
Skeletal Deficits in Male and Female down Syndrome Model Mice Arise Independent of Normalized Dyrk1a Expression in Osteoblasts

Abstract
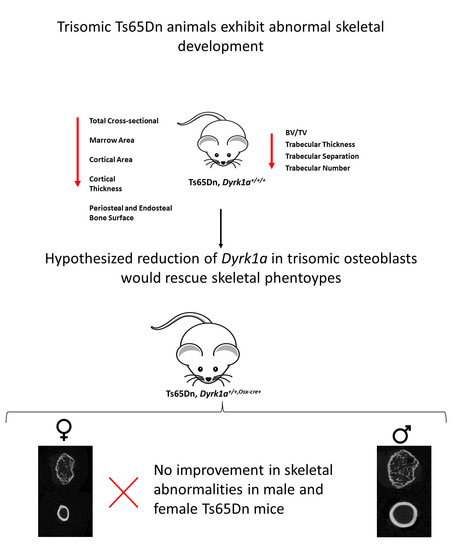
:
1. Introduction
1.1. Skeletal Abnormalities in DS
1.2. Development of Skeletal Abnormalities in Individuals with DS
1.3. Associated Skeletal Abnormalities from DS Mouse Models
1.4. DYRK1A Gene Dosage Contributes to the Development of Skeletal Phenotypes in Ts21
1.5. Hypothesis; Reduction of Dyrk1a in Osteoblasts Will Improve Skeletal Deficits Associated with Ts21 in Ts65Dn Male and Female Mice
2. Materials and Methods
2.1. Animals
2.2. Genotyping
2.3. Microcomputed Tomography (µCT)
2.4. Mechanical Testing
2.5. Statistical Analysis
3. Results
3.1. Suspected Perinatal Death of Ts65Dn,Dyrk1a+/+/Osx-Cre Male Mice
3.2. Trabecular Deficits in Female and Trisomic Mice
3.3. Skeletal Alterations in Cortical Architecture in Trisomic Mice
3.4. Dyrk1a Overexpression in Osteoblasts Does Not Impair Whole Bone or Material Properties as Demonstrated by Mechanical Testing
3.4.1. Whole Bone Mechanical Properties
3.4.2. Material Level Properties
4. Discussion
4.1. Bone Deficits in Ts65Dn Female Mice
4.2. Effects of Dyrk1a Copy Number Reduction in Osteoblasts
4.3. Potential Mechanisms for Dyrk1a Copy Number in Ts65Dn Mice
4.4. Limitations: Potential Uncharacterized Effects of DYRK1A and Low Sample Size of Male Ts65Dn,Dyrk1a+/+/Osx-Cre on Differences Skeletal Parameters
4.5. Limitation: Potential off Target Effects of Osx-Cre Transgene
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angelopoulou, N.; Souftas, V.; Sakadamis, A.; Mandroukas, K. Bone mineral density in adults with Down’s syndrome. Eur. Radiol. 1999, 9, 648–651. [Google Scholar] [CrossRef]
- De Moraes, M.E.; Tanaka, J.L.; de Moraes, L.C.; Filho, E.M.; de Melo Castilho, J.C. Skeletal age of individuals with Down syndrome. Spec. Care Dentist. 2008, 28, 101–106. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, G.; Buckley, F.; Skotko, B.G. Estimation of the number of people with Down syndrome in the United States. Genet. Med. 2017, 19, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.J. Down Syndrome (Trisomy 21). In The Online Metabolic and Molecular Bases of Inherited Disease; Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K.M., Mitchell, G., Eds.; The McGraw-Hill Companies, Inc.: New York, NY, USA, 2014. [Google Scholar]
- Parker, S.E.; Mai, C.T.; Canfield, M.A.; Rickard, R.; Wang, Y.; Meyer, R.E.; Anderson, P.; Mason, C.A.; Collins, J.S.; Kirby, R.S.; et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Carfi, A.; Liperoti, R.; Fusco, D.; Giovannini, S.; Brandi, V.; Vetrano, D.L.; Meloni, E.; Mascia, D.; Villani, E.R.; Manes Gravina, E.; et al. Bone mineral density in adults with Down syndrome. Osteoporos. Int. 2017, 28, 2929–2934. [Google Scholar] [CrossRef]
- Costa, R.; Gullon, A.; De Miguel, R.; de Asua, D.R.; Bautista, A.; Garcia, C.; Suarez, C.; Castaneda, S.; Moldenhauer, F. Bone Mineral Density Distribution Curves in Spanish Adults with Down Syndrome. J. Clin. Densitom. 2018, 21, 493–500. [Google Scholar] [CrossRef]
- Tang, J.Y.M.; Luo, H.; Wong, G.H.Y.; Lau, M.M.Y.; Joe, G.M.; Tse, M.A.; Ip, P.; Wong, I.C.K.; Lum, T.Y.S. Bone mineral density from early to middle adulthood in persons with Down syndrome. J. Intellect. Disabil. Res. 2019, 643, 936–946. [Google Scholar] [CrossRef]
- Rarick, G.L.; Rapaport, I.F.; Seefeldt, V. Long bone growth in Down’s syndrome. Am. J. Dis. Child. 1966, 112, 566–571. [Google Scholar] [CrossRef]
- Bittles, A.H.; Bower, C.; Hussain, R.; Glasson, E.J. The four ages of Down syndrome. Eur. J. Public Health 2007, 17, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Bittles, A.H.; Glasson, E.J. Clinical, social, and ethical implications of changing life expectancy in Down syndrome. Dev. Med. Child Neurol. 2004, 46, 282–286. [Google Scholar] [CrossRef]
- Esbensen, A.J. Health conditions associated with aging and end of life of adults with Down syndrome. Int. Rev. Res. Ment. Retard. 2010, 39, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center, J.; Beange, H.; McElduff, A. People with mental retardation have an increased prevalence of osteoporosis: A population study. Am. J. Ment. Retard. AJMR 1998, 103, 19–28. [Google Scholar] [CrossRef]
- Costa, R.; De Miguel, R.; Garcia, C.; de Asua, D.R.; Castaneda, S.; Moldenhauer, F.; Suarez, C. Bone Mass Assessment in a Cohort of Adults with Down Syndrome: A Cross-Sectional Study. Intellect. Dev. Disabil. 2017, 55, 315–324. [Google Scholar] [CrossRef]
- Baptista, F.; Varela, A.; Sardinha, L.B. Bone mineral mass in males and females with and without Down syndrome. Osteoporos. Int. 2005, 16, 380–388. [Google Scholar] [CrossRef]
- Garcia Hoyos, M.; Humbert, L.; Salmon, Z.; Riancho, J.A.; Valero, C. Analysis of volumetric BMD in people with Down syndrome using DXA-based 3D modeling. Arch. Osteoporos. 2019, 14, 98. [Google Scholar] [CrossRef]
- Garcia-Hoyos, M.; Garcia-Unzueta, M.T.; de Luis, D.; Valero, C.; Riancho, J.A. Diverging results of areal and volumetric bone mineral density in Down syndrome. Osteoporos. Int. 2017, 28, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Aguero, A.; Vicente-Rodriguez, G.; Gomez-Cabello, A.; Casajus, J.A. Cortical and trabecular bone at the radius and tibia in male and female adolescents with Down syndrome: A peripheral quantitative computed tomography (pQCT) study. Osteoporos. Int. 2013, 24, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Guijarro, M.; Valero, C.; Paule, B.; Gonzalez-Macias, J.; Riancho, J.A. Bone mass in young adults with Down syndrome. J. Intellect. Disabil. Res. 2008, 52, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.R.; LaCombe, J.; Long, R.; Lana-Elola, E.; Watson-Scales, S.; Wallace, J.M.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Roper, R.J. Interaction of sexual dimorphism and gene dosage imbalance in skeletal deficits associated with Down syndrome. Bone 2020, 136, 115367. [Google Scholar] [CrossRef]
- McKelvey, K.D.; Fowler, T.W.; Akel, N.S.; Kelsay, J.A.; Gaddy, D.; Wenger, G.R.; Suva, L.J. Low bone turnover and low bone density in a cohort of adults with Down syndrome. Osteoporos. Int. 2013, 24, 1333–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelopoulou, N.; Matziari, C.; Tsimaras, V.; Sakadamis, A.; Souftas, V.; Mandroukas, K. Bone mineral density and muscle strength in young men with mental retardation (with and without Down syndrome). Calcif. Tissue Int. 2000, 66, 176–180. [Google Scholar] [CrossRef] [PubMed]
- LaCombe, J.M.; Roper, R.J. Skeletal dynamics of Down syndrome: A developing perspective. Bone 2020, 133, 115215. [Google Scholar] [CrossRef]
- Blazek, J.D.; Malik, A.M.; Tischbein, M.; Arbones, M.L.; Moore, C.S.; Roper, R.J. Abnormal mineralization of the Ts65Dn Down syndrome mouse appendicular skeleton begins during embryonic development in a Dyrk1a-independent manner. Mech. Dev. 2015, 136, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.W.; McKelvey, K.D.; Akel, N.S.; Vander Schilden, J.; Bacon, A.W.; Bracey, J.W.; Sowder, T.; Skinner, R.A.; Swain, F.L.; Hogue, W.R.; et al. Low bone turnover and low BMD in Down syndrome: Effect of intermittent PTH treatment. PLoS ONE 2012, 7, e42967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimwood, J.S.; Kumar, A.; Bickerstaff, D.R.; Suvarna, S.K. Histological assessment of vertebral bone in a Down’s syndrome adult with osteoporosis. Histopathology 2000, 36, 279–280. [Google Scholar] [CrossRef]
- Lee, Y.; Ha, J.; Kim, H.J.; Kim, Y.S.; Chang, E.J.; Song, W.J.; Kim, H.H. Negative feedback Inhibition of NFATc1 by DYRK1A regulates bone homeostasis. J. Biol. Chem. 2009, 284, 33343–33351. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.K.; Parham, S.G.; Schryver, E.; Akel, N.S.; Shelton, R.S.; Webber, J.; Swain, F.L.; Schmidt, J.; Suva, L.J.; Gaddy, D. Sclerostin Antibody Treatment Stimulates Bone Formation to Normalize Bone Mass in Male Down Syndrome Mice. JBMR Plus 2018, 2, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Woodward, J.F.; Jan, S.; Ciccarelli, M.R. Guidelines for Care of Adults with Down Syndrome. JAMA 2020, 324, 1509–1511. [Google Scholar] [CrossRef]
- Abeysekera, I.; Thomas, J.; Georgiadis, T.M.; Berman, A.G.; Hammond, M.A.; Dria, K.J.; Wallace, J.M.; Roper, R.J. Differential effects of Epigallocatechin-3-gallate containing supplements on correcting skeletal defects in a Down syndrome mouse model. Mol. Nutr. Food Res. 2016, 60, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Blazek, J.D.; Abeysekera, I.; Li, J.; Roper, R.J. Rescue of the abnormal skeletal phenotype in Ts65Dn Down syndrome mice using genetic and therapeutic modulation of trisomic Dyrk1a. Hum. Mol. Genet. 2015, 24, 5687–5696. [Google Scholar] [CrossRef] [Green Version]
- Kamalakar, A.; Harris, J.R.; McKelvey, K.D.; Suva, L.J. Aneuploidy and skeletal health. Curr. Osteoporos. Rep. 2014, 12, 376–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazek, J.D.; Gaddy, A.; Meyer, R.; Roper, R.J.; Li, J. Disruption of bone development and homeostasis by trisomy in Ts65Dn Down syndrome mice. Bone 2011, 48, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.D.; Mjaatvedt, C.H.; Moore, C.S. Characterization of the cardiac phenotype in neonatal Ts65Dn mice. Dev. Dyn. 2008, 237, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Lana-Elola, E.; Watson-Scales, S.; Slender, A.; Gibbins, D.; Martineau, A.; Douglas, C.; Mohun, T.; Fisher, E.M.; Tybulewicz, V. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. eLife 2016, 5, e11614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, W.; Sippl, W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef]
- Tejedor, F.J.; Hammerle, B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Fotaki, V.; Dierssen, M.; Alcantara, S.; Martinez, S.; Marti, E.; Casas, C.; Visa, J.; Soriano, E.; Estivill, X.; Arbones, M.L. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002, 22, 6636–6647. [Google Scholar] [CrossRef] [Green Version]
- Dowjat, W.K.; Adayev, T.; Kuchna, I.; Nowicki, K.; Palminiello, S.; Hwang, Y.W.; Wegiel, J. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci. Lett. 2007, 413, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, M.F.; Meijer, L. Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Thompson, B.J.; Bhansali, R.; Diebold, L.; Cook, D.E.; Stolzenburg, L.; Casagrande, A.S.; Besson, T.; Leblond, B.; Desire, L.; Malinge, S.; et al. DYRK1A controls the transition from proliferation to quiescence during lymphoid development by destabilizing Cyclin D3. J. Exp. Med. 2015, 212, 953–970. [Google Scholar] [CrossRef] [Green Version]
- Reinholdt, L.G.; Ding, Y.; Gilbert, G.J.; Czechanski, A.; Solzak, J.P.; Roper, R.J.; Johnson, M.T.; Donahue, L.R.; Lutz, C.; Davisson, M.T. Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm. Genome 2011, 22, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodda, S.J.; McMahon, A.P. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 2006, 133, 3231–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, A.G.; Clauser, C.A.; Wunderlin, C.; Hammond, M.A.; Wallace, J.M. Structural and Mechanical Improvements to Bone Are Strain Dependent with Axial Compression of the Tibia in Female C57BL/6 Mice. PLoS ONE 2015, 10, e0130504. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.M.; Golcuk, K.; Morris, M.D.; Kohn, D.H. Inbred strain-specific response to biglycan deficiency in the cortical bone of C57BL6/129 and C3H/He mice. J. Bone Miner. Res. 2009, 24, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.M.; Rajachar, R.M.; Chen, X.D.; Shi, S.; Allen, M.R.; Bloomfield, S.A.; Les, C.M.; Robey, P.G.; Young, M.F.; Kohn, D.H. The mechanical phenotype of biglycan-deficient mice is bone- and gender-specific. Bone 2006, 39, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.S.; Park, J.H.; Ryu, Y.S.; Choi, S.H.; Yoon, S.H.; Kwen, M.Y.; Oh, J.Y.; Song, W.J.; Chung, S.H. Regulation of RCAN1 protein activity by Dyrk1A protein-mediated phosphorylation. J. Biol. Chem. 2011, 286, 40401–40412. [Google Scholar] [CrossRef] [Green Version]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, J.J.; Genesca, L.; Kingsbury, T.J.; Cunningham, K.W.; Perez-Riba, M.; Estivill, X.; de la Luna, S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum. Mol. Genet. 2000, 9, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Jeong, B.C.; Nam, K.I.; Kim, K.K.; Molkentin, J.D.; Kim, N. RCANs regulate the convergent roles of NFATc1 in bone homeostasis. Sci. Rep. 2016, 6, 38526. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C. Molecular mechanisms of osteoblast-specific transcription factor Osterix effect on bone formation. Beijing Da Xue Xue Bao Yi Xue Ban 2012, 44, 659–665. [Google Scholar]
- Larsen, L.J.; Moller, L.B. Crosstalk of Hedgehog and mTORC1 Pathways. Cells 2020, 9, 2316. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lauth, M. Emerging Roles of DYRK Kinases in Embryogenesis and Hedgehog Pathway Control. J. Dev. Biol. 2017, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T. Regulation of osteoblast differentiation by Runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of osteoblast differentiation by transcription factors. J. Cell Biochem. 2006, 99, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Rutkovskiy, A.; Stenslokken, K.O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckx, C.; Benitez-Burraco, A. Osteogenesis and neurogenesis: A robust link also for language evolution. Front. Cell. Neurosci. 2015, 9, 291. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shi, Y.; Regan, J.; Karuppaiah, K.; Ornitz, D.M.; Long, F. Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PLoS ONE 2014, 9, e85161. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Olsen, B.R. Skeletal defects in Osterix-Cre transgenic mice. Transgenic Res. 2015, 24, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhang, Z.; Feng, J.Q.; Dusevich, V.M.; Sinha, K.; Zhang, H.; Darnay, B.G.; de Crombrugghe, B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc. Natl. Acad. Sci. USA 2010, 107, 12919–12924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, R.A.; Clarke, M.V.; Sastra, S.; Skinner, J.P.; Chiang, C.; Anderson, P.H.; Zajac, J.D. Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res. 2012, 21, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Song, W.J.; Chung, K.C. Function and regulation of Dyrk1A: Towards understanding Down syndrome. Cell. Mol. Life Sci. 2009, 66, 3235–3240. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eu,Dyrk1a+/+ | Eu,Dyrk1a+/Osx-Cre | Ts65Dn,Dyrk1a+/+/+ | Ts65Dn,Dyrk1a+/+/Osx-Cre | p-Value | |||
|---|---|---|---|---|---|---|---|
| Female | (n = 20) | (n = 11) | (n = 9) | (n = 10) | Dyrk1a Normalization | Ploidy | Dyrk1a Normalization * Ploidy |
| Yield Force (N) a,b,* | 6.63 (0.36) | 5.80 (0.14) | 5.10 (0.87) | 5.25 (0.45) | 0.5106 | 0.0455 | 0.3327 |
| Ultimate Force (N) b,& | 11.78 (0.46) | 9.15 (0.19) | 9.19 (0.62) | 9.97 (0.63) | 0.1032 | 0.1191 | 0.0035 |
| Displacement to Yield (µm) a,& | 106.16 (4.14) | 153.50 (9.50) | 113.69 (15.05) | 101.72 (3.73) | 0.1384 | 0.0655 | 0.0149 |
| Postyield Displacement (µm) a,* | 978.55 (84.18) | 1376.44 (155.13) | 663.44 (117.96) | 935.20 (130.64) | 0.0776 | 0.0473 | 0.7354 |
| Total Displacment (µm) a,* | 1084.71 (83.85) | 1529.94 (161.00) | 777.14 (107.12) | 1036.92 (131.25) | 0.0694 | 0.0402 | 0.6271 |
| Stiffness (N/mm) & | 67.62 (2.57) | 45.77 (2.01) | 47.73 (3.79) | 55.83 (3.41) | 0.0614 | 0.1774 | 0.0001 |
| Work to Yield (mJ) a | 0.38 (0.04) | 0.50 (0.04) | 0.37 (0.13) | 0.29 (0.03) | 0.7784 | 0.1165 | 0.1805 |
| Postyield Work (mJ) b,* | 8.18 (0.61) | 7.07 (0.50) | 4.64 (0.52) | 6.35 (0.68) | 0.7095 | 0.0109 | 0.0845 |
| Total Work (mJ) b | 8.56 (0.62) | 7.57 (0.52) | 5.00 (0.45) | 6.64 (0.70) | 0.6959 | 0.0085 | 0.1143 |
| Yield Stress (MPa) a | 82.19 (2.20) | 87.97 (2.76) | 79.15 (9.48) | 78.71 (5.91) | 0.6257 | 0.2647 | 0.5704 |
| Ultimate Stress (MPa) | 147.48 (3.37) | 137.67 (2.74) | 147.94 (5.91) | 149.29 (5.75) | 0.4145 | 0.2453 | 0.2828 |
| Strain to Yield (µε) a | 28,589 (1277) | 35,512 (1962) | 28,788 (4329) | 25,495 (932) | 0.5195 | 0.0859 | 0.0743 |
| Total Strain (µε) a,* | 291,841 (23,106) | 358,541 (35,955) | 191,803 (24,831) | 255,989 (29,761) | 0.1438 | 0.0259 | 0.9773 |
| Modulus (GPa) | 3.17 (0.09) | 2.86 (0.09) | 3.08 (0.16) | 3.35 (0.16) | 0.9107 | 0.1822 | 0.0583 |
| Resilience (MPa) a,b | 1.27 (0.09) | 1.76 (0.14) | 1.40 (0.42) | 1.08 (0.12) | 0.6964 | 0.2334 | 0.0807 |
| Toughness (MPa) a | 28.97 (1.94) | 26.54 (1.62) | 20.84 (2.37) | 25.36 (2.85) | 0.0745 | 0.0973 | 0.2127 |
| Eu,Dyrk1a+/+ | Eu,Dyrk1a+/Osx-Cre | Ts65Dn,Dyrk1a+/+/+ | Ts65Dn,Dyrk1a+/+/Osx-Cre | p-Value | |||
|---|---|---|---|---|---|---|---|
| Male | (n = 16) | (n = 7) | (n = 13) | (n = 8) | Dyrk1a Normalization | Ploidy | Dyrk1a Normalization * Ploidy |
| Yield Force (N) a,b,* | 7.95 (0.74) | 8.32 (1.33) | 7.21 (0.92) | 5.56 (1.06) | 0.1016 | 0.0471 | 0.4843 |
| Ultimate Force (N) b,* | 12.45 (0.52) | 11.54 (1.31) | 10.33 (0.74) | 9.56 (1.24) | 0.3476 | 0.0267 | 0.9329 |
| Displacement to Yield (µm) a | 187.06 (23.34) | 245.07 (47.74) | 231.57 (42.75) | 149.73 (18.03) | 0.2032 | 0.5269 | 0.1982 |
| Postyield Displacement (µm) a | 690.33 (116.99) | 385.26 (114.53) | 577.24 (135.32) | 783.98 (250.40) | 0.8848 | 0.3856 | 0.1562 |
| Total Displacment (µm) a | 877.40 (112.20) | 630.33 (88.40) | 808.81 (122.85) | 933.71 (246.17) | 0.6786 | 0.4344 | 0.2188 |
| Stiffness (N/mm) | 49.35 (3.46) | 38.75 (8.39) | 61.72 (25.69) | 38.33 (3.81) | 0.5931 | 0.1162 | 0.5161 |
| Work to Yield (mJ) a | 0.90 (0.15) | 1.21 (0.35) | 1.09 (0.27) | 0.47 (0.13) | 0.2256 | 0.2385 | 0.2757 |
| Postyield Work (mJ) b | 6.81 (0.93) | 3.95 (1.16) | 4.75 (0.94) | 5.88 (1.49) | 0.5343 | 0.9722 | 0.1374 |
| Total Work (mJ) b | 7.71 (0.91) | 5.16 (0.93) | 5.84 (0.80) | 6.36 (1.51) | 0.3604 | 0.7713 | 0.1784 |
| Yield Stress (MPa) a | 91.06 (9.01) | 104.85 (14.81) | 105.63 (13.18) | 79.95 (11.57) | 0.1264 | 0.6647 | 0.2141 |
| Ultimate Stress (MPa) | 140.68 (3.46) | 140.90 (6.28) | 146.48 (4.90) | 140.53 (8.88) | 0.6097 | 0.6287 | 0.5822 |
| Strain to Yield (µε) a | 45,435 (5358) | 56,656 (10,416) | 52,890 (10,025) | 32,576 (5181) | 0.15 | 0.3139 | 0.1814 |
| Total Strain (µε) a | 214,883 (27,918) | 147,951 (20,679) | 183,407 (25,559) | 200,766 (51,810) | 0.4693 | 0.7536 | 0.2239 |
| Modulus (GPa) | 2.27 (0.13) | 1.87 (0.32) | 3.46 (1.12) | 2.72 (0.19) | 0.3271 | 0.0605 | 0.3552 |
| Resilience (MPa) a,b | 2.53 (0.43) | 3.73 (1.06) | 3.63 (0.87) | 1.48 (0.40) | 0.201 | 0.4356 | 0.1628 |
| Toughness (MPa) a | 21.32 (2.47) | 14.97 (2.63) | 19.62 (3.12) | 19.45 (4.04) | 0.3274 | 0.6715 | 0.3618 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, J.R.; Sloan, K.; Cave, K.; Wallace, J.M.; Roper, R.J. Skeletal Deficits in Male and Female down Syndrome Model Mice Arise Independent of Normalized Dyrk1a Expression in Osteoblasts. Genes 2021, 12, 1729. https://doi.org/10.3390/genes12111729
Thomas JR, Sloan K, Cave K, Wallace JM, Roper RJ. Skeletal Deficits in Male and Female down Syndrome Model Mice Arise Independent of Normalized Dyrk1a Expression in Osteoblasts. Genes. 2021; 12(11):1729. https://doi.org/10.3390/genes12111729
Chicago/Turabian StyleThomas, Jared R., Kourtney Sloan, Kelsey Cave, Joseph M. Wallace, and Randall J. Roper. 2021. "Skeletal Deficits in Male and Female down Syndrome Model Mice Arise Independent of Normalized Dyrk1a Expression in Osteoblasts" Genes 12, no. 11: 1729. https://doi.org/10.3390/genes12111729