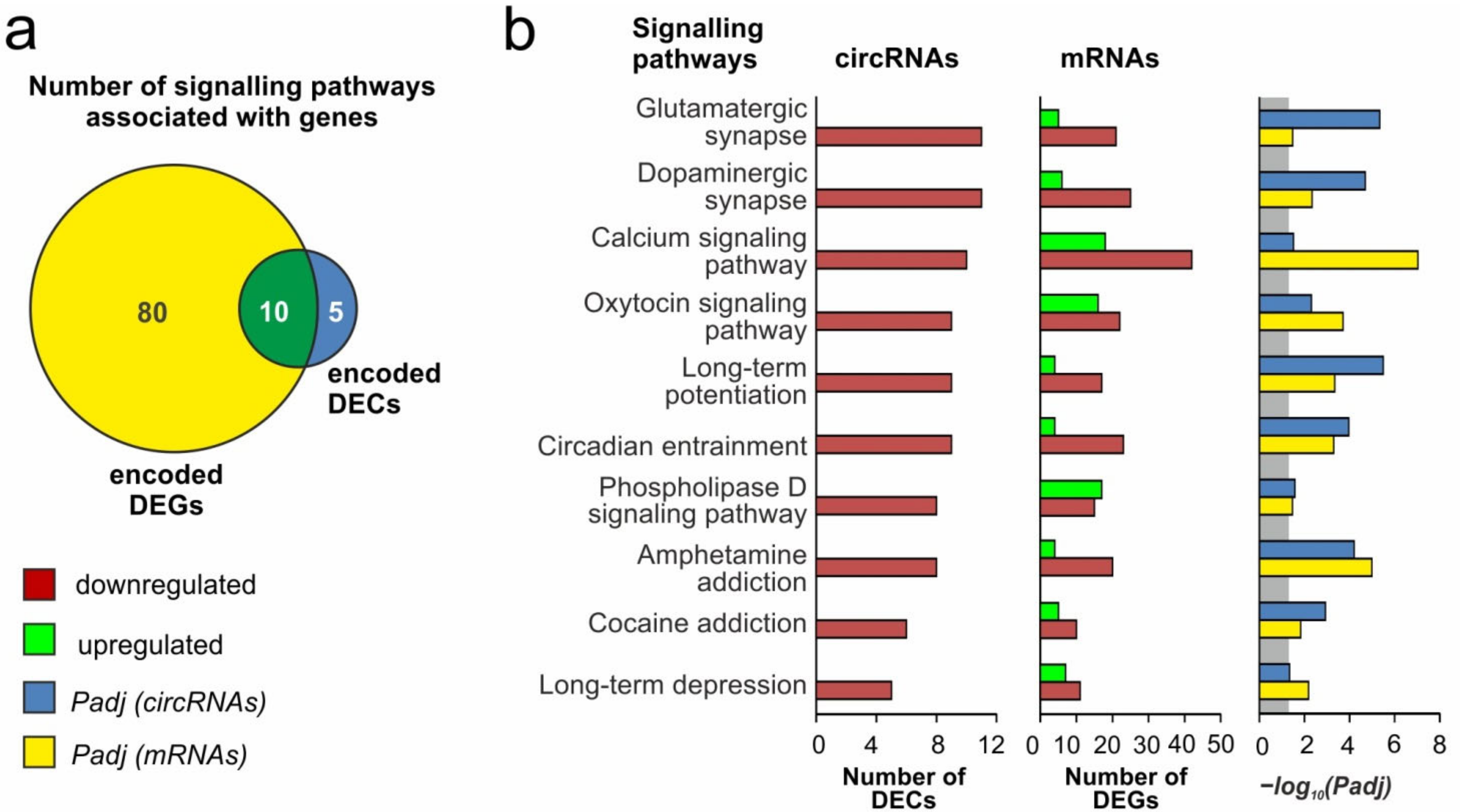
3.5. Differentially Expressed circRNAs Associated with Neurotransmitter Signaling Pathways at 24 h after tMCAO
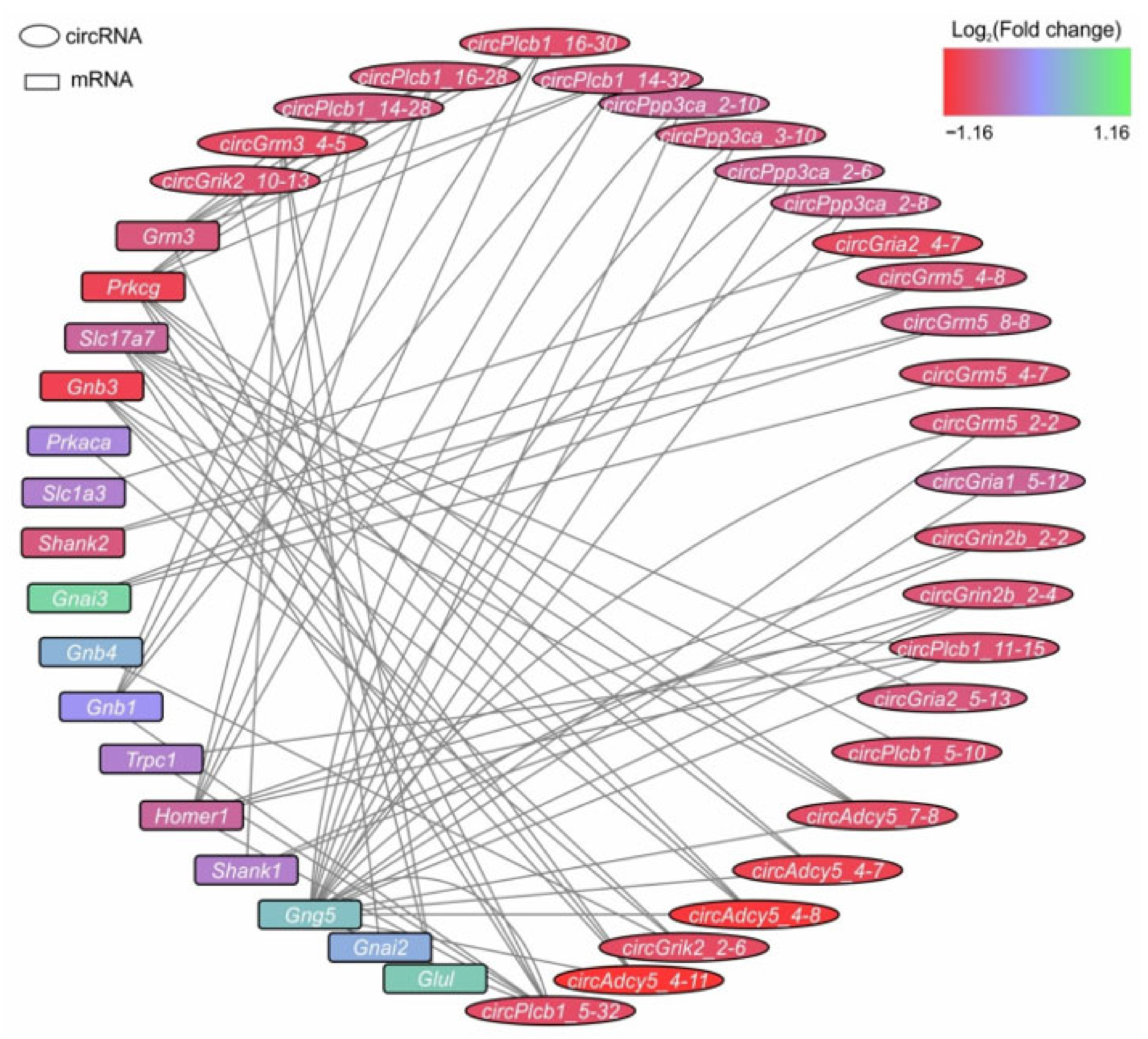
3.6. Analysis of circRNA–miRNA–mRNA Networks Associated with the ‘Glutamatergic Synapse’ Signaling Pathway at 24 h after tMCAO
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mukherjee, D.; Patil, C.G. Epidemiology and the global burden of stroke. World Neurosurg. 2011, 76, S85–S90. [Google Scholar] [CrossRef]
- GBD 2019 Stroke Collaborators Global, regional, and national burden of stroke and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet. Neurol. 2021, 12, 941–948.
- Arboix, A.; Jiménez, C.; Massons, J.; Parra, O.; Besses, C. Hematological disorders: A commonly unrecognized cause of acute stroke. Expert Rev. Hematol. 2016, 9, 891–901. [Google Scholar] [CrossRef]
- Wang, C.; Liu, M.; Pan, Y.; Bai, B.; Chen, J. Global gene expression profile of cerebral ischemia-reperfusion injury in rat MCAO model. Oncotarget 2017, 8, 74607–74622. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, G.A.; Estrada, E.Y.; Dencoff, J.E. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 1998, 29, 2189–2195. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Ford, G.; Xu, Z.; Gates, A.; Jiang, J.; Ford, B.D. Expression Analysis Systematic Explorer (EASE) analysis reveals differential gene expression in permanent and transient focal stroke rat models. Brain Res. 2006, 1071, 226–236. [Google Scholar] [CrossRef]
- DeGracia, D.J. Regulation of mRNA following brain ischemia and reperfusion. Wiley Interdiscip. Rev. RNA 2017, 8, e1415. [Google Scholar] [CrossRef]
- Yang, P.; Qiu, Z.; Jiang, Y.; Dong, L.; Yang, W.; Gu, C.; Li, G.; Zhu, Y. Silencing of cZNF292 circular RNA suppresses human glioma tube formation via the Wnt/β-catenin signaling pathway. Oncotarget 2016, 7, 63449–63455. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Dong, Y.; Huang, Z.; Kuang, Q.; Wu, Y.; Li, Y.; Li, M. Computational identifying and characterizing circular RNAs and their associated genes in hepatocellular carcinoma. PLoS ONE 2017, 12, e0174436. [Google Scholar] [CrossRef]
- Xia, W.; Qiu, M.; Chen, R.; Wang, S.; Leng, X.; Wang, J.; Xu, Y.; Hu, J.; Dong, G.; Xu, P.L.; et al. Circular RNA has_circ_0067934 is upregulated in esophageal squamous cell carcinoma and promoted proliferation. Sci. Rep. 2016, 6, 35576. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yao, Y.; Zhong, X.; Leng, K.; Qin, W.; Qu, L.; Cui, Y.; Jiang, X. Downregulated circular RNA hsa_circ_0001649 regulates proliferation, migration and invasion in cholangiocarcinoma cells. Biochem. Biophys. Res. Commun. 2018, 496, 455–461. [Google Scholar] [CrossRef]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [Green Version]
- Filippenkov, I.B.; Sudarkina, O.Y.; Limborska, S.A.; Dergunova, L.V. Multi-step splicing of sphingomyelin synthase linear and circular RNAs. Gene 2018, 654, 14–22. [Google Scholar] [CrossRef]
- Chen, B.J.; Yang, B.; Janitz, M. Region-specific expression of circular RNAs in the mouse brain. Neurosci. Lett. 2018, 666, 44–47. [Google Scholar] [CrossRef]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [Green Version]
- Filippenkov, I.B.; Sudarkina, O.Y.; Limborska, S.A.; Dergunova, L. V Circular RNA of the human sphingomyelin synthase 1 gene: Multiple splice variants, evolutionary conservatism and expression in different tissues. RNA Biol. 2015, 12, 1030–1042. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-P.; Ye, S.; Long, Y.; Fan, Y.; Mao, H.-F.; Chen, M.-T.; Ma, Q.-J. Circular RNA expression alterations are involved in OGD/R-induced neuron injury. Biochem. Biophys. Res. Commun. 2016, 471, 52–56. [Google Scholar] [CrossRef]
- Mehta, S.L.; Pandi, G.; Vemuganti, R. Circular RNA Expression Profiles Alter Significantly in Mouse Brain after Transient Focal Ischemia. Stroke 2017, 48, 2541–2548. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, C.; Yang, J.; Geng, X.; Du, H.; Ji, X.; Zhao, H. Screening circular RNA expression patterns following focal cerebral ischemia in mice. Oncotarget 2017, 8, 86535–86547. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Li, L.; Gan, J.; Peng, C.; Wang, X.; Chen, W.; Peng, D. Identification and functional analysis of circular RNAs induced in rats by middle cerebral artery occlusion. Gene 2019, 701, 139–145. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Wang, Y.-R.; Li, F.; Liu, X.-L.; Zhang, H.; Zhu, Z.-Z.; Huang, H.; Xu, X.-H. Circ-camk4 involved in cerebral ischemia/reperfusion induced neuronal injury. Sci. Rep. 2020, 10, 7012. [Google Scholar] [CrossRef]
- Han, B.; Zhang, Y.; Zhang, Y.; Bai, Y.; Chen, X.; Huang, R.; Wu, F.; Leng, S.; Chao, J.; Zhang, J.H.; et al. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142 -TIPARP: Implications for cerebral ischemic stroke. Autophagy 2018, 14, 1164–1184. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Zhang, Y.; Han, B.; Yang, L.; Chen, X.; Huang, R.; Wu, F.; Chao, J.; Liu, P.; Hu, G.; et al. Circular RNA DLGAP4 Ameliorates Ischemic Stroke Outcomes by Targeting miR-143 to Regulate Endothelial-Mesenchymal Transition Associated with Blood-Brain Barrier Integrity. J. Neurosci. 2018, 38, 32–50. [Google Scholar] [CrossRef]
- Tang, C.; Ou, J.; Kou, L.; Deng, J.; Luo, S. Circ_016719 plays a critical role in neuron cell apoptosis induced by I/R via targeting miR-29c/Map2k6. Mol. Cell. Probes 2020, 49, 101478. [Google Scholar] [CrossRef]
- Dai, Q.; Ma, Y.; Xu, Z.; Zhang, L.; Yang, H.; Liu, Q.; Wang, J. Downregulation of circular RNA HECTD1 induces neuroprotection against ischemic stroke through the microRNA-133b/TRAF3 pathway. Life Sci. 2021, 264, 118626. [Google Scholar] [CrossRef]
- Chen, L.; Luo, W.; Zhang, W.; Chu, H.; Wang, J.; Dai, X.; Cheng, Y.; Zhu, T.; Chao, J. circDLPAG4/HECTD1 mediates ischaemia/reperfusion injury in endothelial cells via ER stress. RNA Biol. 2020, 17, 240–253. [Google Scholar] [CrossRef]
- Zhu, X.; Ding, J.; Wang, B.; Wang, J.; Xu, M. Circular RNA DLGAP4 is down-regulated and negatively correlates with severity, inflammatory cytokine expression and pro-inflammatory gene miR-143 expression in acute ischemic stroke patients. Int. J. Clin. Exp. Pathol. 2019, 12, 941–948. [Google Scholar]
- Dergunova, L.V.; Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Mozerov, S.A.; Gubsky, L.V.; Limborska, S.A. Genome-wide transcriptome analysis using RNA-Seq reveals a large number of differentially expressed genes in a transient MCAO rat model. BMC Genom. 2018, 19, 655. [Google Scholar] [CrossRef] [Green Version]
- Dergunova, L.V.; Dmitrieva, V.G.; Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Sevan’kaeva, L.E.; Valieva, L.V.; Sudarkina, O.Y.; Gubsky, L.V.; et al. The Peptide Drug ACTH(4-7)PGP (Semax) Suppresses mRNA Transcripts Encoding Proinflammatory Mediators Induced by Reversible Ischemia of the Rat Brain. Mol. Biol. 2021, 55, 402–411. [Google Scholar]
- Sudarkina, O.Y.; Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Sevan’kaeva, L.E.; Valieva, L.V.; Remizova, J.A.; Dmitrieva, V.G.; Gubsky, L.V.; et al. Brain Protein Expression Profile Confirms the Protective Effect of the ACTH(4-7)PGP Peptide (Semax) in a Rat Model of Cerebral Ischemia-Reperfusion. Int. J. Mol. Sci. 2021, 22, 6179. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Izuogu, O.G.; Alhasan, A.A.; Alafghani, H.M.; Santibanez-Koref, M.; Elliott, D.J.; Elliot, D.J.; Jackson, M.S. PTESFinder: A computational method to identify post-transcriptional exon shuffling (PTES) events. BMC Bioinformatics 2016, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. NCBI Reference Sequence Databases for Rnor_5.0. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=Rnor_5.0 (accessed on 1 September 2021).
- National Center for Biotechnology Information. NCBI Reference Sequence Databases for Rnor_6.0. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001895.5/ (accessed on 1 September 2021).
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. CircBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Wu, H.; Wang, T.; Yu, S.; Han, Z.; Zhang, W.; Mu, L.; Wang, H.; Na, M.; Wang, H.; et al. Profiling Analysis of Circular RNA and mRNA in Human Temporal Lobe Epilepsy with Hippocampal Sclerosis ILAE Type 1. Cell. Mol. Neurobiol. 2021, 12, 697. [Google Scholar] [CrossRef]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [Green Version]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. RNA Sequencing Data in the Sequence Read Archive Database under Accession Code PRJNA523319. Available online: https://www.ncbi.nlm.nih.gov/sra/PRJNA523319 (accessed on 18 February 2020).
- National Center for Biotechnology Information. The Annotated Sequence in the GenBank with the Accession Number MK520929. Available online: https://www.ncbi.nlm.nih.gov/nuccore/MK520929 (accessed on 19 February 2020).
- Nieswandt, B.; Kleinschnitz, C.; Stoll, G. Ischaemic stroke: A thrombo-inflammatory disease? J. Physiol. 2011, 589, 4115–4123. [Google Scholar] [CrossRef]
- Ryang, Y.-M.; Dang, J.; Kipp, M.; Petersen, K.-U.; Fahlenkamp, A.V.; Gempt, J.; Wesp, D.; Rossaint, R.; Beyer, C.; Coburn, M. Solulin reduces infarct volume and regulates gene-expression in transient middle cerebral artery occlusion in rats. BMC Neurosci. 2011, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.D.; Piccini, J.P.; Hylek, E.M.; Granger, C.B.; Alexander, J.H. Antithrombotic therapy in atrial fibrillation: Guidelines translated for the clinician. J. Thromb. Thrombolysis 2008, 26, 167–174. [Google Scholar] [CrossRef]
- Alexandrov, A.V.; Hall, C.E.; Labiche, L.A.; Wojner, A.W.; Grotta, J.C. Ischemic stunning of the brain: Early recanalization without immediate clinical improvement in acute ischemic stroke. Stroke 2004, 35, 449–452. [Google Scholar] [CrossRef] [Green Version]
- Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Sevan’kaeva, L.E.; Sudarkina, O.Y.; Dmitrieva, V.G.; Gubsky, L.V.; Myasoedov, N.F.; Limborska, S.A.; et al. Novel insights into the protective properties of acth(4-7)pgp (semax) peptide at the transcriptome level following cerebral ischaemia– reperfusion in rats. Genes 2020, 11, 681. [Google Scholar] [CrossRef]
- Khvorykh, G.; Khrunin, A.; Filippenkov, I.; Stavchansky, V.; Dergunova, L.; Limborska, S. A Workflow for Selection of Single Nucleotide Polymorphic Markers for Studying of Genetics of Ischemic Stroke Outcomes. Genes 2021, 12, 328. [Google Scholar] [CrossRef]
- Feng, D.; Li, Z.; Wang, G.; Yao, J.; Li, Y.; Qasim, W.; Zhao, Y.; Tian, X. Microarray Analysis of Differentially Expressed Profiles of Circular RNAs in a Mouse Model of Intestinal Ischemia/Reperfusion Injury with and Without Ischemic Postconditioning. Cell. Physiol. Biochem. 2018, 48, 1579–1594. [Google Scholar] [CrossRef]
- Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Ivanova, K.A.; Limborska, S.A.; Dergunova, L.V. Experimental Cerebral Ischemia Affects the Expression of Circular RNA Genes of Metabotropic Glutamate Receptors mGluR3 and mGluR5 in Rat Brain. Russ. J. Bioorganic Chem. 2018, 44, 302–309. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Piwecka, M.; Glažar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef] [Green Version]
- Akhter, R. Circular RNA and Alzheimer’s Disease. Circ. RNA 2018, 239–243. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, L.; Wang, S.; Hong, Q. Circular RNA circDLGAP4 exerts neuroprotective effects via modulating miR-134-5p/CREB pathway in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2020, 522, 388–394. [Google Scholar] [CrossRef]
- Chen, H.; Liu, T.; Liu, J.; Feng, Y.; Wang, B.; Wang, J.; Bai, J.; Zhao, W.; Shen, Y.; Wang, X.; et al. Circ-ANAPC7 is Upregulated in Acute Myeloid Leukemia and Appears to Target the MiR-181 Family. Cell. Physiol. Biochem. 2018, 47, 1998–2007. [Google Scholar] [CrossRef]
- Li, F.; Zhang, L.; Li, W.; Deng, J.; Zheng, J.; An, M.; Lu, J.; Zhou, Y. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/β-catenin pathway. Oncotarget 2015, 6, 6001–6013. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kong, F.; Lou, S.; Yang, D.; Gu, L. Global identification of circular RNAs in chronic myeloid leukemia reveals hsa_circ_0080145 regulates cell proliferation by sponging miR-29b. Biochem. Biophys. Res. Commun. 2018, 504, 660–665. [Google Scholar] [CrossRef]
- Sun, S.; Kong, Q.; Cai, Z.; Wang, M.; Zhao, H.; Zhao, C. circ-Grm1 promotes pulmonary artery smooth muscle cell proliferation and migration via suppression of GRM1 expression by FUS. Int. J. Mol. Med. 2021, 48, 1–12. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, Y.; Xiong, W.; Zhang, Z.; Wang, Z.; Lv, L.; Liu, C.; Hu, Z.; Zheng, Y.-T.; Lu, L.; et al. CircGRIA1 shows an age-related increase in male macaque brain and regulates synaptic plasticity and synaptogenesis. Nat. Commun. 2020, 11, 3594. [Google Scholar] [CrossRef]
- Lu, J.; Li, Y. Circ_0079593 facilitates proliferation, metastasis, glucose metabolism and inhibits apoptosis in melanoma by regulating the miR-516b/GRM3 axis. Mol. Cell. Biochem. 2020, 475, 227–237. [Google Scholar] [CrossRef]
- Fang, G.; Wu, Y.; Zhang, X. CircASXL1 knockdown represses the progression of colorectal cancer by downregulating GRIK3 expression by sponging miR-1205. World J. Surg. Oncol. 2021, 19, 176. [Google Scholar] [CrossRef]
- Du, H.; He, Z.; Feng, F.; Chen, D.; Zhang, L.; Bai, J.; Wu, H.; Han, E.; Zhang, J. Hsa_circ_0038646 promotes cell proliferation and migration in colorectal cancer via miR-331-3p/GRIK3. Oncol. Lett. 2020, 20, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; He, J.; Wang, B. Circular RNA circ_HECTD1 regulates cell injury after cerebral infarction by miR-27a-3p/FSTL1 axis. Cell Cycle 2021, 20, 914–926. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Zhang, X.; Zhai, H.; Sun, X.; Li, Y. Circ_0091579 Serves as a Tumor-Promoting Factor in Hepatocellular Carcinoma Through miR-1225-5p/PLCB1 Axis. Dig. Dis. Sci. 2021, 1–13. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Jiang, W.; Zhang, H.; Wen, Z.; Su, Y.; Wu, F.; Zhi, Z.; Shen, Q.; Li, H.; et al. Down-regulation of circ-PRKCI inhibits cell migration and proliferation in Hirschsprung disease by suppressing the expression of miR-1324 target PLCB1. Cell Cycle 2018, 17, 1092–1101. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.-F.; Cheng, Y.; Dong, Q.-R.; Zheng, M.-Q. MicroRNA-675-3p regulates IL-1β-stimulated human chondrocyte apoptosis and cartilage degradation by targeting GNG5. Biochem. Biophys. Res. Commun. 2020, 527, 458–465. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. RNA-Seq Information in NCBI Those are Different Sequencing Data Search Results in NCBI. Available online: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP148632 (accessed on 6 February 2020).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Valieva, L.V.; Remizova, J.A.; Mozgovoy, I.V.; Zaytceva, E.I.; Gubsky, L.V.; Limborska, S.A.; Dergunova, L.V. Genome-Wide RNA-Sequencing Reveals Massive Circular RNA Expression Changes of the Neurotransmission Genes in the Rat Brain after Ischemia–Reperfusion. Genes 2021, 12, 1870. https://doi.org/10.3390/genes12121870
Filippenkov IB, Stavchansky VV, Denisova AE, Valieva LV, Remizova JA, Mozgovoy IV, Zaytceva EI, Gubsky LV, Limborska SA, Dergunova LV. Genome-Wide RNA-Sequencing Reveals Massive Circular RNA Expression Changes of the Neurotransmission Genes in the Rat Brain after Ischemia–Reperfusion. Genes. 2021; 12(12):1870. https://doi.org/10.3390/genes12121870
Chicago/Turabian StyleFilippenkov, Ivan B., Vasily V. Stavchansky, Alina E. Denisova, Liya V. Valieva, Julia A. Remizova, Ivan V. Mozgovoy, Elizaveta I. Zaytceva, Leonid V. Gubsky, Svetlana A. Limborska, and Lyudmila V. Dergunova. 2021. "Genome-Wide RNA-Sequencing Reveals Massive Circular RNA Expression Changes of the Neurotransmission Genes in the Rat Brain after Ischemia–Reperfusion" Genes 12, no. 12: 1870. https://doi.org/10.3390/genes12121870